

Abstract
Homeobox transcription factor Nkx2-5, highly expressed in heart, is a critical factor during early embryonic cardiac development. In this study, using tamoxifen-inducible Nkx2-5 knockout mice, we demonstrate the role of Nkx2-5 in conduction and contraction in neonates within 4 days after perinatal tamoxifen injection. Conduction defect was accompanied by reduction in ventricular expression of the cardiac voltage-gated Na+ channel pore-forming α-subunit (Nav1.5-α), the largest ion channel in the heart responsive for rapid depolarization of the action potential, which leads to increased intracellular Ca2+ for contraction (conduction-contraction coupling). In addition, expression of ryanodine receptor 2, through which Ca2+ is released from sarcoplasmic reticulum, was substantially reduced in Nkx2-5 knockout mice. These results indicate that Nkx2-5 function is critical not only during cardiac development but also in perinatal hearts, by regulating expression of several important gene products involved in conduction and contraction.
Keywords: conduction, contraction, gene targeting, transcription
Coordinated conduction and contraction is critical for cardiac function. Cardiac conduction is initiated by a spontaneous wave of electricity (action potential) that arises from sinoatrial (SA) nodal cells located in the upper right atrium, followed by sequential spreading of action potential to the atria, atrioventricular (AV) node, His bundles, peripheral Purkinje fiber, and the ventricles. In the SA and AV nodes, inward Ca2+ currents are primarily responsible for slow depolarization of the action potential, whereas Na+ currents are primarily responsible for rapid depolarization of the action potential in other cardiomyocytes.1-5
In both neonatal and adult mouse cardiomyocytes, robust Na+ currents producing the rapid upstroke action potential are tetrodotoxin-resistant.6,7 In the mouse heart, tetrodotoxin-resistant Nav1.5 is the primary isoform of the Na+ channel mRNA throughout development and is most highly expressed in adults; embryonic day (ED)12.5 50% to 100%, neonatal 150% to 200%, and adult 500% to 550%, in comparison to a house keeping gene.8 Other Na+ channels, mostly tetrodotoxin-sensitive, are also expressed at lower levels in the heart at the 3 stages.8 Among them, the highest expression was demonstrated for Nav2.3 and Nav1.4 in the adult heart but at 25% to 30% at most compared to the house keeping gene.8 In adult mouse ventricular cardiomyocytes, 16% of Na+ channel transcripts are other than Nav1.5, expressed in the order of Nav1.4>Nav1.3>Nav1.2>Nav1.1>Nav1.6.4
After membrane depolarization, L-type Ca2+ channels are activated, followed by Ca2+ release from intracellular Ca2+ stores in sarcoplasmic reticulum (SR) through the cardiac isoform of the ryanodine receptor (RyR2) (Ca2+-induced Ca2+ release). Sufficient intracellular Ca2+ allows actomyosin interactions resulting in cardiac contraction (excitation-contraction coupling).1-5 Presumably, ion channels preferentially expressed in heart, such as Nav1.5 and RyR2, are transcribed in a cardiac-specific manner.
Nkx2-5, a homeodomain-containing transcription factor, is identified as a mammalian homolog of Drosophila tinman,9 critical for cardiac mesoderm formation. Nkx2-5 is highly conserved among species and is among the earliest cardiogenic markers,10,11 with its expression continuing throughout adulthood.12-14 Targeted disruption of Nkx2-5 in mice causes embryonic lethality around ED10.5, with retarded cardiac development.15,16 Ventricular-restricted Nkx2-5 knockout around ED8.0 to ED8.5, created by crossing floxed-Nkx2-5 mice with myosin light chain 2v-Cre knock-in mice, survive and demonstrate progressive and advanced conduction defects and left ventricular hypertrophy postnatally.17 In humans, mutations in NKX2-5 cause various cardiac anomalies in an autosomal dominant fashion, as well as progressive conduction defects and occasional left ventricular dysfunction, which become apparent after birth.18-21
We hypothesize that Nkx2-5 actively regulates a critical set of genes in postnatal cardiomyocytes to maintain proper cardiac function. We used tamoxifen-inducible knockout mice to examine our hypothesis and to elucidate Nkx2-5-dependent regulatory pathways in the perinatal hearts. We describe its effects on cardiomyocyte function, which differ from recently published results on formation of cardiac conduction systems.22,23
Materials and Methods
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org and describes the following: inducible Nkx2-5 knockout mice; Southern, Northern, and Western blotting and immunostaining; surface ECG recording and ultrasound imaging; histological analyses; gene expression profiling; measurement of sodium current and simultaneous recording of cardiac contraction and Ca2+; real-time RT-PCR; reporter assays; and statistical analyses.
