

Abstract
VEGF and TGF-β1 induce angiogenesis but have opposing effects on vascular endothelial cells: VEGF promotes survival; TGF-β1 induces apoptosis. We have previously shown that TGF-β1 induces endothelial cell apoptosis via up-regulation of VEGF expression and activation of signaling through VEGF receptor-2 (flk-1). In context with TGF-β1, VEGF signaling is transiently converted from a survival into an apoptotic one. VEGF promotes cell survival in part via activation of PI3K/Akt by a mechanism dependent on the formation of a multi-protein complex that includes flk-1 and the adherens junction proteins VE-cadherin and β-catenin. Here we report that TGF-β1 induces rearrangement of the adherens junction complex by separating flk-1 from VE-cadherin and increasing β-catenin association with both flk-1 and VE-cadherin. This rearrangement is caused neither by changes in adherens junction mRNA or protein expression nor by post-translational modification, and requires VEGF signaling through flk-1. These results show that the adherens junction is an important regulatory component of TGF-β1-VEGF interaction in endothelial cells.
Keywords: TGF-beta1, VEGF, VEGF receptor-2, VE-cadherin, beta-catenin, endothelial cells, adherens junction
Angiogenesis, the formation of new capillaries from pre-existing blood vessels, occurs in many physiological and pathological processes such as wound healing, embryonic development, and tumor growth. Angiogenesis is dependent on endothelial cell proliferation, migration, and apoptosis, processes that are regulated by cytokine and growth factor signaling, particularly VEGF and TGF-β1. VEGF increases vascular permeability and stimulates endothelial cell proliferation and angiogenesis (Keck et al., 1989), (Ferrara and Henzel, 1989), (Plouet and Gospodarowicz, 1989). Heterozygous deficiency of VEGF in mice results in embryonic lethality with delayed endothelial cell differentiation (Plouet and Gospodarowicz, 1989). Two tyrosine kinase receptors, VEGF-R1/flt-1 and VEGF-R2/flk-1, mediate signaling induced by VEGF (Plouet and Gospodarowicz, 1989), (Robinson and Stringer, 2001), (Ferrara et al., 2003), (Neufeld et al., 1999). VEGF-R2 is the primary mediator of the mitogenic and angiogenic properties of VEGF, while VEGF-R1 may perform an inhibitory role in vivo, sequestering soluble VEGF (Robinson and Stringer, 2001), (Neufeld et al., 1999). Genetic deletion of either VEGF receptor is embryonic lethal. VEGF-R2-deficient mice fail to develop sufficient populations of differentiated endothelial cells by E9.5; conversely, in VEGF-R1-deficient mouse embryos endothelial cell differentiation occurs but endothelial cells fail to assemble into functional vascular networks (Neufeld et al., 1999), (Breen, 2007). FGF-2, another endothelial cell mitogen, is not essential for embryonic development, and FGF-2−/− mice are viable and fertile with neuronal and wound-healing defects (Breen, 2007).
TGF-β1 is a multifunctional cytokine with cell type- and context-specific properties (Massague et al., 2000). In endothelial cells, TGF-β1 is an inhibitor of proliferation and migration, opposing the activity of VEGF. TGF-β1 induces endothelial cell apoptosis, and down-regulates expression of VEGF-R2 in endothelial cells (Maharaj et al., 2006), although it promotes angiogenesis in vivo and in vitro (Carmeliet et al., 1996). TGF-β1 induction of angiogenesis requires endothelial cell apoptosis, which occurs via autocrine/paracrine stimulation of VEGF expression and signaling through VEGF-R2 (Ferrara et al., 1996). This mechanism relies on molecular cross-talk between the VEGF and TGF-β1 signaling pathways, which results in converting a survival signal into an apoptotic one, in part via downstream activation of the MAP kinase p38 α (Hyman et al., 2002) (Ferrari, unpublished results).
VEGF-R2 can associate with a membrane multiprotein complex at the endothelial cell adherens junction. The adherens junction is responsible for homotypic cell-cell adhesion by linking actin filaments of adjacent cells, and in endothelial cells it consists of members of the catenin family associated directly and indirectly with VE-cadherin (Terman et al., 1991). Both VEGF and TGF-β1 have been shown to control adherens junction formation and protein-protein association (Matthews et al., 1991), (Quinn et al., 1993).
β-catenin is an armadillo family member that binds intracellularly to the VE-cadherin cytoplasmic domain and the TCF/Lef-1 family of transcription factors. β-catenin has two primary known functions: it is a structural protein involved in cell-cell adhesion, and an effector of canonical Wnt signaling that translocates to the nucleus, inducing proliferative genes, which in vivo can stimulate tumorigenesis (Waltenberger et al., 1994), (Cross and Claesson-Welsh, 2001), (Liebner et al., 2006). β-catenin is essential in endothelial cells for normal vascular patterning. Conditional deletion under control of the Tie-2 promoter is embryonic lethal and results in abnormal vascular development, particularly in the yolk sac and head, and in altered cell junctions devoid of α-catenin (Cattelino A, 2003).
