

Abstract
Oculodentodigital syndrome (ODD) is a rare, usually autosomal-dominant disorder that is characterized by developmental abnormalities of the face, eyes, teeth, and limbs. The most common clinical findings include a long, narrow nose, short palpebral fissures, type III syndactyly, and dental abnormalities including generalized microdontia and enamel hypoplasia. Recently, it has been shown that mutations in the gene GJA1, which encodes the gap junction protein connexin 43, underlie oculodentodigital syndrome. Gap junction communication between adjacent cells is known to be vital during embryogenesis and subsequently for normal tissue homeostasis. Here, we report 8 missense mutations in the coding region of GJA1, 6 of which have not been described previously, in ten unrelated families diagnosed with ODD. In addition, immunofluorescence analyses of a developmental series of mouse embryos and adult tissue demonstrates a strong correlation between the sites of connexin 43 expression and the clinical phenotype displayed by individuals affected by ODD.
Keywords: Oculodentodigital syndrome, ODD, GJA1, Connexin 43
INTRODUCTION
Oculodentodigital syndrome (ODD; MIM no. 164200) is a distressing congenital disorder that affects the development of the face, eyes, limbs, and dentition. The ODD phenotype encompasses a wide range of features, including a long, thin nose, hypoplastic alae, short palpebral fissures, microcornea, and type III syndactyly. Dental abnormalities are a common finding, affected individuals presenting with a range of anomalies such as microdontia, hypodontia, enamel hypoplasia, multiple caries, and early tooth loss; these abnormalities have been reported in both the deciduous and permanent dentitions (Sugar et al., 1966; Patton and Laurence, 1985). To date, several studies have shown that mutations in GJA1 underlie ODD (Paznekas et al., 2003; Kjaer et al., 2004; Pizzuti et al., 2004; Richardson et al., 2004; Debeer et al., 2005; van Steensel et al., 2005; Vasconcellos et al., 2005; Vitiello et al., 2005; Kelly et al., 2006; Richardson et al., 2006; van Es et al., 2007; de la Parra and Zenteno, 2007; Vreeburg et al., 2007).
GJA1 encodes the gap junction protein connexin 43, which is part of a family composed of over 20 different members in humans. It is well-documented that gap junction communication plays a vital role during embryogenesis and, subsequently, in normal cellular homeostasis (Richard, 2003). Connexin 43 has previously been shown to be vital for normal murine heart development, as demonstrated by Gja1 knockout mice, which die at birth as the result of a severe heart malformation (Reaume et al., 1995). To form a functioning gap junction channel, 6 connexin subunits form a hemichannel, or connexon, in the plasma membrane of a cell. Two connexons from adjacent cells dock to form an entire channel, which allows for the passage of molecules up to 1.2 kDa between the cytoplasm of the 2 cells (Bennett et al., 1991). In light of existing studies, we hypothesized that mutations in GJA1 would underlie the phenotype(s) observed in an additional cohort of ODD families, and that there would be strong correlation between the sites of connexin 43 expression and the tissues affected in this condition.
MATERIALS & METHODS
Families
All ten families were referred by clinical geneticists or ophthalmologists after a diagnosis of ODD had been made. Samples were collected with informed consent under appropriate ethical approval.
Mutation Analysis
The coding region of GJA1 was amplified with primers described previously (Richardson et al., 2004, 2006). After PCR amplification, the products were excised from a 1% agarose gel and sequenced directly by dye primer chemistry.
Immunofluorescence Analysis
Mice were housed and killed in accordance with the UK Animals (Scientific Procedures) Act, 1986. At specific ages, embryos were dissected from time-mated, wild-type females, fixed in Bouin’s fixative overnight, dehydrated through a graded series of ethanol washes, cleared in chloroform, embedded in wax, and sectioned at 6 μm. P0 pups and dissected adult lower jaws were fixed overnight in 4% paraformaldehyde, decalcified for 2 wks in 0.5 M EDTA, dehydrated, cleared, embedded, and sectioned as for embryos. For connexin 43 immunodetection, slides were rehydrated and treated with 10 mM citrate buffer at 96°C for 10 min, incubated with an anti-connexin 43 primary antibody (71-0700, Zymed Laboratories Inc., San Francisco, CA, USA) at a 1:100 dilution, detected with Streptavidin-Cy3 (Sigma, St. Louis, MO, USA), and counterstained with DAPI (Sigma). Negative controls in which the primary antibody was omitted were established for every experiment. Fluorescent staining was visualized by means of a Leica DMRB microscope and fluorescence pack.
