

Abstract
During focal cerebral ischemia, the detachment of astrocytes from the microvascular basal lamina is not completely explained by known integrin receptor expression changes. Here, the impact of experimental ischemia (oxygen–glucose deprivation (OGD)) on dystroglycan expression by murine endothelial cells and astrocytes grown on vascular matrix laminin, perlecan, or collagen and the impact of middle cerebral artery occlusion on αβ-dystroglycan within cerebral microvessels of the nonhuman primate were examined. Dystroglycan was expressed on all cerebral microvessels in cortical gray and white matter, and the striatum. Astrocyte adhesion to basal lamina proteins was managed in part by α-dystroglycan, while ischemia significantly reduced expression of dystroglycan both in vivo and in vitro. Furthermore, dystroglycan and integrin α6β4 expressions on astrocyte end-feet decreased in parallel both in vivo and in vitro. The rapid loss of astrocyte dystroglycan during OGD appears protease-dependent, involving an matrix metalloproteinase-like activity. This may explain the rapid detachment of astrocytes from the microvascular basal lamina during ischemic injury, which could contribute to significant changes in microvascular integrity.
Keywords: astrocytes, dystroglycan, endothelial cells, focal ischemia, microvessels, neurovascular unit
Introduction
Close apposition of the endothelium to astrocyte end-feet seems to be required for the integrity of the capillaries of the neurovascular unit within the central nervous system (CNS) (Risau and Wolburg, 1990; del Zoppo and Mabuchi, 2003). Adhesion to the intervening basal lamina accomplishes this proximity between the two cell types (del Zoppo and Mabuchi, 2003). β1-Integrins are expressed by the endothelium throughout the cerebral microvascular tree (Tagaya et al, 2001), but only integrin α6β4 has so far been identified on the astrocyte end-feet of select microvessels (Wagner et al, 1997). The expression of these two receptor groups is consistent with the presence of their ligands within the basal lamina matrix (Hamann et al, 1996; Wagner et al, 1997). Focal cerebral ischemia rapidly alters these relationships within cerebral microvessels, decreasing microvascular expression of the β1-integrins and integrin α6β4 as well as their matrix ligands. Hemorrhagic transformation is related to loss of the basal lamina matrix (Hamann et al, 1996), and permeability changes occur very early during focal ischemia (del Zoppo et al, 1986). The loss of integrin expression and matrix integrity also appears directly related to the degree of neuron injury, being greatest in the ischemic core (Tagaya et al, 2001). The ischemic core coincides with low regional cerebral blood flow, decreased metabolism, and neuron demise (Branston et al, 1976; Astrup et al, 1981). Under these conditions, detachment of astrocyte end-feet and astrocyte swelling are seen (Heo et al, 2005). The differences in integrin receptor expression between the two cell compartments suggest that other nonintegrin adhesion receptors could participate in maintaining microvessel integrity, particularly along the glial end-feet.
Recent evidence indicates that the adhesion receptor dystroglycan is also associated with cerebral or cerebellar microvessels (Zaccaria et al, 2001). The dystroglycan complex, an αβ-heterodimeric glycoprotein transmembrane receptor unrelated to the integrin family, forms a physical link between the intracellular cytoskeleton of select cells and the extracellular matrix (ECM). The α-subunit, a 120 to 190 kDa glycoprotein, is formed by posttranslational cleavage of the much longer β-subunit precursor, and interacts noncovalently with the 40 to 50 kDa transmembrane β-subunit product to form the active receptor (Durbeej and Campbell, 1999; Esapa et al, 2003). α-dystroglycan binds to the laminin α1 and α2 chains, domain V of perlecan (a heparin sulfate proteoglycan (HSPG)), agrin, and biglycan (Tian et al, 1996; Talts and Timpl, 1999; Andac et al, 1999; Friedrich et al, 1999; Montanaro and Carbonetto, 2003). Furthermore, dystroglycan can bind to a subset of the laminin–neurexin–sex hormone-binding globulin/laminin G domains of neurexins, which could mediate cell–cell adhesion (Sugita et al, 2001). Laminin serves as the substrate for dystroglycan receptor on Schwann cells in the peripheral nervous system and vascular smooth muscle cells (Yamada et al, 1996). The cytoplasmic tail of β-dystroglycan has been shown to interact with the C-terminus of the cytoplasmic proteins dystrophin and utrophin (Ervasti and Campbell, 1993; James et al, 1996).
Within the CNS, the precise functions of the αβ-dystroglycan complex have not been fully defined. In the mouse striatum, αβ-dystroglycan appears on neurons, glial cells, and blood vessels (Tian et al, 1996; Zaccaria et al, 2001). β-Dystroglycan has been found in the end-feet of Müller glial cells and the astrocyte end-feet of capillaries of the retinal outer plexiform layer (Koulen et al, 1998). But, it is currently unclear whether dystroglycan is expressed by all endothelial cells within the CNS. Recently, glial-selective loss of αβ-dystroglycan expression was shown to produce defects in the glia limitans (Moore et al, 2002). Those studies point to the potential importance of dystroglycan in intercellular relationships that involve astrocytes within the CNS. While alterations in dystrophin have been associated with cognitive dysfunction in muscular dystrophy (North et al, 1996), the potential role of microvessel dystroglycan in vascular dementias has not been entertained.
In view of the expression of dystroglycan in cerebral blood vessels and its receptor ligands laminin and HSPG within the basal lamina, we tested the hypothesis that αβ-dystroglycan could play a functional role in maintaining cerebral microvessel structural integrity, and that the dystroglycan complex could display, like select integrins within the cerebral microvasculature, sensitivity to focal ischemia. αβ-Dystroglycan expressions in the non-human primate and the mouse CNS were related to quantitative αβ-dystroglycan expression on primary murine astrocytes and endothelial cells. The relationship of astrocyte-associated dystroglycan to the normal astrocyte expression of the integrin β4 subunit was examined. In consequence, the impact of focal cerebral ischemia on microvessel αβ-dystroglycan expression in vivo was confirmed by experiments that detailed the effects of oxygen–glucose deprivation (OGD) on isolated astrocytes and endothelial cells in vitro. Taken together, this is the first demonstration that the astrocyte-associated dystroglycan complex participates in the integrity of select CNS microvessels (within the ‘neurovascular unit’), displays characteristic matrix ligand selectivity, and responds coordinately to focal ischemia by protease cleavage.
Materials and methods
All experimental procedures employed here were approved by the institutional (TSRI) Animal Care and Use Committee and performed according to the standards published by the National Research Council and the US Department of Agriculture Animal Welfare Act. All primates were judged free of infection or known inflammatory disorders before entry into any experiments.
Model of Focal Cerebral Ischemia
Samples of cerebral tissues from nine adolescent male baboons (Papio anubis/cynocephalus), prepared for other studies, were used (Hamann et al, 1996; Wagner et al, 1997; Tagaya et al, 2001). The surgical approach to the awake non-human primate stroke model has been previously described in detail. Brain tissues under normoxia, after 2 h middle cerebral artery occlusion (MCAO), and 7 days MCAO were removed after transcardiac perfusion with isosmotic heparinized perfusate under thiopental Na+, prepared for frozen and paraffin sections, and archived. Archived materials were used here.