VE-cadherin is an integral membrane glycoprotein expressed exclusively in endothelial cells (Cross et al., 2003), (Shalaby, 2002), (Fong et al., 1995). Unlike other cadherin family members VE-cadherin expression at cell junctions is independent of β-catenin binding, which appears to be required only for junction stabilization (Karsan et al., 1997). VE-cadherin clusters at cell junctions and mediates cell adhesion in a calcium-dependent manner, inhibits cell proliferation, and decreases cell permeability and migration when over-expressed in various cell types (Massague et al., 2000), (Ortega et al., 1998), (Bertolino et al., 2005), (Mandriota et al., 1996). Additionally, VE-cadherin inhibits VEGF-R2 phosphorylation and internalization and is required for normal vascular integrity (Lampugnani et al., 2006), (Hofer and Schweighofer, 2007), (Madri et al., 1988), (Segura et al., 2002). Genetic deletion or cytoplasmic truncation of VE-cadherin in mice results in embryonic lethality by E9.5, with endothelial cells failing to undergo remodeling into vascular structures (Carmeliet et al., 1996). The endothelial cells derived from these mice exhibit disruption of a protein complex at the adherens junction consisting of β-catenin, VE-cadherin, and VEGF-R2. These endothelial cells also exhibit a high apoptotic rate in culture, a result of dismantling of the VEGF survival signal normally propagated through VEGF-R2 and PI3K/Akt activation (Carmeliet et al., 1996). We reasoned that TGF-β1 might influence the composition of the adherens junction complex. Here we show that TGF-β1 induction of rearrangement of the endothelial cell adherens junction is mediated by VEGF.
MATERIALS & METHODS
Materials
Pooled HUVEC from multiple donors were obtained from Cascade Biologics (Portland, OR) and grown until the 5th passage in culture. BCE cells were isolated and cultured as described (Seghezzi et al., 1998), (Folkman et al., 1979) and used until passage 22. Human recombinant VEGF and TGF-β1 were obtained from PeproTech (Rocky Hill, NJ), and TNF-α was obtained from Invitrogen (Portland, OR). Anti-VEGF neutralizing antibody was purchased from R&D Systems (Minneapolis, MN). The following antibodies were obtained as follows: VEGF-R2/flk-1 (rabbit polyclonal serum from Calbiochem, and goat polyclonal from R&D Systems); VE-cadherin (goat polyclonal C-19 from Santa Cruz Biotechnology, Santa Cruz, CA, and mouse monoclonal from Chemicon, Pittsburgh, PA); β-catenin (Cell Signaling Technology, Danvers, MA); phospho-β-catenin (ser 33/37/thr 41, Cell Signaling Technology); phosphotyrosine (Santa Cruz Biotechnology); ERK-2 (Santa Cruz Biotechnology), and p120 (H-90; Santa Cruz). Anti-plakoglobin mouse ascites fluid was generously provided by Dr. P. Cowin (NYU School of Medicine).
Cell Culture
Endothelial cells were grown onto 2% gelatin-coated culture dishes. HUVEC were grown in 200 Medium supplemented with LSGS endothelial cell supplement (Cascade Biologics) and 10% fetal bovine serum (FBS). BCE cells were grown in alpha-MEM (Cellgro, Herndon, VA) supplemented with 5% donor calf serum (DCS). Treatments with growth factors/cytokines and inhibitors were carried out in cells starved overnight with 0.5% serum-containing medium. Cells were treated for 6 h with either 1 ng/ml of TGF-β1 or 1ng/ml of TNF-α and 1 μg/ml of cycloheximide. Cells were treated with 50 ng/ml of VEGF for 30 min to activate VEGF-R2. Endogenous VEGF activity was blocked by treating cells with 10 μg/ml of anti-VEGF antibody for 6 h.
Immunoprecipitation and Western Blotting
Cells were washed twice with ice-cold PBS containing 100 mg/l of Mg2+ and Ca2+. Cells were lysed in buffer containing 150 mM NaCl, 10 mM Tris pH 7.4, and 1% Triton-X-100, supplemented with Pefabloc (Roche, 1 mM), leupeptin (1 mM), Na3VO4 (1 mM), and 2 mM CaCl2. For phosphorylation studies the lysis buffer also contained 100 μM peroxyvanadate. For immunoprecipitation, 100 μg of protein was pre-cleared by rocking at 4°C for 30 min with 0.5 μg of non-immune IgG coupled to 10 μl of protein A+G agarose beads (Santa Cruz Biotechnology) that had been reconstituted by washing three times in lysis buffer. Pre-cleared extracts were centrifuged for 60 sec at 300×g, and the supernatant was immunoprecipitated by rocking overnight at 4°C with 10 μl of reconstituted protein A+G agarose beads and 0.75 μg of antibody per 100 μg of protein. The samples were centrifuged at 300×g and the pelleted beads were washed three times with lysis buffer, and boiled in reducing sample buffer. Samples were loaded onto SDS-PAGE gels and separated electrophoretically. Proteins were transferred onto nitrocellulose PVDF membranes (Immobilon-P, Millipore, Bedford, MA), and blots were incubated with antibodies as indicated. In cases where whole cell extracts were examined, the lysates were measured for protein concentration and 25 μg of protein was loaded onto the gel.