RESULTS
Mutation Analysis
Mutation analysis of the entire coding region of GJA1 revealed heterozygous missense mutations in all those individuals diagnosed with ODD who were analyzed in the current study (Fig. 1; Table). The same changes were not detected in unaffected family members or in 100 normal chromosomes. The mutations were distributed throughout the connexin 43 protein; 1 mutation (nt7 G>A, resulting in D3N) was detected in the amino terminus, 1 mutation (nt253 G>A, resulting in V85M) was found in the second transmembrane domain, 4 mutations (nt412 G>A, resulting in G138S; nt413 G>A, resulting in G138D; nt428 G>A, resulting in G143D; nt443 G>A, resulting in R148Q) were detected in the cytoplasmic loop of connexin 43, and the final 2 mutations (nt602 G>A, resulting in S201Y; nt605 G>A, resulting in R202H) were detected in the second extracellular domain of connexin 43 (Fig. 1; Table).
Figure 1.
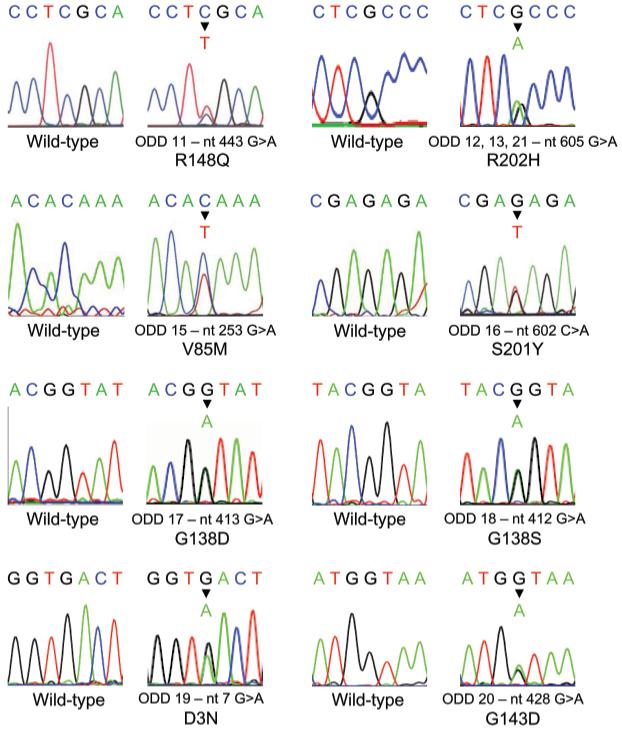
Mutation analysis in ODD. Partial sequence chromatograms demonstrating the 8 different missense mutations in GJA1 detected in the current study. In each case, a partial sequence chromatogram showing the wild-type sequence is displayed alongside. The chromatogram shown for ODD families 11, 15, and 16 is the non-coding strand of GJA1; all other chromatograms depict the coding strand.
Table.