Antibodies
The following antibodies were used in immunocytochemistry and function-blocking studies. Monoclonal antibodies (MoAb) raised against the human β4-integrin subunit (clone 439-9B, BD Pharmingen, La Jolla, CA, USA), the α-dystroglycan subunit (clone 11H6C4, IgM, Upstate Cell Signaling Solutions, Lake Placid, NY, USA), and the β-dystroglycan subunit (clone 43DAG/8D5, Novocastra, Newcastle, UK) were used. Polyclonal antibodies against laminin and glial fibrillary acidic protein glial fibrillary acidic protein were obtained from Sigma (St Louis, MO, USA). Antibodies to perlecan (rat monoclonal A7L6 and the mouse monoclonal 7E12) were obtained from Chemicon (Temecula, CA, USA). A polyclonal antibody against aquaporin-4 raised in rabbit (AB 3594, Chemicon) was employed for select colocalization experiments. The FITC-conjugated secondary antibodies for these studies were obtained from Jackson Immuno-Research Laboratories (West Grove, USA).
Cell Culture
Primary cultures of murine brain endothelial cells were prepared according to the method of Sapatino et al (1993) with modifications applied by this laboratory. The exact methods have been described recently by us (Milner et al, 2008). The brains of 2- to 3-month-old mice were removed, cleaned of meninges and external blood vessels, minced, and dissociated for 1 h in an enzymatic solution containing 30 U/mL papain (Worthington, Lakewood, NJ, USA), 0.24 mg/mL l-cysteine (Sigma), and 40 μg/mL DNAase I type IV (Sigma) in 1 mL MEM-HEPES (Milner and ffrench-Constant, 1994). Vascular tubes and cells were centrifuged away from other brain tissue products, then filtered through a 40 μm cell strainer (Falcon) to separate the vascular tubes. These were washed, centrifuged in MEM-HEPES, then resuspended in endothelial cell growth media (ECGM; Hams F12, supplemented with 10% fetal calf serum, heparin, ascorbic acid, l-glutamine (Sigma) and endothelial cell growth supplement (ECGS; Upstate Cell Signaling Solutions)). The vascular tubes were then added to type I collagen-coated T25 flasks and cultured at 37°C under 5% CO2. After 2 to 3 days, endothelial cells had dispersed out of the vascular tubes forming small colonies. Cell culture medium was replaced every 3 days and confluent endothelial cell cultures were obtained after 7 days. Cells passaged at a ratio of 1:3 were used for the first two passages only. These cell cultures were determined to be greater than 98% pure endothelial cells by flow cytometry using antibodies for the endothelial markers CD31 (PECAM-1) and CD105 (endoglin).
Nearly pure cultures of mouse astrocytes were obtained as described previously (Milner and ffrench-Constant, 1994) and by us (Milner et al, 2008). Brains from postnatal mice pups (less than 2 days old) were cleaned of meninges and external blood vessels, minced, and dissociated for 1 h in the same enzymatic solution as for endothelial cells. After trituration, cells were centrifuged and resuspended in DMEM containing 10% fetal calf serum, 4 mmol/L l-glutamine, penicillin, and streptomycin (Sigma), plated onto poly-D-lysine coated T75 flasks (Falcon), and cultured at 37°C and 5% CO2. After 10 days in culture, astrocyte monolayers were confluent and the flasks were mechanically shaken at 250 r.p.m. overnight to remove any attached microglia or oligodendrocytes. Astrocytes were used for the first two passages only. The purity of these astrocyte cultures was assessed by immunostaining with antibodies for glial fibrillary acidic protein (Sigma) and determined as greater than 98% pure.
Identification of Cells in Culture
For assessment of endothelial cell purity, endothelial cells were cultured on collagen I- or fibronectin-coated glass coverslips in ECGM. Cells were blocked in 5% normal goat serum (NGS) in phosphate-buffered saline (PBS) for 30 mins and then live-labeled with CD31-specific MoAb for 1 h at 20°C. Cells were washed in PBS, incubated with an FITC-conjugated antibody for 30 mins at 20°C, washed again, and fixed in acetic acid:ethanol (95:5, vol:vol) at −20°C for 30 mins. Cell nuclei were labeled with Hoechst stain (Sigma). Coverslips were washed, mounted in Aqua Poly/Mount (Polysciences Inc., Warrington, PA, USA), and the cell populations tabulated using video-imaging microscopy (see below). Astrocyte purity was assessed by staining fixed cell cultures with a glial fibrillary acidic protein-specific polyclonal antibody (Sigma).
Flow Cytometry
Mouse brain capillary endothelial cells or astrocytes were cultured in ECM-coated six-well plates (Nunc) with 10% fetal calf serum-supplemented or ECGM-supplemented DMEM, respectively. The ECM substrates (all Sigma) were prepared by coating individual wells with 10 μg/mL laminin, 10 μg/mL collagen IV, or 1 μg/mL perlecan for 2 h at 37°C, before being washed in PBS. After reaching confluence, cells were removed from the culture plates using a cell lifter, thereby avoiding the use of enzymes, such as trypsin, that cleave cell surface proteins. Cells were first fixed and permeabilized in Cytofix/Cytoperm (BD Pharmingen, La Jolla, CA, USA) in order for the β-dystroglycan MoAb to access the cytoplasmic binding site. Cells were then blocked in suspension in 5% normal goat serum in PBS for 30mins on ice, then transferred to wells of a 96-round-bottom-well plate (Nunc), and incubated for 1 h on ice with the MoAb 43DAG/8D5. Cells were washed twice in the blocking buffer and labeled with anti-mouse-FITC for 1 h on ice, then washed twice with blocking buffer before being resuspended in 2% formaldehyde in PBS. The fluorescent intensity of labeled cells was then analyzed on a Becton Dickinson FACScan machine (San Diego, CA, USA), with 10,000 events recorded for each condition. To investigate the influence of OGD on dystroglycan expression, the mean fluorescent intensity of cells exposed to OGD was compared with the control state (normoxia) and expressed as the percentage change relative to the control condition.
Cell Adhesion Assays
Using 24-well tissue culture dishes (Nunc) the central area of each well was coated with a 25 μL drop of fibronectin or laminin (10 μg/mL in PBS; both from Sigma) for 2 h at 37°C. The wells were washed twice with serum-free DMEM medium immediately before the addition of cells to remove unbound ECM. Astrocytes were centrifuged, resuspended in DMEM medium, and applied to the substrates in a 25 μL drop for 1 h at 37°C. The assays were stopped by adding 1 mL of DMEM to each well, washing off loosely attached cells, and fixing in 4% paraformaldehyde for 20 mins. Adhesion was quantified by counting all attached cells under phase microscopy, with a minimum of 500 cells counted for each substrate. The results were expressed as the percentage of cells adhering under control conditions.
In the antibody-blocking experiments, the antagonist was added to the medium used to resuspend the cells before plating so that it was present when the cells were added to the ECM substrate and throughout the experiment. The MoAb Ha 2/5 and its respective isotype control were used at 5 μg/mL.
Oxygen–Glucose Deprivation
Conditions for ex vivo OGD were standardized, and lasted 18 h. At cell confluence, culture media was changed from normal (high glucose, 4.5 g/L, supplemented) MEM-HEPES to low-glucose (1.0 g/L, supplemented) media. Cells in low-glucose medium were placed into a hypoxia chamber (Billups-Rothenburg, Del Mar, CA, USA), which was flushed through with a mixture of 95% N2 and 5% CO2 for 1 h, and then closed for the duration of the experiment. O2 levels decreased to < 3% after 2 h and < 1% after 4 h, at which they were maintained for the rest of the experiment.