Indirect Immunofluorescence
Cells were grown on gelatin-coated glass coverslips and treated with TGF-β1 as described above. Cells were fixed in 2% paraformaldehyde and permeabilized with 0.5% Triton-X-100 in PBS. Coverslips were incubated with primary antibody for 2 h at 37° C in a humidified chamber. Coverslips were washed several times with 1% BSA-PBS, and primary antibodies were detected with tetramethyl rhodamine isothiocyanate-conjugated goat anti-rabbit or donkey anti-goat secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA), diluted 1:100 in 1% BSA-PBS and incubated at 37° C for 1 h. Nuclei were stained with 4, 6-diamino-2-phenylindole (DAPI; 0.2 μg /ml). Images were acquired and recorded with a Zeiss Axioskop 2 microscope using a Zeiss Axiocam HRc camera and Carl Zeiss Axiovision software.
RT-PCR
Total RNA was extracted from cells using Trizol (Invitrogen) or the RNeasy Micro Kit (Qiagen, Valencia, CA), and reverse transcribed to cDNA using the Superscript II First-Strand kit (Invitrogen). For PCR, 2 μl of cDNA were used in a 25 μl reaction volume with Jump Start Taq polymerase (Sigma, St. Louis, MO), using 18 cycles for β-actin, 23 cycles for β-catenin, 28 cycles for VE-cadherin, and 25 cycles for VEGF-R2 with an annealing temperature of 56° C. The following primers were used: β-actin, forward: 5′-GGA AAT CGT GCG TGA CAT CAA AG-3′; β-actin, reverse: 5′-CGT CAC ACT TCA TGA TGG AAT TG-3′ (239 bp product); VEGF-R2 (bovine), forward: 5′-CAG CTT CCA AGT GGC TAA GGG C-3′; VEGF-R2, reverse: 5′-GTC TGG TAC ATT TCT GGT G-3′ (390 bp product).
RNA Interference
Smartpool siRNA for VEGF-R2 was obtained from Dharmacon (Chicago, IL). BCE cells were grown to 50% confluence in 6-well plates and transfected with VEGF-R2 siRNA using Oligofectamine (Invitrogen) per manufacturer's instructions. When the cells reached confluency approximately 36 h after transfection, they were serum-starved overnight and treated with growth factors/cytokines as described above, and then processed for immunoprecipitation and Western blotting analyses.
RESULTS
Effect of TGF-β1 on Adherens Junction Composition
VEGF exerts its pro-survival effect on endothelial cells in part through rearrangement of the adherens junctions. TGF-β1, a potent inducer of endothelial cell apoptosis, induces endothelial cell expression of VEGF. Therefore, we examined the effects of TGF-β1 on adherens junction composition. For this purpose, we used human and bovine endothelial cells to investigate the changes wrought by TGF-β1 in the interactions among three components of the adherens junction: VEGF receptor-2, β-catenin, and VE-cadherin. In agreement with previous results (Matthews et al., 1991), (Quinn et al., 1993), VEGF stimulated the formation of a tripartite complex consisting of flk-1, β-catenin, and VE-cadherin. In contrast, TGF-β1 treatment induced a substantial increase in the association of VEGF-R2 with β-catenin but not with VE-cadherin (Fig. 1A). TGF-β1 also increased the association of β-catenin with VE-cadherin (Fig. 1B); however, it did not increase VE-cadherin association with plakoglobin or β-catenin association with p120 (Fig. 1 C).
Figure 1. TGF-β1 stimulates rearrangement of endothelial cell adherens junctions and increases association of VE-cadherin with β-catenin.
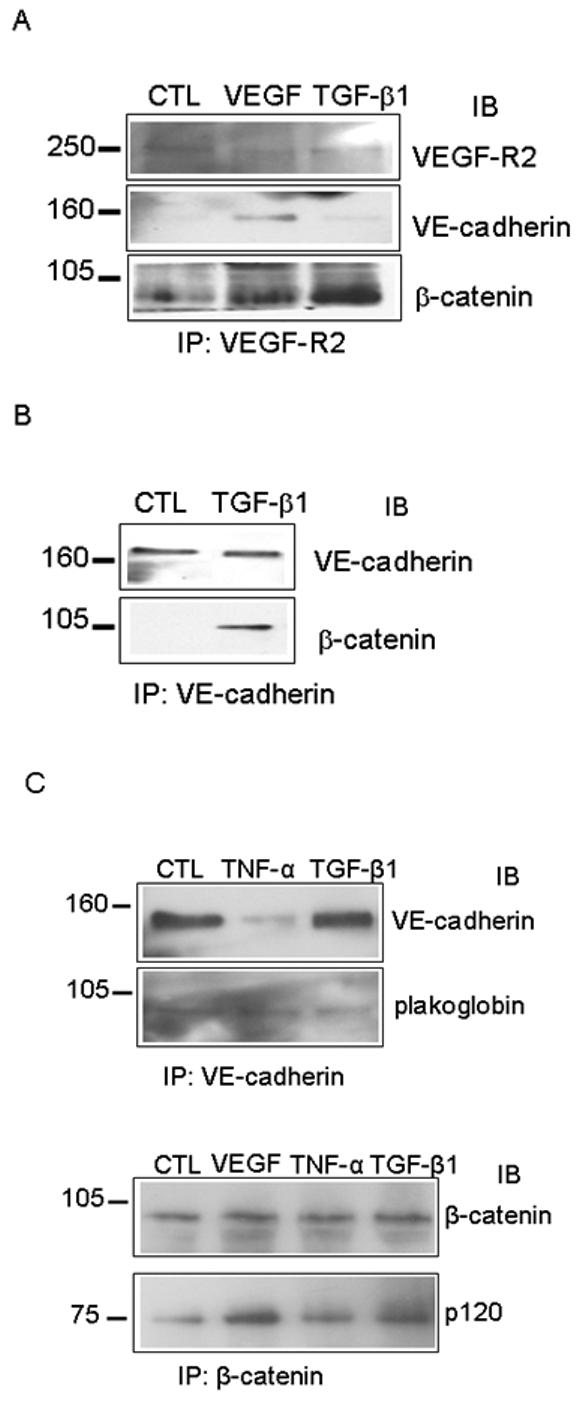