Mutations Identified in GJA1
| Family | GJA1 Mutation | Connexin 43 Mutation | Position in Connexin 43 |
|---|---|---|---|
| ODD19 | nt7 G>A | D3N | Amino terminus |
| ODD15 | nt253 G>A | V85M | Transmembrane domain 2 |
| ODD18 | nt412 G>A | G138S | Cytoplasmic loop |
| ODD17 | nt413 G>A | G138D | Cytoplasmic loop |
| ODD20 | nt428 G>A | G143D | Cytoplasmic loop |
| ODD11 | nt443 G>A | R148Q | Cytoplasmic loop |
| ODD16 | nt602 G>A | S201Y | Extracellular loop 2 |
| ODD12, 13, 21 | nt602 G>A | R202H | Extracellular loop 2 |
Expression Analysis
Immunofluorescence analysis of a series of wild-type mouse embryos and P0 and adult tissue revealed strong connexin 43 expression during development of the teeth, eyes, face, and limbs. At E10.5, connexin 43 expression was observed in the localized epithelial thickenings that presage the sites of tooth development in the maxilla and mandible, and also toward the midline of the fusing mandibular processes (Figs. 2A, 2B); however, by E11.5, connexin 43 expression had shifted to the odontogenic mesenchyme (Fig. 2C). At E12.5, connexin 43 expression persisted in the condensing mesenchyme underlying the invaginating odontogenic epithelium (Fig. 2D). By E13.5, connexin 43 expression was confined to the enamel organ of the bud-stage tooth germs (Fig. 2E). By E15.5, as the tooth germs entered the cap stage of development, connexin 43 expression became restricted to the lateral aspect of the enamel organ of the molar tooth germs, particularly strong expression being detected in the epithelium immediately adjacent to the dental papilla; in contrast, connexin 43 was not expressed in the medial portion of the enamel organ or in the dental papilla (Fig. 2F). In the developing molar tooth germs of P0 mice, differentiated ameloblasts had entered the secretory phase of enamel formation; at this stage of development, strong connexin 43 expression was observed in the ameloblasts, the stratum intermedium, and the stellate reticulum (Figs. 2G, 2H). In the incisors of adult mice, the ameloblasts undergo a constant process of secretion and maturation to produce enamel. In the secretory zone, strong punctate expression of connexin 43 was observed throughout the ameloblasts, with expression also detected in the stratum intermedium (Fig. 2I). In the maturation zone of the adult incisor teeth, connexin 43 expression was restricted to the distal junctional complex of the ameloblasts, while the adjacent stratum intermedium displayed high levels of expression (Fig. 2J).
Figure 2.
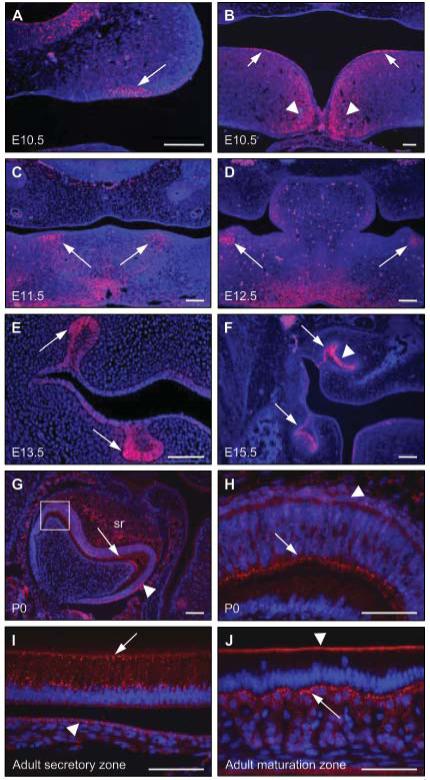
Connexin 43 expression in the developing tooth germs of E10.5 through adult wild-type mice. (A,B) At E10.5, connexin 43 expression was observed in specific regions of the ventral epithelium of the developing maxilla (A; arrowed), in distinct regions of the dorsal epithelium of the developing mandible (B; arrowed), and also in the mesenchyme around the midline of the fusing mandibular processes (B; arrowheads). (C) At E11.5, connexin 43 expression was observed in the odontogenic mesenchyme of the mandibular processes (arrowed). (D) At E12.5, connexin 43 expression persisted in the condensing mesenchyme adjacent to the invaginating odontogenic epithelium (arrowed). (E) At E13.5, connexin 43 expression was detected in the lateral aspect of the bud-stage molar tooth germs (arrowed). (F) By E15.5, connexin 43 expression was observed in the lateral epithelium (arrowed) and in the enamel organ (arrowhead). (G,H) At P0, strong connexin 43 expression was apparent in the secretory ameloblasts (arrowed) and also in the stratum intermedium (arrowhead); weaker expression of connexion 43 was also detected in the stellate reticulum (sr). The boxed region in G is shown at a higher magnification in H. (I) In adult incisors, the secretory ameloblasts demonstrated strong, punctate expression of connexin 43 (arrowed); weaker expression was also detected in the stratum intermedium (arrowhead). (J) In the maturation zone of adult incisors, connexin 43 expression is restricted to the distal junctional complex of the ameloblasts (arrowhead), with strong expression observed in the stratum intermedium (arrowed). Scale bars in A-G = 100 μm; scale bars in H-J = 50 μm.