Cell Viability Assay
Endothelial cells and astrocytes cultured on laminincoated glass cover slips for 3 days were either maintained under standard growth conditions or exposed to OGD for 18 h. Afterward, the cells were incubated with 1 μg/mL propidium iodide (Sigma) for 15 mins at 37°C, washed in PBS and fixed at −20°C for 20 mins, before labeling with Hoechst stain for 5 mins. The percentage of dead or dying cells was assessed by calculating the total cell proportion (Hoechst positive) that were also propidium iodide positive.
Cell Lysates
Astrocyte and endothelial cell lysates were prepared using lysis buffer containing 50 mmol/L Tris-Cl, 1% Triton X-100, 0.005% Brij35, 150 mmol/L NaCl, 0.05% sodium azide, 0.1mol/L l-arginine, 10 mmol/L EDTA, and working concentrations of protease inhibitor (Sigma). Cell lysates were subsequently aliquoted and stored at −80°C. Protein concentration was determined using the BioRad DC Protein Assay according to the manufacturer's instructions.
Electrophoresis and Immunoblots
Cell lysates were reduced with 1 × dithiothreitol (Invitrogen, Carlsbad, CA, USA) and loaded at 3 μg total protein/lane. Samples were run on precast 12% Tris-Glycine (Invitrogen) at 200 V for 1 h. Proteins were electroblotted for 1 h onto a polyvinylidene fluoride membrane (Millipore), blocked for 1 h with 1% BSA in PBS containing 0.1% Tween 20 (Sigma), and probed with 43DAG/8D5 primary antibody against the C-terminus of β-dystroglycan (Novocastra) and secondary antibody peroxidase-conjugated AffiniPure goat anti-mouse-IgG F(ab')2 specific antibody (Jackson Immuno-Research Laboratories) for 1 h. Polyvinylidene fluoride membranes were washed extensively in PBS-T, and the proteins were identified with an enhanced chemiluminescence detection system (ECL; Pierce) according to the manufacturer's instructions.
RNA Extraction and Quantitative Real-Time RT-PCR
Total RNA was extracted from astrocytes subjected to normoxia or OGD using the RNeasy Plus Mini kit (Qiagen, Valencia, CA, USA). Eluted RNA was treated with DNA-free DNase (Ambion, Austin, TX, USA). The RT-PCR reaction was set up according to the SuperScript III Platinum One-Step Quantitative RT-PCR System protocol (Invitrogen). The final reaction conditions for RT-PCR were 200 nmol/L per primer, 100 nmol/L per probe, 200 μmol/L of each dNTP, 5 mmol/L MgSO4, and 100 ng total RNA. The PCR cycling conditions were 45 cycles of 95°C for 30 secs and 60°C for 30 secs. The mouse dystroglycan sequence information was obtained from the GenBank database (BC007150). Primers and TaqMan probes were designed with primer3 software. Quantification of the amount of target in the unknown samples was accomplished using a standard curve to determine the starting concentration of the target. Gene expression was normalized through dividing the starting concentration of the target by the starting concentration of constitutively expressed beta-actin. The standard curve was generated using QPCR mouse reference total RNA (Stratagene).
Immunohistochemistry
Display of antigens of interest in the cerebral microvessels of 10 μm frozen sections of brain tissues from the non-human primate and the mouse was achieved as previously reported (Tagaya et al, 2001; Fukuda et al, 2004).
Analyses of Data
All data derived from tissue experiments and the cell culture experiments are presented as the mean ± s.d.. For flow cytometry studies, each experiment was repeated a minimum of four times and the data expressed as mean ± s.d. For the adhesion assays within each experiment each condition was performed in duplicate; the results represent the mean ± s.e.m. of three experiments. Comparisons were made using the Student's paired or unpaired t-test, where appropriate. Significance was set at P < 0.05 or 2P < 0.05 where appropriate.
Results
Dystroglycan Expression in the Cerebral Microvasculature
The α- and β-subunits of dystroglycan are both expressed throughout the entire cerebral microvasculature of the normal corpus striatum (Figure 1), and the cerebral cortex of the non-human primate and the mouse. Although both subunits appeared on equivalent numbers of immunoreactive microvessels, the intensity of immunoreactivity of the transmembrane β-dystroglycan routinely exceeded that of the α-subunit. Hence, data from the β-subunit are shown.
Figure 1.
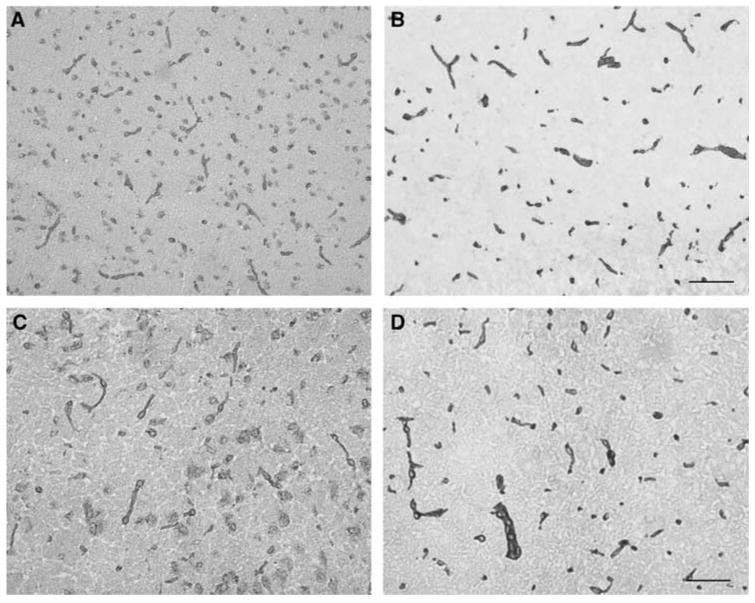
Expression of α-dystroglycan (A and C) and β-dystroglycan (B and D) on normal microvessels within the striatum of the non-human primate (A and B) and C57Bl/6 mice (C and D). Scale bars = 100 μm (A and B) and 50 μm (C and D).
Within the cerebral cortex, the microvessel density (microvessels/mm2) expressing β-dystroglycan was greater in the gray matter than white matter (Figures 3 and 4). In the cortical gray matter (primate), immunoreactive β-dystroglycan was uniformly found on capillaries (95.3% ± 0.6% of microvessels 4.0 to 7.5 μm in diameter), with the remainder primarily associated with 7.5 to 30.0 μm diameter microvessels (Figure 4A). This is in keeping within the known distribution of microvessels within these territories.
Figure 3.
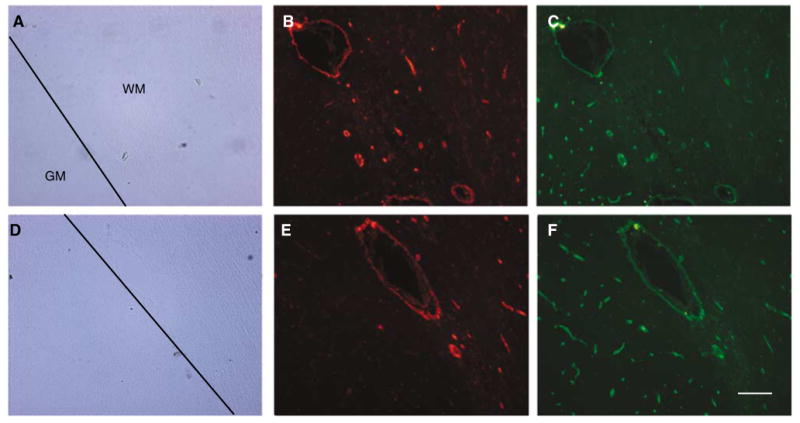
Vascular coexpression of β-dystroglycan and the integrin β4 subunit in cortical gray matter (GM) and white matter (WM). Two representative fields (A to C and D to F) show the appearance of β-dystroglycan (C and F) in both GM and WM, but β4 (B and E) primarily in WM. Scale bar = 100 μm.