(A) Co - immunopreciptitation analysis of VEGF-R2 interaction with adherens junction proteins. HUVEC were treated with VEGF for 30 min to induce VEGF-R2 activation, or TGF-β1 for 6 h to induce apoptosis, and cell lysates were subjected to immunoprecipitation using VEGF-R2 antibody. Precipitates were analyzed by Western blotting for VEGF-R2, and for co-precipitated β-catenin or VE-cadherin. (B) HUVEC cells were serum-starved (CTL) and treated with TGF-β1 for 6 h, then cell lysates were subjected to immunoprecipitation with VE-cadherin antibody. Precipitates were analyzed by Western blotting for VE-cadherin and β-catenin. (C) BCE cells were serum-starved (CTL) and treated with TNF-α or TGF-β1 for 6 h. Cell lysates were subjected to immunoprecipitation with antibody to VE-cadherin (top panel) or β-catenin (bottom panel). Precipitates were analyzed by Western blotting for VE-cadherin and plakoglobin (top panel), and for β-catenin and p120 (bottom panel). Only the second largest of the four p120 isoforms is associated with β-catenin, while the full-length protein is not.
Effect of TGF-β1 on β-catenin localization
Since β-catenin was increasingly, but separately, associated with two cell-membrane proteins, VEGF-R2 and VE-cadherin, under conditions of TGF-β1 treatment, it was important to determine whether β-catenin nuclear translocation and activation of proliferative genes were altered by TGF-β1 treatment. Immunofluorescence analysis showed that TGF-β1 treatment did not significantly alter β-catenin localization, which was concentrated along the cell membrane in endothelial cells both in the presence and in the absence of TGF-β1; similarly, TGF-β1 did not alter VE-cadherin localization (Fig. 2). β-catenin activity was measured using a luciferase reporter assay, which did not show a consistent change in response to TGF-β1 treatment (data not shown).
Figure 2. TGF β-1 does not alter subcellular localization of β-catenin.
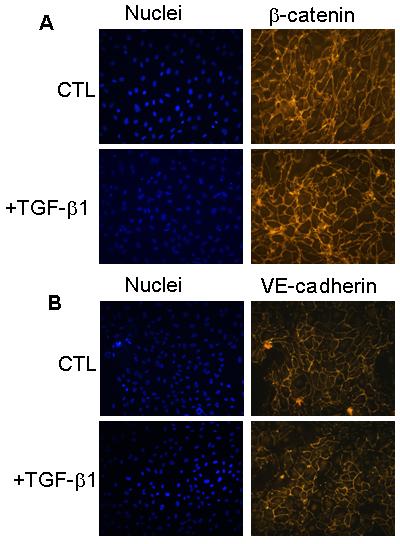
BCE cells were treated as above, fixed in paraformaldehyde, and analyzed by immunofluorescence with β-catenin or VE-cadherin antibody, which was visualized with a TRITC-conjugated secondary antibody; nuclei were stained with DAPI (blue).
TGF-β1 does not alter expression of adherens junction proteins in endothelial cells
Because β-catenin localization did not appear to be affected by TGF-β1, we reasoned that TGF-β1-induced adherens junction rearrangement could be due to an increase in cellular expression of β-catenin or of another adherens junction protein. Therefore, we examined the protein expression of β-catenin and VE-cadherin in response to TGF-β1. The results show that TGF-β1 had no effect on the expression of these proteins (Fig. 3). In contrast, TNF-α, which induces endothelial cell apoptosis by a VEGF-independent mechanism (Ferrari et al., 2006), down-regulated expression of VE-cadherin (Hofmann S, 2002 ) and decreased the protein level of β-catenin (Fig. 3). TGF-β1 did not affect the expression of other proteins at the adherens junction that are not directly involved in endothelial cell survival signaling, such as p120 catenin (data not shown). The analysis of caspase-3 activation confirmed that both TNF-α and TGF-β1 induced endothelial cell apoptosis. TNF-α downregulated VE-caherin and β-catenin levels, whereas TGF-β1 had no such effect (Fig 3).
Figure 3. TGF-β1 does not alter expression of adherens junction proteins in endothelial cells.