Strong connexin 43 expression was also detected at other sites affected by the ODD phenotype. At E9.5, expression was observed throughout the internal surface of the developing optic vesicles; at this stage of development, this layer is continuous with the neuroepithelium of the developing brain, which also exhibited strong connexin 43 expression (Fig. 3A). At E10.5, connexin 43 expression persisted in the epithelium of the optic cup and was also detected in the epithelium of the invaginating lens vesicle (Fig. 3B). At E13.5, strong connexin 43 expression was detected in the outer surface of the optic cup, which ultimately forms the pigmented layer of the retina, and in the developing optic nerve (Fig. 3C). By E15.5, connexin 43 expression was localized to the anterior portion of the developing retina, which ultimately forms the ciliary process, and persisted in the optic nerve (Fig. 3D). In P0 mice, strong connexin 43 expression was observed in the ciliary process of the eye (Fig. 3E), and in an arc around the optic nerve (Fig. 3F). In the developing limbs at E9.5, connexin 43 expression was detected throughout the epithelium and mesenchyme of the apical region of the limb bud (Fig. 3E); however, by E12.5, connexin 43 expression became restricted to the apical ectodermal ridge (Fig. 3F). At E14.5, when the paired secondary palatal shelves are fusing above the tongue, connexin 43 expression was observed in the midline epithelial seam and in distinct regions of the nasal epithelium (Fig. 3G). Strong connexin 43 expression was also observed in the follicles of the developing vibrissae (Fig. 3H).
Figure 3.
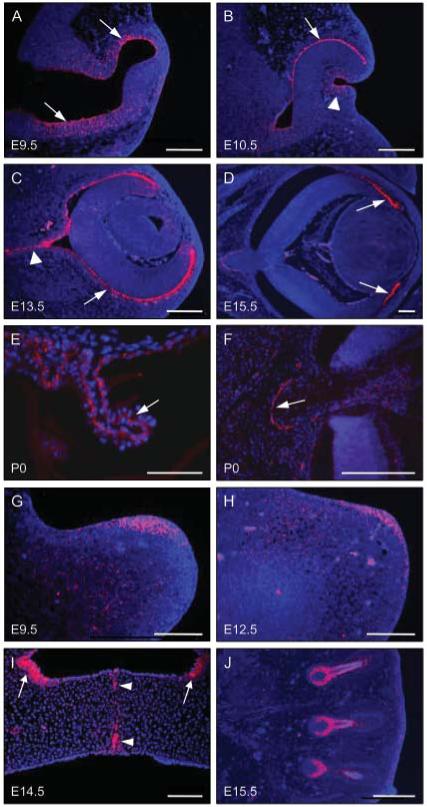
Connexin 43 expression at non-dental sites in developing embryos and P0 mice. (A-F) Coronal sections through the heads of E9.5 - P0 wild-type mice showing the development of the eyes. (A) At E9.5, connexin 43 staining was observed throughout the epithelium of the developing eye, extending into the neuroepithelium (arrowed). (B) At E10.5, connexin 43 expression was detected in the epithelium of the optic cup (arrowed) and in the invaginating lens vesicle (arrowhead). (C) At E13.5, connexin 43 expression persisted in the epithelium (arrowed) and was also detected in the developing optic nerve (arrowhead). (D) At E15.5, connexin 43 expression became localized to the area that will form the ciliary process (arrowed). (E) At P0, strong expression of connexin 43 was observed in the ciliary process of the eye (arrowed). (F) At P0, a strong arc of connexin 43 was also detected in the epithelium around the optic nerve (arrowed). (G,H) Longitudinal sections through the developing limb buds. (G) At E9.5, connexin 43 expression was detected on the dorsal surface of the limb bud in a diffuse pattern throughout the apical epithelium and mesenchyme. (H) At E12.5, connexin 43 expression was restricted to the epithelium of the apical ectodermal ridge. (I) Strong connexin 43 expression was also observed in the midline epithelial seam of the fusing secondary palate at E14.5 (arrowheads) and in nasal epithelium (arrowed). (J) Connexin 43 expression was also observed in the developing follicles of the vibrissae. Scale bars in A-D, F, H-J = 100 μm; scale bars in E and G = 50 μm.