Figure 4.

Microvessel distributions of β-dystroglycan and the integrin β4 subunit expression in the cortical gray matter (A) and white matter (B), and the striatum (C) of the non-human primate. β-Dystroglycan is found on all microvessels in the white and gray matter, whereas integrin β4 is on all microvessels in the white matter only (B). Solid line = β-dystroglycan, broken line = integrin subunit β4.
In striatal microvessels, immunoreactive β-dystroglycan was found on the abluminal surface of the basal lamina, suggesting that it is expressed mostly at the astrocyte end-feet (Figures 2A to 2C). This is supported by the nearly identical microvessel expression of immunoreactive β-dystroglycan and aquaporin-4 on the microvasculature of normoxic gray matter (Figures 2J to 2L).
Figure 2.
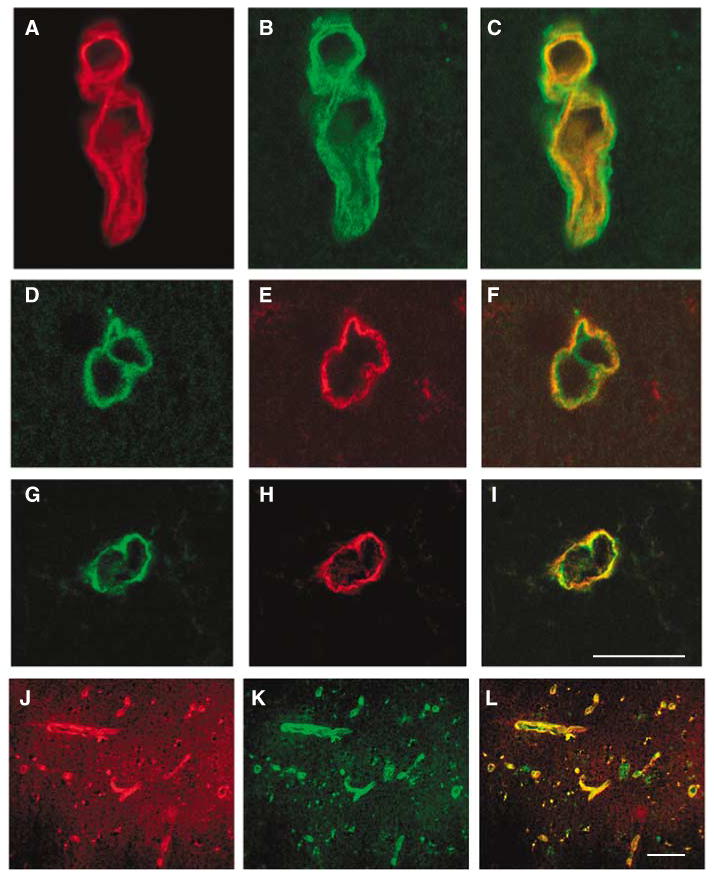
Expression of β-dystroglycan and the integrin β4 subunit relative to HSPG (perlecan) on select cerebral microvessels of the nonhuman primate in normoxic gray matter. Individual fluorescence channels of confocal microscopy are merged in (C), (F), (I), and (L). The expression of β-dystroglycan (B) appears outside HSPG (A), and subunit β4 (E) appears outside HSPG (D), in representative experiments. Both β-dystroglycan (H) and subunit β4 (G) are coexpressed by the same vascular structures. Generally, aquaporin-4 (J) and β-dystroglycan (K) appear to be colocalized to most microvessels in the gray matter. Scale bars = 50 μm.
Regional Specialization of Integrin β4 and β-Dystroglycan Coexpression
As integrin α6β4 is expressed on astrocyte end-feet in striatal tissue (Wagner et al, 1997), the relative expressions of astrocyte dystroglycan and α6β4 integrin are of interest (Figures 2D to 2I and Figure 3). As shown in Figures 3 and 4, there are significant differences in the relative distribution of β-dystroglycan and the integrin α6β4. β-dystroglycan was expressed by microvessels throughout both the white and gray matter, but integrin α6β4 was coexpressed with β-dystroglycan by nearly all microvessels only in the white matter. There was nearly 88.9% concordance in the expression of β-dystroglycan and integrin β4 on all capillaries in cortical white matter (Figure 4B). In cortical gray matter, β-dystroglycan was found on 80.5% of capillaries, whereas integrin β4 appeared on only 45.8% of capillaries (Figures 3 and 4C), primarily on penetrating vessels of the outer cortex. The coexpression in the striatum was similar to cortical gray matter. This differential distribution shows a significant regional specialization of astrocyte matrix receptor expression.
Cell Selectivity of Dystroglycan Expression
β-Dystroglycan was expressed on both murine primary astrocytes and cerebral endothelial cells grown separately on type IV collagen, HSPG, or laminin, all components of the cerebral microvascular basal lamina. However, β-dystroglycan was expressed consistently at 3.3- to 4.1-fold higher levels on astrocytes than endothelial cells on all three matrix substrates, as detected by flow cytometry (Figure 5).
Figure 5.

Expression of β-dystroglycan by cultured murine cerebral endothelial cells and astrocytes on collagen type IV (solid bar), HSPG/perlecan (hatched), and laminin (reverse hatched). For each matrix ligand, astrocytes expressed 3.3- to 4.1-fold more dystroglycan than endothelium (P < 0.0001 each). Inset: primary astrocytes contained both 51 and 31 kDa forms of β-dystroglycan, whereas endothelial cells expressed the 51 kDa form predominantly.
In the absence of injury, astrocytes expressed slightly more of the 51 kDa form of β-dystroglycan than endothelial cells, but considerably more of the 31 kDa form. Quantification of immunoblots showed that overall expression of β-dystroglycan by primary astrocytes exceeded that of endothelial cells by 2.4-fold (Figure 5, inset). Fractionation studies showed that 95.9% to 99.7% of the β-dystroglycan on endothelial cells or astrocytes was found in the membrane fraction (data not shown).
β-Dystroglycan on Astrocytes is a Functional Laminin Receptor
The extracellular α-dystroglycan subunit is required for successful interaction of this receptor with its matrix ligands (Montanaro and Carbonetto, 2003). To determine whether astrocyte adhesion to laminin could be mediated by dystroglycan, adhesion assays for α-dystroglycan function were performed. Blockade of α-dystroglycan significantly reduced astrocyte adhesion to laminin (Figure 6) but had no effect on adhesion to fibronectin, a non-dystroglycan ligand. Furthermore, astrocyte adhesion to laminin was only partly mediated by α-dystroglycan, consistent with the participation of other (unknown) adhesion receptors in binding astrocyte end-feet to the microvascular basal lamina.
Figure 6.
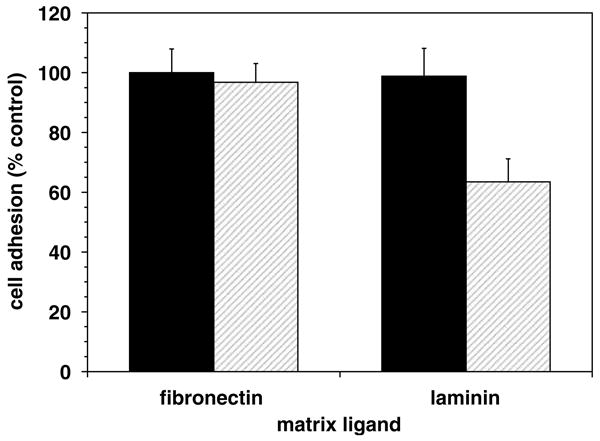
Inhibition of α-dystroglycan-mediated astrocyte–matrix adhesion. The anti-α-dystroglycan MoAb 11H6C4 significantly reduced astrocyte adhesion to laminin (P = 0.034; n = 3), but not to fibronectin.