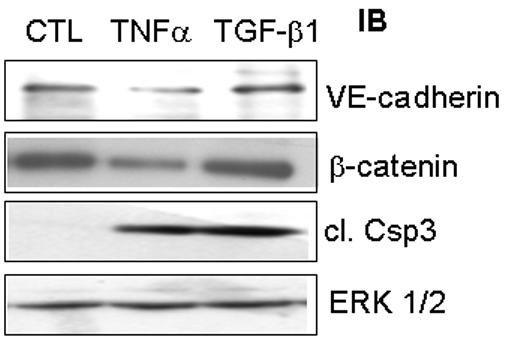
Serum-starved BCE cells were treated with TGF-β1 for 6 h, and cell extracts were analyzed by Western blotting for VE-cadherin and β-catenin. As a control, cells were treated with TNFα, a known inducer of apoptosis, and extracts were analyzed by Western blotting for cleaved caspase-3 to measure the induction of apoptosis. ERK-2 served as a protein loading control.
Tyrosine Phosphorylation of Adherens Junction Proteins
Because TGF-β1 did not affect adherens junction protein expression, we examined whether it influenced post-translational modification of one or more of the junction components. Association of β-catenin with VE-cadherin is regulated in part by tyrosine phosphorylation of both proteins; therefore, we used co-immunoprecipitation experiments to see if TGF-β1 altered VE-cadherin and β-catenin phosphorylation. Cells were treated with TGF-β1, extracts were subjected to immunoprecipitation for the protein of interest, and the precipitates were analyzed by Western blotting with phosphotyrosine antibody. TGF-β1 did not induce a significant change in the tyrosine phosphorylation of either β-catenin (Fig. 4A) or VE-cadherin (Fig. 4B) as compared to control, serum-starved cells.
Figure 4. Tyrosine phosphorylation of adherens junction proteins is unaffected by TGF-β1.
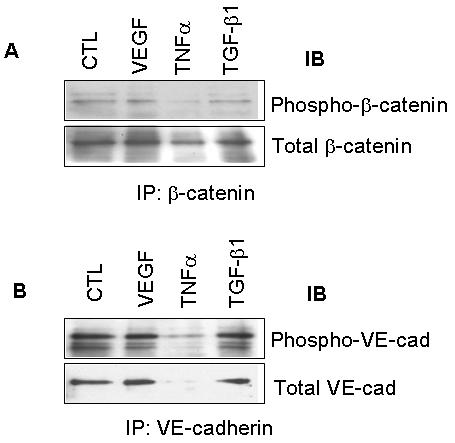
BCE cells were treated as before with VEGF, TNFα, or TGF-β1 and lysates were subjected to immunoprecipitation with antibodies to the indicated proteins. TNFα reduces expression of adherens junction proteins, particularly of VE-cadherin. A) Precipitated β-catenin was analyzed by Western blotting with monoclonal antibody to phospho-tyrosine. B) Precipitated VE-cadherin was analyzed by Western blotting for phospho-tyrosine.
TGF-β1-mediated rearrangement of the adherens junction and its protein association is dependent on VEGF signaling
We next examined the contribution of VEGF signaling through VEGF-R2 to TGF-β1-induced adherens junction rearrangement. For this purpose we used inhibition, and transient knockdown of different molecules in the VEGF signaling pathway, as described (Ferrari et al., 2006). Inhibition of VEGF signaling by neutralizing anti-VEGF antibody blocked TGF-β1-induced increase in β-catenin/VEGF-R2 association (Fig. 5A). When VEGF-R2 expression was downregulated by specific siRNAs (Fig. 5B) TGF-β1 failed to increase β-catenin/VE-cadherin association compared to untreated cells lacking VEGF-R2 (Fig. 5C). These results showed that the TGF-β1-mediated rearrangement of the adherens junction and its protein association is dependent on VEGF signaling through VEGF-R2, as a component of the autocrine/paracrine loop essential for TGF-β1-mediated endothelial cell apoptosis.
Figure 5. TGF-β1-mediated association of β-catenin with VEGF-R2 is dependent on VEGF.
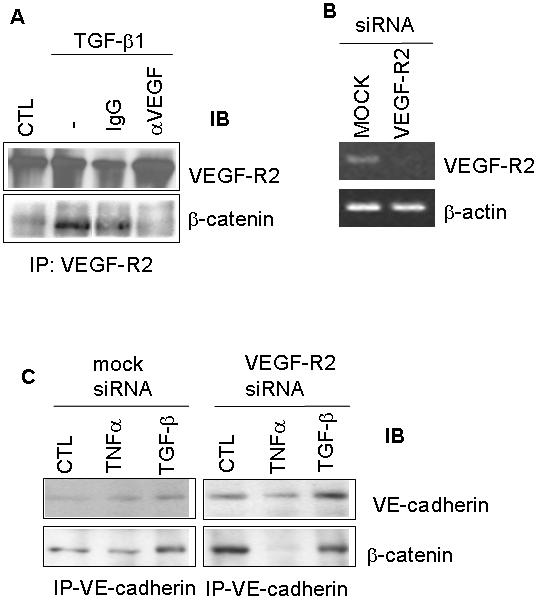
(A) HUVEC were serum-starved and treated with TGF-β1 for 6 h alone, or in the presence of neutralizing VEGF antibody. Non-immune IgG was used as a control. Cell lysates were subjected to immunoprecipitation with a VEGF-R2 antibody, and analyzed by Western blotting for VEGF-R2 and β-catenin. (B) VEGF-R2 expression was transiently inhibited by siRNA in BCE cells and was measured by RT-PCR. (C) ) Transient knockdown of VEGF-R2 blocked the ability of TGF-β1 to stimulate β-catenin/VE-cadherin association compared to mock-transfected cells, as measured by immunoprecipitation and Western blotting.