DISCUSSION
In the current study, we present ten previously unreported ODD families exhibiting 8 different missense mutations in connexin 43, 6 of which (D3N, V85M, G138S, G138D, G143D, S201Y) have not been described previously. Connexin 43—which consists of 4 transmembrane spanning domains, 2 extracellular domains, a single cytoplasmic loop, and carboxy and amino termini located within the cytoplasm—is typical of the connexin family (Martin and Evans, 2004). The short, intracellular amino terminus, where only 4 mutations in connexin 43 (G2V, D3N, Y17S, S18P) have been reported to date, is very highly conserved between connexins and is thought to be important for membrane insertion and/or trafficking; indeed, mutations in the amino terminus of connexin 32, which underlie X-linked Charcot-Marie-Tooth disease, have been shown to cause intracellular accumulation of the mutant protein, suggesting that this region is vital for the correct transport of the connexin into the plasma membrane (Martin et al., 2000). Seven mutations (G21R, G22E, K23T, S27P, I31M, R33X, A40V) have been reported in the first transmembrane domain of connexin 43, 2 (V85M, L90V) in the second transmembrane domain, and 1 (V216L) in the fourth transmembrane domain; the 4 transmembrane domains are predominantly α-helical and between them are thought to be responsible for anchoring the connexon in the plasma membrane and determining the properties of the channel (Rabionet et al., 2002; Skerrett et al., 2002; Richard, 2003). Six mutations (Q49K, F52dup, P59H, S69Y, R76S, R76H) have been reported in the first extracellular loop of connexin 43 and 3 (H194P, S201Y, R202H) in the second; the extracellular domains are thought to be crucial for the correct docking of 2 adjacent connexons, and it is therefore possible that the substitution of an amino acid in either of the extracellular domains may prevent connexon interaction (Foote et al., 1998; Evans and Martin, 2002; Martin and Evans, 2004). The cytoplasmic loop runs from amino acid residues 97 to 167, and the 13 mutations (Y98C, K102N, L113P, I130T, K134E, K134N, G138R, G138D, G138S, G143D, G143S, R148Q, A154T) described in this region of the protein are spread throughout the domain; it has been suggested that the cytoplasmic loop is involved in the chemical gating through phosphorylation of the gap junction, and its variability is thought to determine the various functions of each connexin (Anumonwo et al., 2001; Richard, 2003). Overall, the mutations described in this manuscript are entirely consistent with those described previously, in that they are missense changes that are spread throughout the functional domains of the amino-terminal two-thirds of connexin 43.