Cerebral Microvascular Dystroglycan Expression is Lost during Focal Ischemia
Based on the premise that α-dystroglycan is a functional astrocyte-vascular matrix receptor, the response of the dystroglycan complex to focal cerebral ischemia was determined. Within 2 h after MCAO, expression of both α- and β-dystroglycan decreased significantly on cerebral capillaries in the primate striatum (a 74.6% ± 26.2% reduction in capillary expression; data not shown) (Figure 7).
Figure 7.
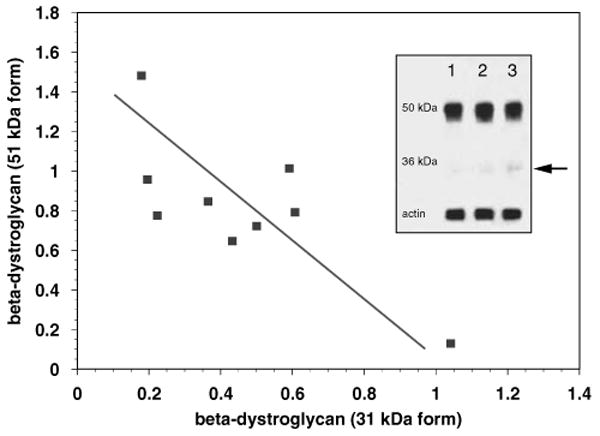
A direct inverse correlation between the loss of the 51 kDa β-dystroglycan and the appearance of the 31 kDa form was observed in the striatum after 2 h MCAO and 7 days MCAO compared with nonischemic tissue (r = −0.7570, P = 0.009; n = 3 each). Inset: immunoblot of striatum at 0 h (lane 1), 2 h (lane 2), and 7 days (lane 3) after MCAO (see Materials and methods). Arrow = 31 kDa form.
Furthermore, within the ischemic tissue, a significant reduction in the 51 kDa β-dystroglycan beginning at 2 h MCAO inversely correlated with the appearance of the 31 kDa form (Figure 7, inset). This is consistent with previous data showing that after tissue injury of the 51 kDa form, β-dystroglycan is cleaved to the 31 kDa form (Agrawal et al, 2006).
OGD Reduces Astrocyte Dystroglycan Expression
To determine whether ischemia per se drives the loss in microvessel dystroglycan expression, both primary murine astrocytes and endothelial cells were subjected to standardized experimental ischemia (OGD). A significant differential sensitivity to the ischemic insult was seen between endothelial cells and astrocytes (Figure 8). Astrocytes grown on type IV collagen, perlecan, or laminin showed a significant loss of β-dystroglycan expression during OGD (Figures 8 and 9A), compared with the minor loss of β-dystroglycan expression from endothelial cells. Cell survival experiments showed that the loss of β-dystroglycan on astrocytes during OGD was not because of cell death (Figure 8, inset), suggesting selective receptor sensitivity to ischemia. Interestingly, in response to OGD, primary astrocytes grown on perlecan or laminin displayed no change in expression of the 31 kDa form of β-dystroglycan, whereas a nearly three-fold increase in this form was observed when cells were grown on collagen IV (Figure 9B).
Figure 8.
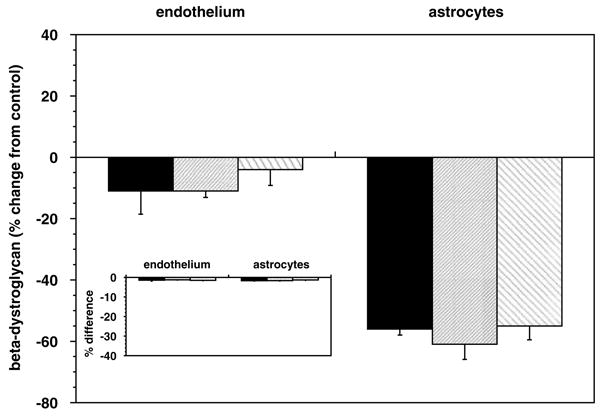
β-Dystroglycan expression on astrocytes was significantly reduced by OGD when the cells were grown on collagen type IV (black), HSPG (fine hatched), or laminin (coarse hatched). A nonsignificant reduction in endothelial cell expression was seen (n = 3 each). Inset: no substantial evidence of cell demise (by propidium iodide) was observed between cells subject to OGD and normoxia.
Figure 9.
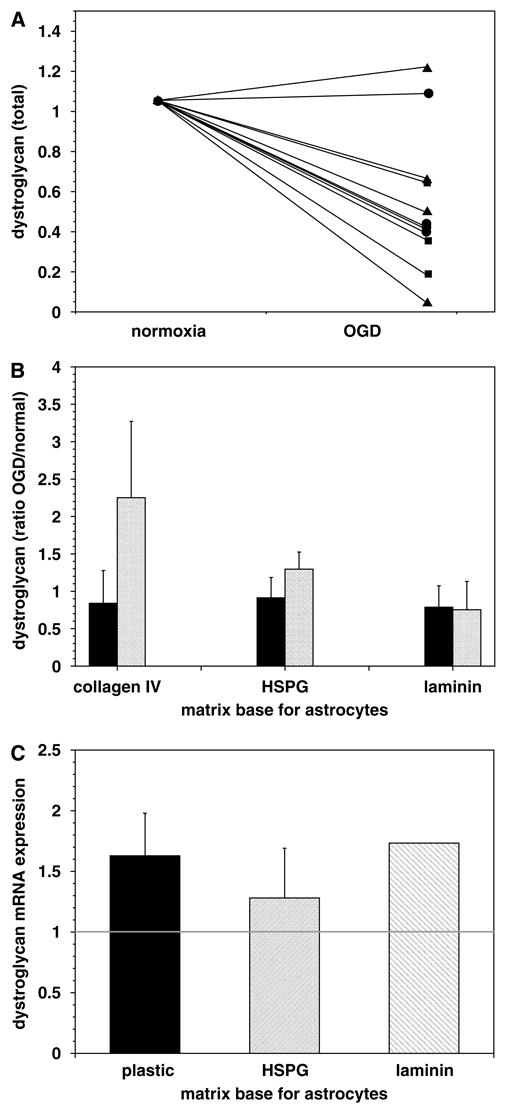
Impact of OGD on astrocyte expression of β-dystroglycan protein and transcripts. Change in astrocyte β-dystroglycan content because of OGD when cells were grown on collagen IV (squares), HSPG (circles), or laminin (triangles), from immunoblots (n = 4 each) (A). OGD produced a significant increase in the appearance of the 31 kDa form of β-dystroglycan by astrocytes grown on collagen IV compared with normoxia (P = 0.046) (depicted are the ratios OGD/normoxia of each form; solid = 51 kDa form, hatched = 31 kDa form) (B). Increase in the relative transcription of β-dystroglycan mRNA induced in astrocytes grown on plastic and laminin, which were subject to OGD (2P = 0.007 and 0.006, respectively; n = 6 each) (ratio = OGD/normoxia) (C).