DISCUSSION
In endothelial cells, β-catenin and VE-cadherin associate to form a multiprotein complex with other catenin family members and VEGF-R2, which promotes endothelial cell survival (Carmeliet et al., 1996). VEGF activation of VEGF-R2 results in tyrosine phosphorylation and activation of β-catenin, in addition to the generation of survival signaling through the PI3 kinase/Akt pathway. Conversely, when VE-cadherin is truncated at its carboxyl terminus its association with β-catenin and VEGF-R2 is lost, resulting in endothelial cell apoptosis (Carmeliet et al., 1996). We have shown that TGF-β1 induces endothelial cell apoptosis through VEGF activation of VEGF-R2. Therefore, we reasoned that TGF-β1 might affect the composition of the adherens junction complex. We found that TGF-β1 induces adherens junction rearrangement in endothelial cells and that this effect is mediated by VEGF activation of flk-1.
TGF-β1 induced an increase in the association of β-catenin independently with VEGF-R2 and VE-cadherin. This change in protein-protein interaction was specific for β-catenin, as association with plakoglobin or p120 did not change. However, we were unable to observe a change in β-catenin activity or intracellular localization. It is possible that a change in β-catenin localization, if any, would only occur in those cells that undergo apoptosis in response to TGF-β1 (approximately 15-20 percent of the cell population in culture).
The association of β-catenin with VE-cadherin was not caused by changes in protein or mRNA expression. Although in our experiments TGF-β1 did not appear to alter tyrosine phosphorylation of either β-catenin or VE-cadherin, the association of β-catenin with VE-cadherin has been shown to be regulated by tyrosine phosphorylation of both proteins. Inhibition of tyrosine phosphatases results in decreased electrical conductivity across adherens junctions, and in stabilization of β-catenin and VE-cadherin phosphorylation, which disrupts junction integrity (Staddon et al., 1995), (Behrens et al., 1993), (Matsuyoshi et al., 1992). Also, VE-cadherin tyrosine phosphorylation is up-regulated in sparsely seeded cell cultures and colocalizes preferentially with β-catenin, rather than plakoglobin (Lampugnani et al., 1997). Src kinases, particularly c-Src, have been identified as the likely mediators of adherens junction protein tyrosine phosphorylation, and Src-dependent VE-cadherin phosphorylation prevents binding of both β-catenin and p120 (Wallez et al., 2006), (Potter et al., 2005), (Ali et al., 2006). Individual residues important for protein association have been identified; for instance, interaction between β-catenin and E-cadherin in epithelial cells is disrupted by EGFR-dependent modification of Y654 (Miravet et al., 2003). It will be interesting to determine whether the interaction between β-catenin and VE-cadherin is controlled via the same residue, but perhaps in a TGF-β1 - VEGFR2-dependent manner. VE-cadherin Y731 phosphorylation disrupts its interaction with β-catenin in CHO cells overexpressing VE-cadherin, and phospho-mimetic mutant constructs restore the migratory capability of these cells in culture (Potter et al., 2005). However, it remains to be determined whether or not this is an important regulatory mechanism for endogenous VE-cadherin interactions in endothelial cells. Future studies with VE-cadherin mutants compromised for tyrosine phosphorylation or lacking the cytoplasmic domain will help determine whether these mechanisms control the ability of TGF-β1 to rearrange adherens junctions and induce apoptosis. Other adherens junction proteins, including p120 and plakoglobin, are also phosphorylated on tyrosine residues. This post-translational modification functions as an important regulatory mechanism in other cell types, and for the interactions of other adherens junction proteins, for instance mediating the association of β-catenin with E-cadherin and α-catenin (Miravet et al., 2003), (Roura et al., 1999), (Piedra et al., 2003). Finally, VEGF induces tyrosine phosphorylation of VE-cadherin via signaling through VEGF-R2 (Esser et al., 1998), and endothelial cell apoptosis induced by growth factor withdrawal is mediated by dephosphorylation of membrane proteins, which can be blocked by peroxyvanadate, a potent phosphatase inhibitor (Yang et al., 1996).
Recent data have shown that VE-cadherin clustering at endothelial cell adherens junctions is required for TGF-β1 signaling, and that VE-cadherin is associated with the TGF-β receptor complex (Rudini et al., 2008) . Therefore, the endothelial cell adherens junction includes a large protein complex consisting of VE-cadherin, flk-1, TGF-β receptors and the catenins. Our data show that TGF-β1 disrupts the adherens junction complex of VE-cadherin with flk-1. Based on these findings, we speculate that TGF-β1 binding to the TGF-β receptor/VE cadherin complex causes VE-cadherin disassociation from flk-1. It is possible that flk-1 and TGF-β receptors compete for binding to VE-cadherin, and that TGF-β1 binding to its receptors increases VE-cadherin clustering with TGF-β receptors by removing VE-cadherin from flk-1. This hypothesis warrants further investigation.