The expression pattern of Gja1, previously delineated by whole-mount in situ hybridization (Ruangvoravat and Lo, 1992; Richardson et al., 2004; Pemberton et al., 2007), is supported by the protein expression pattern of connexin 43 presented here. Connexin 43 expression was detected throughout murine tooth development. At E10.5, distinct regions of connexin 43 expression were seen in the ventral epithelium of the maxillary processes and the dorsal epithelium of the mandibular processes, which correspond to the earliest stages of tooth development (Jernvall and Thesleff, 2000; Miletich and Sharpe, 2003). During E11 and E12, connexin 43 expression shifted to the odontogenic mesenchyme, corresponding to the signaling potential that drives development of the tooth germs at this stage of embryogenesis (Jernvall and Thesleff, 2000; Miletich and Sharpe, 2003). At E13.5, connexin 43 expression reverted to the enamel organ, which ultimately forms the enamel. In P0 molar tooth germs, the secretory ameloblasts and the stratum intermedium stained strongly for connexin 43. At this stage of development, the ameloblasts actively secrete matrix proteins, which contribute to enamel formation. In adult incisors, secretory ameloblasts also exhibited strong connexin 43 expression, with lower levels being detected in the stratum intermedium. In contrast, although strong connexin 43 expression was observed throughout the stratum intermedium underlying the maturation-stage ameloblasts, staining in the ameloblasts themselves was restricted to the distal junctional complex. Although the precise function of the stratum intermedium is not known, it is believed to be essential for normal enamel formation, and both this layer and the ameloblasts are known to exhibit a high degree of organization and cell communication (Kagayama et al., 1995). In this context, mice heterozygous for either a G60S or G138R mutation in connexin 43 exhibit small, fragile incisors that are covered with hypoplastic enamel that is prone to faster abrasion (Flenniken et al., 2005; Dobrowolski et al., 2008).
Strong connexin 43 expression was also detected during the development of the eyes. At E9.5, expression was observed in the optic cup and in the neuroepithelium of the brain, both of which are continuous at this stage. Interestingly, loss of connexin 43 during brain development in the mouse has been shown to result in abnormalities very similar to those exhibited by a subset of persons with ODD (Wiencken-Barger et al., 2007). Connexin 43 expression persisted in the outer epithelium of the optic cup, which will form the pigmented layer of the retina, and was also detected in the invaginating lens vesicle at E10.5. Expression was also seen in the developing optic nerve and, later in development, in the ciliary body, suggesting an important role for connexin 43 throughout development of the eye; indeed, it has recently been suggested that loss of connexin 43 expression in the ciliary body results in abnormal aqueous humor production (Calera et al., 2006). Analysis of the developing limbs demonstrated diffuse connexin 43 expression throughout the epithelium and mesenchyme in the apical region of the forelimb bud at E9, but by E12, this pattern is narrowed to the apical tip of the limb bud corresponding to the apical ectodermal ridge, which plays a central role in the signaling events driving limb development. Connexin 43 and gap junction communication have been shown to be vital for the correct development of the limbs, indicating the importance of this protein during embryogenesis (Makarenkova and Patel, 1999). Strong connexin 43 expression was also detected in the fusing secondary palate and hair follicles, development of which is also affected in ODD (Sugar et al., 1966; Patton and Laurence, 1985).
The strong correlation between the sites of Gja1 expression during mouse embryonic development and the ODD phenotype provides further support for mutations in GJA1 underlying ODD. Taken together, these results indicate that connexin 43 plays a key role in normal facial and limb development, and that mutation of GJA1 results in craniofacial anomalies and limb abnormalities. These results also confirm a role for connexin 43 throughout the development of the dentition, from the initiation of the tooth germs to the maintenance of adult teeth.
ACKNOWLEDGMENTS
We thank all the families and the clinicians for their involvement in this project. This research was funded by the Wellcome Trust (075945).