To determine whether reduced transcription decreased dystroglycan expression, β-dystroglycan mRNA levels were examined by real-time RT-PCR. Transcription of the β-dystroglycan gene increased when astrocytes cultured on HSPG, laminin, or plastic were exposed to OGD (Figure 9C).
Cleavage of Dystroglycan during OGD
To test whether the rapid loss of dystroglycan from microvessels could be explained by proteolytic cleavage, primary astrocytes grown on collagen IV or laminin were exposed to the nonspecific matrix metalloproteinase (MMP) inhibitor GM6001 during OGD. A dose-dependent complete inhibition of the loss in β-dystroglycan expression compared with OGD alone was observed (data not shown). Extended studies show that inhibition of serine proteases (aprotinin) or cysteine proteases (E64c, E64d) had no effect on the loss of β-dystroglycan expression, whereas both GM6001 and 1,10-phenanthroline (an MMP inhibitor) reversed the loss of β-dystroglycan expression (Figure 10).
Figure 10.
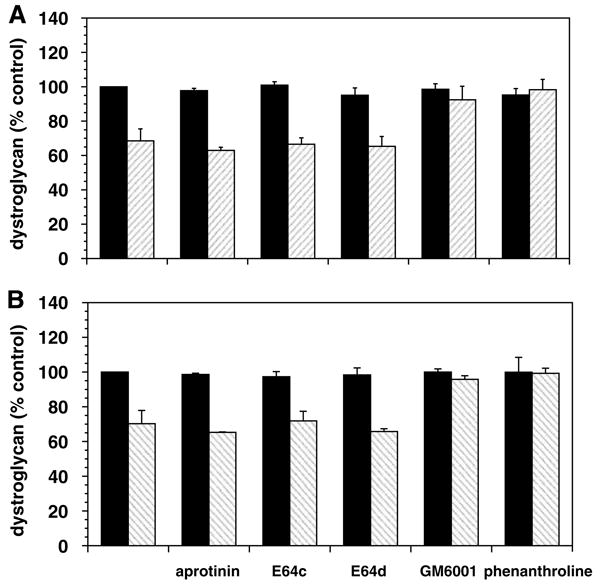
Preservation of β-dystroglycan expression by astrocytes exposed to various protease inhibitors (n = 3 each). Cells grown on collagen IV (A) or laminin (B) were maintained under normoxia (black bars) or exposed in parallel to OGD (hatched bars), in the absence (first data pair) or presence (subsequent pairs) of each protease inhibitor. OGD significantly decreased astrocyte β-dystroglycan expression relative to normoxia (collagen IV, P = 0.015; laminin, P < 0.01) for all inhibitors except GM6001 and 1,10-phenanthroline (P > 0.10 each).
Discussion
Focal cerebral ischemia rapidly disturbs the ultrastructure and functional integrity of microvessels within the injured vascular territory (del Zoppo and Mabuchi, 2003; Heo et al, 2005). These alterations coincide with the early evidence of neuron dysfunction and metabolic disturbances within the ischemic regions (Branston et al, 1976; Astrup et al, 1981; Tagaya et al, 2001). Tagaya et al (2001) showed the rapid disappearance of β1-integrin immunoreactivity from cerebral microvascular endothelium 1 to 2 h after the onset of MCAO within the ischemic core. Simultaneously, the expression of integrin α6β4, found exclusively on astrocyte end-feet of select microvessels, also decreases in this interval, coinciding with the detachment of astrocyte end-feet from the basal lamina matrix early after MCAO (Wagner et al, 1997). However, the distribution of integrin α6β4 on only a subset of microvessels suggests the participation of other adhesion receptor families in the binding of astrocytes to the cerebrovascular matrix (e.g., laminins and HSPG). This raises questions about their identity, localization and density, the relative sensitivity of their functional activity to ischemia, and the mechanisms of receptor loss.
This study confirms that αβ-dystroglycan is expressed on resting cerebral microvessels, indicating that astrocytes are the primary cellular source of this receptor. Furthermore, astrocyte-associated dystroglycan and the integrin α6β4 are found on overlapping microvessel subsets depending on tissue localization. Dystroglycan is principally localized to astrocyte end-feet in both cortical and striatal gray matter, where it is positioned to assist adherence of astrocytes to the vascular basal lamina matrix, and could assist in microvascular integrity. To support this notion, we show (i) the exact location of the dystroglycan complex in cerebral tissue, and that (ii) astrocyte-associated α-dystroglycan acts as a functional laminin receptor, (iii) focal cerebral ischemia causes a significant loss of astrocyte dystroglycan expression, (iv) this loss is mimicked by primary astrocytes subject to OGD, and (v) the loss in astrocyte dystroglycan expression involves metalloproteinase-like activities.
Based on a survey of integrin subunit expression within cerebral microvessels of the non-human primate, it was presumed that β1-integrins mediate endothelial adhesion to the basal lamina matrix, while integrin α6β4 binds astrocyte end-feet to the basal lamina (e.g., laminin-5) in cerebral microvessels (Wagner et al, 1997; Tagaya et al, 2001). But, two additional findings are important: (i) the rapid loss of integrin β1 from endothelial cells does not cause detectable detachment of the endothelium from the microvascular matrix (Tagaya et al, 2001), and (ii) integrin α6β4 is found on a minority of microvessels of capillary diameter (Wagner et al, 1997). Both observations suggest that other matrix receptors could be involved in the adhesion of endothelial cells and astrocyte end-feet to the basal lamina in cerebral capillaries.
The dystroglycan complex appears throughout the cortical and striatal microvasculature of normal non-human primate brain, providing another adhesion receptor family, in addition to the well-characterized integrins. Both α- and β-dystroglycan are expressed by endothelial cells and astrocytes in vitro as well. The finding that β-dystroglycan is expressed on astrocyte end-feet in all cerebral microvessels whereas α6β4-integrin is expressed on only a fraction suggests that the dystroglycan complex could be the principal adhesion receptor system for anchoring astrocytes to the vascular basal lamina. In terms of cell compartment, β-dystroglycan is expressed at four-fold higher levels on cultured murine astrocytes than endothelial cells, in keeping with observations of its microvascular expression in cerebral tissues of both species. These findings clarify recent reports of αβ-dystroglycan in cerebral microvessels (Zaccaria et al, 2001). The receptor is associated variably with astrocytes and endothelial cells in the CNS. These studies do not clarify to what degree dystroglycan is effective in maintaining adherence of endothelial cells to the subtending basal lamina matrix. Confocal microscopy confirmed the association of dystroglycan with astrocyte end-feet, mostly exterior to perlecan within larger cerebral microvessels. Dystroglycan and integrin α6β4 are differentially expressed on astrocyte end-feet in a very specific manner. Whereas dystroglycan is found in all microvessel classes in gray and white matter, integrin α6β4 is found on all microvessels in the white matter, but on only a small proportion in the gray matter. The reasons for this distribution are not yet known.
Dystroglycan shares laminin and HSPG (perlecan) as primary vascular matrix ligands with β1-integrins and integrin α6β4 (laminin-5) (Gesemann et al, 1996; Wagner et al, 1997; Talts et al, 1999; Tagaya et al, 2001). α-Dystroglycan binds to the C-terminal domain V of perlecan (Friedrich et al, 1999), to the L4 and L5 modules of the mouse laminin α1 chain (Andac et al, 1999), and to the L3 module of the laminin α2 chain (Jucker et al, 1996; Talts and Timpl, 1999). The latter is independent of β1-integrin binding. In the studies reported here, rapid loss of the dystroglycan complex after MCAO implies its potential role in astrocyte adhesion to the matrix. Astrocyte-associated α-dystroglycan behaves as a functional laminin receptor, although functional blockade of the α-dystroglycan subunit only partly prevented astrocyte adherence to laminin in vitro in our hands. A similar adhesion function may exist for αβ-dystroglycan on endothelial cells, but given the lower receptor density and the role of β1-integrins as laminin receptors on microvascular endothelial cells, their functions may be different.