Our experiments using inhibition, and siRNA-mediated mRNA knockdown of molecules in the VEGF signaling pathway have shown that TGF-β1-mediated adherens junction rearrangement requires VEGF signaling through VEGF-R2. This identifies the endothelial cell adherens junction as potentially responsible for the autocrine/paracrine loop involved in VEGF-mediated effects of TGF-β1 on endothelial cells. These results also suggest that TGF-β1-induced adherens junction rearrangement could be controlled downstream of VEGF-R2 by signaling through p38MAPK (Hyman et al., 2002), (Ferrari et al., 2006). Although VE-cadherin/VEGF-R2 interaction at the adherens junction provides endothelial cells with survival signaling through the PI3K/Akt pathway, TGF-β1 might not alter the activation of this pathway. In contrast, activation of the p38MAPK signaling pathway through flk-1 is required for TGF-β1 induction of endothelial cell apoptosis (Ferrari et al., 2006).
While our studies show some of the early biological consequences of TGF-β1 treatment, at a later time cultured endothelial cells become refractory to TGF-β1 induction of cell death (Ferrari et al., 2006). In addition, TGF-β1-mediated apoptosis only affects a subpopulation of cultured endothelial cells, usually amounting to about 20 percent. This contrasts with the widespread apoptosis induced in serum-starved cultures by inhibition of FGF-2, a growth factor that protects endothelial cells from apoptosis in a VEGF-independent manner (Karsan et al., 1997), (Korff and Augustin, 1998). These observations suggest that VEGF-dependent apoptosis induced by TGF-β1 could serve as a mechanism for pruning a subpopulation of endothelial cells, then providing the surviving cells with stimuli for the migration and proliferation required to form new capillary sprouts. These stimuli are likely represented by VEGF and FGF-2, whose endothelial cell expression is upregulated by TGF-β1 (Ferrari et al., 2006). This hypothesis raises the question of whether or not the adherens junction rearrangement induced by TGF-β1 is similarly short-lived and only necessary for the initial winnowing of the cell population, before the survival and angiogenic functions of VEGF and/or FGF-2 come to dominate, resulting in a re-activated adherens junction protein complex.
ACKNOWLEDGMENTS
This work was supported by grants NIH R01 HL070203 and R01 HL070203-03S1 to P.M., and by funds from the Department of Cardiothoracic Surgery of NYU School of Medicine. We thank Dr. Pamela Cowin (NYU School of Medicine) for the generous gift of anti-plakoglobin mouse ascitic fluid..
Contract grant sponsor: NIH; Contract grant number: R01 HL070203 and R01 HL070203S.
REFERENCES
- Ali N, Yoshizumi M, Yano S, Sone S, Ohnishi H, Ishizawa K, Kanematsu Y, Tsuchiya K, Tamaki T. The novel Src kinase inhibitor M475271 inhibits VEGF-induced vascular endothelial-cadherin and beta-catenin phosphorylation but increases their association. J Pharmacol Sci. 2006;102:112–20. doi: 10.1254/jphs.fp0060357. [DOI] [PubMed] [Google Scholar]
- Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest. 2005;128:585S–590S. doi: 10.1378/chest.128.6_suppl.585S. [DOI] [PubMed] [Google Scholar]
- Breen EC. VEGF in biological control. J Cell Biochem. 2007;102:1358–67. doi: 10.1002/jcb.21579. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–9. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Cattelino ALS, Gallini R, Zanetti A, Balconi G, Corsi A, Bianco P, Wolburg H, Moore R, Oreda B, Kemler R, Dejana E. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. Journal of Cell Biology. 2003;162:1111–1122. doi: 10.1083/jcb.200212157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22:201–7. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. VEGF-receptor signal transduction. Trends Biochem Sci. 2003;28:488–94. doi: 10.1016/S0968-0004(03)00193-2. [DOI] [PubMed] [Google Scholar]
- Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998;111(Pt 13):1853–65. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161:851–8. doi: 10.1016/0006-291x(89)92678-8. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P. VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induce endothelial cell apoptosis. Proc Natl Acad Sci U S A. 2006;103:17260–5. doi: 10.1073/pnas.0605556103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J, Haudenschild CC, Zetter BR. Long-term culture of capillary endothelial cells. Proc Natl Acad Sci U S A. 1979;76:5217–21. doi: 10.1073/pnas.76.10.5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- Hofer E, Schweighofer B. Signal transduction induced in endothelial cells by growth factor receptors involved in angiogenesis. Thromb Haemost. 2007;97:355–63. [PMC free article] [PubMed] [Google Scholar]
- Hofmann SGH, Jung P, Bidlingmaier M, Vlotides J, Janssen OE, Landgraf R. The tumour necrosis factor-alpha induced vascular permeability is associated with a reduction of VE-cadherin expression. Eur J Med Res. 2002;7:171–6. [PubMed] [Google Scholar]
- Hyman KM, Seghezzi G, Pintucci G, Stellari G, Kim JH, Grossi EA, Galloway AC, Mignatti P. Transforming growth factor-beta1 induces apoptosis in vascular endothelial cells by activation of mitogen-activated protein kinase. Surgery. 2002;132:173–9. doi: 10.1067/msy.2002.125304. [DOI] [PubMed] [Google Scholar]