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License http://creativecommons.org/licenses/by-nc-nd/3.0/us/, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
REFERENCES
- Anumonwo JM, Taffet SM, Gu H, Chamson M, Moreno AP, Delmar M. The carboxyl terminal domain regulates the unitary conductance and voltage dependence of connexin 40 gap junction channels. Circ Res. 2001;88:666–673. doi: 10.1161/hh0701.088833. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC. Gap junctions: new tools, new answers, new questions. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- Calera MR, Topley HL, Liao Y, Duling BR, Paul DL, Goodenough DA. Connexin43 is required for production of the aqueous humor in the murine eye. J Cell Sci. 2006;119(Pt 21):4510–4519. doi: 10.1242/jcs.03202. [DOI] [PubMed] [Google Scholar]
- de la Parra DR, Zenteno JC. A new GJA1 (connexin 43) mutation causing oculodentodigital dysplasia associated to uncommon features. Ophthalmic Genet. 2007;28:198–202. doi: 10.1080/13816810701538620. [DOI] [PubMed] [Google Scholar]
- Debeer P, Van Esch H, Huysmans C, Pijkels E, De Smet L, Van de Ven W, et al. Novel GJA1 mutations in patients with oculo-dento-digital dysplasia (ODDD) Eur J Med Genet. 2005;48:377–387. doi: 10.1016/j.ejmg.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Dobrowolski R, Sasse P, Schrickel JW, Watkins M, Kim JS, Rackauskas M, et al. The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum Mol Genet. 2008;17:539–554. doi: 10.1093/hmg/ddm329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans WH, Martin PEM. Gap junctions: structure and function. Mol Membr Biol. 2002;19:121–136. doi: 10.1080/09687680210139839. [DOI] [PubMed] [Google Scholar]
- Flenniken AM, Osborne LR, Anderson N, Ciliberti N, Fleming C, Gittens JE, et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132:4375–4386. doi: 10.1242/dev.02011. [DOI] [PubMed] [Google Scholar]
- Foote CI, Zhou L, Zhu X, Nicholson BJ. The pattern of disulphide linkages in the extracellular loop regions of connexin 32 suggests a model for the docking interface of gap junctions. J Cell Biol. 1998;140:1187–1197. doi: 10.1083/jcb.140.5.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernvall J, Thesleff I. Reiterative signalling and patterning during mammalian tooth morphogenesis. Mech Dev. 2000;92:19–29. doi: 10.1016/s0925-4773(99)00322-6. [DOI] [PubMed] [Google Scholar]
- Kagayama M, Akita H, Sasano Y. Immunohistochemical localization of connexin 43 in the developing tooth germ of rat. Anat Embryol (Berl) 1995;191:561–568. doi: 10.1007/BF00186744. [DOI] [PubMed] [Google Scholar]
- Kelly SC, Ratajczak P, Keller M, Purcell SM, Griffin T, Richard G. A novel GJA 1 mutation in oculo-dento-digital dysplasia with curly hair and hyperkeratosis. Eur J Dermatol. 2006;16:241–245. [PubMed] [Google Scholar]
- Kjaer KW, Hansen L, Eiberg H, Leicht P, Opitz JM, Tommerup N. Novel Connexin 43 (GJA1) mutation causes oculo-dento-digital dysplasia with curly hair. Am J Med Genet A. 2004;127:152–157. doi: 10.1002/ajmg.a.20614. [DOI] [PubMed] [Google Scholar]
- Makarenkova H, Patel K. Gap junction signalling mediated through connexin-43 is required for chick limb development. Dev Biol. 1999;207:380–392. doi: 10.1006/dbio.1998.9171. [DOI] [PubMed] [Google Scholar]
- Martin PEM, Evans WH. Incorporation of connexins into plasma membranes and gap junctions. Cardiovasc Res. 2004;62:378–387. doi: 10.1016/j.cardiores.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Martin PEM, Mambetisaeva ET, Archer DA, George CH, Evans WH. Analysis of gap junction assembly using mutated connexins detected in Charcot Marie Tooth X linked disease. J Neurochem. 2000;74:711–720. doi: 10.1046/j.1471-4159.2000.740711.x. [DOI] [PubMed] [Google Scholar]
- Miletich I, Sharpe PT. Normal and abnormal dental development. Hum Mol Genet. 2003;12:69–73. doi: 10.1093/hmg/ddg085. [DOI] [PubMed] [Google Scholar]