Focal ischemia interferes with astrocyte–basal lamina apposition, resulting in rapid detectable swelling of the end-feet and its detachment from the matrix (Heo et al, 2005). The mechanisms for end-foot detachment are unknown, although we had formerly proposed that loss of integrin α6β4 was a necessary event (Wagner et al, 1997). The predominant expression of the dystroglycan complex on astrocyte end-feet of capillaries and its sensitivity to ischemia offer another possibility, particularly in view of the reported codistribution of dystroglycan, aquaporin-4, and Kir 4.1 in the perivascular domain (Guadagno and Moukhles, 2004; Warth et al, 2005). The general colocalization of β-dystroglycan and aquaporin-4 on the normoxic microvasculature, seen here (Figures 2J to 2L), supports this relationship. Murine astrocytes grown on type IV collagen, laminin, or perlecan suffer a significant loss of αβ-dystroglycan expression when exposed to glucose and O2 deprivation in vitro, whereas endothelial cells displayed little change in dystroglycan expression on any substrate, in keeping with their relative resistance to ischemia. In vivo, end-foot swelling and detachment could represent separate but related effects of ischemia on aquaporin-4, Kir 4.1, and/or elements of dystrophin–β-dystroglycan–α-syntrophin in the dystrophin-associated protein complex in astrocytes (Neely et al, 2001). The ultrastructural relation of aquaporin-4 to α-syntrophin (and dystroglycan) in astrocytes, but not endothelial cells (Bragg et al, 2006), could underlie the altered water regulation of this part of the neurovascular unit. The impact of dystroglycan loss on the endothelial permeability barrier is not known.
The loss of dystroglycan expression after MCAO coincides with the rapid appearance of the activities of several protease families, including MMPs, cathepsin L, and heparanase in vivo (Fukuda et al, 2004). The loss of astrocyte-associated β-dystroglycan was inhibited by GM6001 and 1,10-phenanthroline, but not other protease inhibitors, suggesting the involvement of MMP-like activity in the loss of dystroglycan immunoreactivity. Recently, Yamada et al (2001) reported that β-dystroglycan on skeletal muscle cells is processed to the shorter 31 kDa form by a metalloproteinase-like activity. Very recently, Agrawal et al (2006) showed the ability of macrophage-associated proteases, inhibitable by 1,10-phenanthroline, to cleave astrocyte-associated β-dystroglycan to the 31 kDa product (Agrawal et al, 2006). Herzog et al (2004) showed that β-dystroglycan is shed from cutaneous epithelial cells by a gelatinase activity consistent with either active MMP-2 or MMP-9. The nature of this MMP-like activity and its source(s) have not been identified. However, pro-MMP-2, pro-MMP-9, MMP-14, MMP-16, and the activators of the latent MMPs (Chang et al, 2003), as well as MMP-3 (Rosenberg et al, 2001), are rapidly expressed in the ischemic regions after MCAO in several model systems.
After MCAO, an inverse relation between the intact β-dystroglycan subunit and the 31 kDa fragment was observed in samples of striatal tissue. Importantly, during OGD, the 31 kDa form of β-dystroglycan on astrocytes was significantly lower when the cells were grown on the α-dystroglycan ligands laminin and HSPG, compared with collagen IV (Figure 9). It has been suggested that binding of the dystroglycan complex to its native ligands protects the β-subunit from proteolytic cleavage (Herzog et al, 2004). This would explain the matrix dependence of β-dystroglycan cleavage from cultured astrocytes in these studies. But, it does not explain the general loss of surface expression in response to OGD, which appears to be not because of downregulation of transcription. Conversely, the protection of β-dystroglycan cleavage could explain the modest generation of the 31 kDa form observed in striatal tissue at 2 h and later after MCAO, when microvessel perlecan is more rapidly degraded than the abundant laminin and collagen.
Taken together, these studies show that dystroglycan is a principal matrix adhesion receptor on astrocyte end-feet within cerebral microvessels, and that its expression is exquisitely sensitive to both focal cerebral ischemia and experimental ischemia. While the time courses of astrocyte dystroglycan and integrin α6β4 loss coincide, their expression patterns in the cerebral microvasculature of gray matter and white matter differ. The purpose of this differential expression is unknown. Loss of β-dystroglycan immunoreactivity from primary astrocytes coincides with the appearance of its 31 kDa form, which is generated when inhibitable metalloproteinase-like activity is detected. Decreased dystroglycan expression and the increase in the 31 kDa form also coincide with detachment of astrocytes from the basal lamina and the appearance of pro-MMP-2 very early during focal ischemia. These events are consistent with a model in which cleavage of the dystroglycan complex leads to disruption of astrocyte–matrix interactions and consequent loss of capillary integrity, which may then contribute to the induction of neuron injury.
Acknowledgments
This is manuscript number 18751-MEM from the Scripps Research Institute. The studies were performed with the kind support of R01 NS26945, R01 NS53716, and R37 NS38710 from the National Institutes of Health. Appreciation is also given to the Stein Endowment Fund which underwrote a portion of the costs of the gene transcription studies.