- Karsan A, Yee E, Poirier GG, Zhou P, Craig R, Harlan JM. Fibroblast growth factor-2 inhibits endothelial cell apoptosis by Bcl-2-dependent and independent mechanisms. Am J Pathol. 1997;151:1775–84. [PMC free article] [PubMed] [Google Scholar]
- Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–12. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- Korff T, Augustin HG. Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J Cell Biol. 1998;143:1341–52. doi: 10.1083/jcb.143.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110(Pt 17):2065–77. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Cavallaro U, Dejana E. The multiple languages of endothelial cell-to-cell communication. Arterioscler Thromb Vasc Biol. 2006;26:1431–8. doi: 10.1161/01.ATV.0000218510.04541.5e. [DOI] [PubMed] [Google Scholar]
- Madri JA, Pratt BM, Tucker AM. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J Cell Biol. 1988;106:1375–84. doi: 10.1083/jcb.106.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj AS, Saint-Geniez M, Maldonado AE, D'Amore PA. Vascular endothelial growth factor localization in the adult. Am J Pathol. 2006;168:639–48. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandriota SJ, Menoud PA, Pepper MS. Transforming growth factor beta 1 down-regulates vascular endothelial growth factor receptor 2/flk-1 expression in vascular endothelial cells. J Biol Chem. 1996;271:11500–5. doi: 10.1074/jbc.271.19.11500. [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Matsuyoshi N, Hamaguchi M, Taniguchi S, Nagafuchi A, Tsukita S, Takeichi M. Cadherin-mediated cell-cell adhesion is perturbed by v-src tyrosine phosphorylation in metastatic fibroblasts. J Cell Biol. 1992;118:703–14. doi: 10.1083/jcb.118.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews W, Jordan CT, Gavin M, Jenkins NA, Copeland NG, Lemischka IR. A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c-kit. Proc Natl Acad Sci U S A. 1991;88:9026–30. doi: 10.1073/pnas.88.20.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miravet S, Piedra J, Castano J, Raurell I, Franci C, Dunach M, Garcia de Herreros A. Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates beta-catenin-mediated transcription. Mol Cell Biol. 2003;23:7391–402. doi: 10.1128/MCB.23.20.7391-7402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. Faseb J. 1999;13:9–22. [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci U S A. 1998;95:5672–7. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedra J, Miravet S, Castano J, Palmer HG, Heisterkamp N, Garcia de Herreros A, Dunach M. p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin Interaction. Mol Cell Biol. 2003;23:2287–97. doi: 10.1128/MCB.23.7.2287-2297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouet J, Gospodarowicz D. Transforming growth factor beta-1 positively modulates the bioactivity of fibroblast growth factor on corneal endothelial cells. J Cell Physiol. 1989;141:392–9. doi: 10.1002/jcp.1041410221. [DOI] [PubMed] [Google Scholar]
- Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–12. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- Quinn TP, Peters KG, Vries CD, Ferrara N, Williams LT. Fetal Liver Kinase 1 is a Receptor for Vascular Endothelial Growth Factor and is Selectively Expressed in Vascular Endothelium. Proceedings of the National Academy of Sciences. 1993;90:7533–7537. doi: 10.1073/pnas.90.16.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–65. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–40. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- Rudini N, Felici A, Giampietro C, Lampugnani M, Corada M, Swirsding K, Garre M, Liebner S, Letarte M, ten Dijke P, Dejana E. VE-cadherin is a critical endothelial regulator of TGF-beta signalling. Embo J. 2008;27:993–1004. doi: 10.1038/emboj.2008.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141:1659–73. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura I, Serrano A, De Buitrago GG, Gonzalez MA, Abad JL, Claveria C, Gomez L, Bernad A, Martinez AC, Riese HH. Inhibition of programmed cell death impairs in vitro vascular-like structure formation and reduces in vivo angiogenesis. Faseb J. 2002;16:833–41. doi: 10.1096/fj.01-0819com. [DOI] [PubMed] [Google Scholar]
- Shalaby JR, Yamaguchi Terry P., Gertsenstein Marina, Wu Xiang-Fu, Breitman Martin L., Schuh Andre C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 2002;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Staddon JM, Herrenknecht K, Smales C, Rubin LL. Evidence that tyrosine phosphorylation may increase tight junction permeability. J Cell Sci. 1995;108(Pt 2):609–19. doi: 10.1242/jcs.108.2.609. [DOI] [PubMed] [Google Scholar]
- Terman BI, Carrion ME, Kovacs E, Rasmussen BA, Eddy RL, Shows TB. Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene. 1991;6:1677–83. [PubMed] [Google Scholar]
- Wallez Y, Vilgrain I, Huber P. Angiogenesis: the VE-cadherin switch. Trends Cardiovasc Med. 2006;16:55–9. doi: 10.1016/j.tcm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–95. [PubMed] [Google Scholar]
- Yang C, Chang J, Gorospe M, Passaniti A. Protein tyrosine phosphatase regulation of endothelial cell apoptosis and differentiation. Cell Growth Differ. 1996;7:161–71. [PubMed] [Google Scholar]