- Patton MA, Laurence KM. Three new cases of oculodentodigital (ODD) syndrome: development of the facial phenotype. J Med Genet. 1985;22:386–389. doi: 10.1136/jmg.22.5.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton TJ, Li FY, Oka S, Mendoza-Fandino GA, Hsu YH, Bringas P, Jr, et al. Identification of novel genes expressed during mouse tooth development by microarray gene expression analysis. Dev Dyn. 2007;236:2245–2257. doi: 10.1002/dvdy.21226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzuti A, Flex E, Mingarelli R, Salpietro C, Zelante L, Dallapiccola B. A homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phenotype. Hum Mutat. 2004;23:286. doi: 10.1002/humu.9220. [DOI] [PubMed] [Google Scholar]
- Rabionet R, López-Bigas N, Lourdes Arbonès M, Estivill X. Connexin mutations in hearing loss, dermatological and neurological disorders. Trends Mol Med. 2002;8:205–212. doi: 10.1016/s1471-4914(02)02327-4. [DOI] [PubMed] [Google Scholar]
- Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, et al. Cardiac malformations in neonatal mice lacking connexin 43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- Richard G. Connexin gene pathology. Clin Exp Dermatol. 2003;28:397–409. doi: 10.1046/j.1365-2230.2003.01312.x. [DOI] [PubMed] [Google Scholar]
- Richardson R, Donnai D, Meire F, Dixon MJ. Expression of GJA1 correlates with the phenotype observed in oculodentodigital syndrome/type III syndactyly. J Med Genet. 2004;41:60–67. doi: 10.1136/jmg.2003.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Joss S, Tomkin S, Ahmed M, Sheridan E, Dixon MJ. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome. J Med Genet. 2006;43:e37. doi: 10.1136/jmg.2005.037655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruangvoravat CP, Lo CW. Connexin 43 expression in the mouse embryo: localisation of transcripts within developmentally significant domains. Dev Dyn. 1992;194:261–281. doi: 10.1002/aja.1001940403. [DOI] [PubMed] [Google Scholar]
- Skerrett IM, Aronowitz J, Shin JH, Cymes G, Kasperek E, Cao FL, et al. Identification of amino acid residues lining the pore of a gap junction channel. J Cell Biol. 2002;159:349–360. doi: 10.1083/jcb.200207060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugar HS, Thompson JP, Davis JD. The oculo-dento-digital dysplasia syndrome. Am J Ophthalmol. 1966;61:1448–1451. doi: 10.1016/0002-9394(66)90484-3. [DOI] [PubMed] [Google Scholar]
- van Es RJ, Wittebol-Post D, Beemer FA. Oculodentodigital dysplasia with mandibular retrognathism and absence of syndactyly: a case report with a novel mutation in the Connexin 43 gene. Int J Oral Maxillofac Surg. 2007;36:858–860. doi: 10.1016/j.ijom.2007.03.004. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Spruijt L, van der Burgt I, Bladergroen RS, Vermeer M, Steijlen PM, et al. A 2-bp deletion in the GJA1 gene is associated with oculo-dento-digital dysplasia with palmoplantar keratoderma. Am J Med Genet A. 2005;132:171–174. doi: 10.1002/ajmg.a.30412. [DOI] [PubMed] [Google Scholar]
- Vasconcellos JP, Melo MB, Schimiti RB, Bressanim NC, Costa FF, Costa VP. A novel mutation in the GJA1 gene in a family with oculodentodigital dysplasia. Arch Ophthalmol. 2005;123:1422–1426. doi: 10.1001/archopht.123.10.1422. [DOI] [PubMed] [Google Scholar]
- Vitiello C, D’Adamo P, Gentile F, Vingolo EM, Gasparini P, Banfi S. A novel GJA1 mutation causes oculodentodigital dysplasia without syndactyly. Am J Med Genet A. 2005;133:58–60. doi: 10.1002/ajmg.a.30554. [DOI] [PubMed] [Google Scholar]
- Vreeburg M, de Zwart-Storm EA, Schouten MI, Nellen RG, Marcus-Soekarman D, Devies M, et al. Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. Am J Med Genet A. 2007;143:360–363. doi: 10.1002/ajmg.a.31558. [DOI] [PubMed] [Google Scholar]
- Wiencken-Barger AE, Djukic B, Casper KB, McCarthy KD. A role for Connexin43 during neurodevelopment. Glia. 2007;55:675–686. doi: 10.1002/glia.20484. [DOI] [PubMed] [Google Scholar]