References
- Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007–19. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andac Z, Sasaki T, Mann K, Brancaccio A, Deutzmann R, Timpl R. Analysis of heparin, alpha-dystroglycan and sulfatide binding to the G domain of the laminin alpha1 chain by site-directed mutagenesis. J Mol Biol. 1999;287:253–64. doi: 10.1006/jmbi.1999.2606. [DOI] [PubMed] [Google Scholar]
- Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12:723–5. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- Bragg AD, Amiry-Moghaddam M, Ottersen OP, Adams ME, Froehner SC. Assembly of a perivascular astrocyte protein scaffold at the mammalian blood–brain barrier is dependent on alpha-syntrophin. Glia. 2006;53:879–90. doi: 10.1002/glia.20347. [DOI] [PubMed] [Google Scholar]
- Branston NM, Symon L, Crockard HA. Recovery of the cortical evoked response following middle cerebral artery occlusion in baboons: relation to local blood flow and PO2. Stroke. 1976;7:151–7. doi: 10.1161/01.str.7.2.151. [DOI] [PubMed] [Google Scholar]
- Chang DI, Hosomi N, Lucero J, Heo JH, Abumiya T, Mazar AP, del Zoppo GJ. Activation systems for matrix metalloproteinase-2 are upregulated immediately following experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:1408–19. doi: 10.1097/01.WCB.0000091765.61714.30. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Copeland BR, Harker LA, Waltz TA, Zyroff J, Hanson SR, Battenberg E. Experimental acute thrombotic stroke in baboons. Stroke. 1986;17:1254–65. doi: 10.1161/01.str.17.6.1254. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–94. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- Durbeej M, Campbell KP. Biochemical characterization of the epithelial dystroglycan complex. J Biol Chem. 1999;274:26609–16. doi: 10.1074/jbc.274.37.26609. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–23. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esapa CT, Bentham GR, Schroder JE, Kroger S, Blake DJ. The effects of post-translational processing on dystroglycan synthesis and trafficking. FEBS Lett. 2003;555:209–16. doi: 10.1016/s0014-5793(03)01230-4. [DOI] [PubMed] [Google Scholar]
- Friedrich MV, Gohring W, Morgelin M, Brancaccio A, David G, Timpl R. Structural basis of glycosaminoglycan modification and of heterotypic interactions of perlecan domain V. J Mol Biol. 1999;294:259–70. doi: 10.1006/jmbi.1999.3259. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL, Jr, del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke. 2004;35:998–1004. doi: 10.1161/01.STR.0000119383.76447.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesemann M, Cavalli V, Denzer AJ, Brancaccio A, Schumacher B, Ruegg MA. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–67. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- Guadagno E, Moukhles H. Laminin-induced aggregation of the inwardly rectifying potassium channel, Kir4.1, and the water-permeable channel, AQP4, via a dystroglycan-containing complex in astrocytes. Glia. 2004;47:138–49. doi: 10.1002/glia.20039. [DOI] [PubMed] [Google Scholar]
- Hamann GF, Okada Y, del Zoppo GJ. Hemorrhagic transformation and microvascular integrity during focal cerebral ischemia/reperfusion. J Cereb Blood Flow Metab. 1996;16:1373–8. doi: 10.1097/00004647-199611000-00036. [DOI] [PubMed] [Google Scholar]
- Heo JH, Han SW, Lee SK. Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med. 2005;39:51–70. doi: 10.1016/j.freeradbiomed.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Herzog C, Has C, Franzke CW, Echtermeyer FG, Schlotzer-Schrehardt U, Kroger S, Gustafsson E, Fassler R, Bruckner-Tuderman L. Dystroglycan in skin and cutaneous cells: beta-subunit is shed from the cell surface. J Invest Dermatol. 2004;122:1372–80. doi: 10.1111/j.0022-202X.2004.22605.x. [DOI] [PubMed] [Google Scholar]
- James M, Nguyen TM, Wise CJ, Jones GE, Morris GE. Utrophin–dystroglycan complex in membranes of adherent cultured cells. Cell Motil Cytoskeleton. 1996;33:163–74. doi: 10.1002/(SICI)1097-0169(1996)33:3<163::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Jucker M, Tian M, Norton DD, Sherman C, Kusiak JW. Laminin α2 is a component of brain capillary basement membrane: reduced expression in dystrophic dy mice. Neuroscience. 1996;71:1153–61. doi: 10.1016/0306-4522(95)00496-3. [DOI] [PubMed] [Google Scholar]
- Koulen P, Blank M, Kroger S. Differential distribution of beta-dystroglycan in rabbit and rat retina. J Neurosci Res. 1998;51:735–47. doi: 10.1002/(SICI)1097-4547(19980315)51:6<735::AID-JNR7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Milner R, ffrench-Constant C. A developmental analysis of oligodendroglial integrins in primary cells: changes in αv-associated β subunits during differentiation. Development. 1994;120:3497–506. doi: 10.1242/dev.120.12.3497. [DOI] [PubMed] [Google Scholar]
- Milner R, Hung S, Wang X, Berg GI, Spatz M, del Zoppo GJ. Responses of endothelial cell and astrocyte matrix–integrin receptors to ischemia mimic those observed in the neurovascular unit. Stroke. 2008 doi: 10.1161/STROKEAHA.107.486134. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanaro F, Carbonetto S. Targeting dystroglycan in the brain. Neuron. 2003;37:193–6. doi: 10.1016/s0896-6273(03)00032-1. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–5. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc Natl Acad Sci USA. 2001;98:14108–13. doi: 10.1073/pnas.241508198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North KN, Miller G, Iannaccone ST, Clemens PR, Chad DA, Bella I, Smith TW, Beggs AH, Specht LA. Cognitive dysfunction as the major presenting feature of Becker's muscular dystrophy. Neurology. 1996;46:461–5. doi: 10.1212/wnl.46.2.461. [DOI] [PubMed] [Google Scholar]
- Risau W, Wolburg H. Development of the blood–brain barrier. Trends Neurosci. 1990;13:174–8. doi: 10.1016/0166-2236(90)90043-a. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Sullivan N, Esiri MM. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. 2001;32:1162–8. doi: 10.1161/01.str.32.5.1162. [DOI] [PubMed] [Google Scholar]
- Sapatino BV, Welsh CJ, Smith CA, Bebo BF, Linthicum DS. Cloned mouse cerebrovascular endothelial cells that maintain their differentiation markers for factor VIII, low density lipoprotein, and angiotensin-converting enzyme. In vitro Cell Dev Biol Anim. 1993;29A:923–8. doi: 10.1007/BF02634230. [DOI] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Sudhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–45. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagaya M, Haring HP, Stuiver I, Wagner S, Abumiya T, Lucero J, Lee P, Copeland BR, Seiffert D, del Zoppo GJ. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. 2001;21:835–46. doi: 10.1097/00004647-200107000-00009. [DOI] [PubMed] [Google Scholar]
- Talts JF, Andac Z, Göhring W, Brancaccio A, Timpl R. Binding of the G domains of laminin alpha1 and alpha2 chains and perlecan to heparin, sulfatides, alpha-dystroglycan and several extracellular matrix proteins. EMBO J. 1999;18:863–70. doi: 10.1093/emboj/18.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talts JF, Timpl R. Mutation of a basic sequence in the laminin alpha2LG3 module leads to a lack of proteolytic processing and has different effects on beta1 integrin-mediated cell adhesion and alpha-dystroglycan binding. FEBS Lett. 1999;458:319–23. doi: 10.1016/s0014-5793(99)01180-1. [DOI] [PubMed] [Google Scholar]
- Tian M, Jacobson C, Gee SH, Campbell KP, Carbonetto S, Jucker M. Dystroglycan in the cerebellum is a laminin alpha 2-chain binding protein at the glial-vascular interface and is expressed in Purkinje cells. Eur J Neurosci. 1996;8:2739–47. doi: 10.1111/j.1460-9568.1996.tb01568.x. [DOI] [PubMed] [Google Scholar]
- Wagner S, Tagaya M, Koziol JA, Quaranta V, del Zoppo GJ. Rapid disruption of an astrocyte interaction with the extracellular matrix mediated by integrin α6β4 during focal cerebral ischemia/reperfusion. Stroke. 1997;28:858–65. doi: 10.1161/01.str.28.4.858. [DOI] [PubMed] [Google Scholar]
- Warth A, Mittelbronn M, Wolburg H. Redistribution of the water channel protein aquaporin-4 and the K+ channel protein Kir4.1 differs in low- and high-grade human brain tumors. Acta Neuropathol (Berl) 2005;109:418–26. doi: 10.1007/s00401-005-0984-x. [DOI] [PubMed] [Google Scholar]
- Yamada H, Denzer AJ, Hori H, Tanaka T, Anderson LV, Fujita S, Fukuta-Ohi H, Shimizu T, Ruegg MA, Matsumura K. Dystroglycan is a dual receptor for agrin and laminin-2 in Schwann cell membrane. J Biol Chem. 1996;271:23418–23. doi: 10.1074/jbc.271.38.23418. [DOI] [PubMed] [Google Scholar]
- Yamada H, Saito F, Fukuta-Ohi H, Zhong D, Hase A, Arai K, Okuyama A, Maekawa R, Shimizu T, Matsumura K. Processing of beta-dystroglycan by matrix metalloproteinase disrupts the link between the extracellular matrix and cell membrane via the dystroglycan complex. Hum Mol Genet. 2001;10:1563–9. doi: 10.1093/hmg/10.15.1563. [DOI] [PubMed] [Google Scholar]
- Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 2001;104:311–24. doi: 10.1016/s0306-4522(01)00092-6. [DOI] [PubMed] [Google Scholar]