Results
Generation of Perinatal Nkx2-5 Knockout Mice
To examine whether Nkx2-5 expression is necessary for perinatal development and maintenance and function of the heart, we generated tamoxifen-inducible Nkx2-5 knockout mice that carry homozygous floxed-Nkx2-5 alleles17 and heterozygous Cre-ER™ transgene under the control of the CMV enhancer and the chicken β-globin promoter (Figure 1A).24 Cre-recombinase activities, including tissue preferences, dose-dependent effects of tamoxifen, were extensively analyzed by Hayashi and McMahon using the R26R reporter mice.24 Heart was demonstrated as among of the most effective organs for Cre-mediated knockout24; however, spatial Cre-recombinase activities in the heart have not yet been analyzed.
Figure 1.
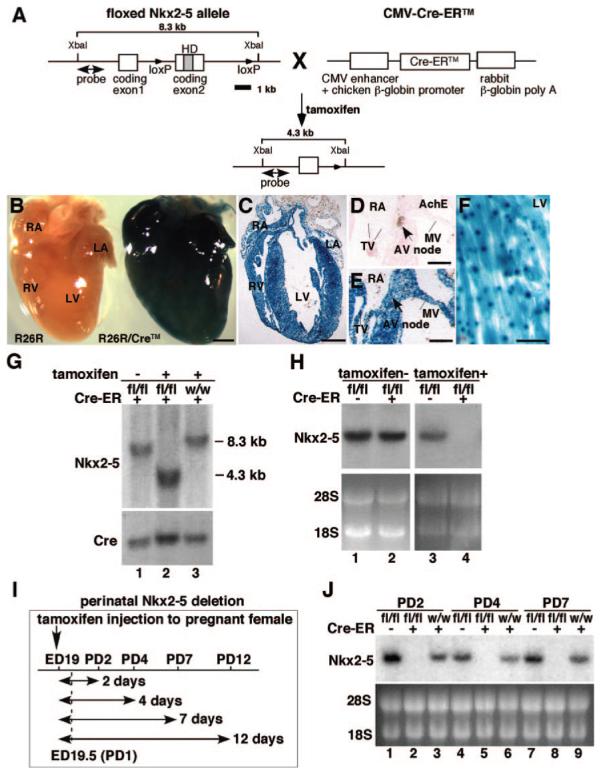
Schematics of generation of tamoxifen-inducible Nkx2-5 knockout mice. A, Homozygous mice for floxed-Nkx2-5 were bred with heterozygous Cre-ER™ transgenic mice. Subsequent matings between offspring generated Nkx2-5flox/flox/Tg-Cre-ER™ (flox/flox/Cre), Nkx2-5wild/wild/Tg-Cre-ER™ (wild/wild/Cre), and Nkx2-5flox/flox (flox/flox). A 8.3-kb XbaI-digested genomic DNA fragments hybridized with the 5′ probe were reduced to 4.3 kb after tamoxifen-induced Cre-recombinase activity. Nkx2-5 gene has 2 coding exons. B through H, Recombination efficiency of Tg-Cre-ER™ in R26R reporter mice. B, Whole-mount staining for β-galactosidase with or without Cre-ER™ transgene. Frozen heart sections of R26R/Tg-Cre-ER™ stained for β-galactosidase. C through F, Four chamber saggital section (C), acetylcholine esterase staining of AV node (D) and adjacent tissue section including AV node (E) and left ventricle (F). Scale bars: 500 μm (B and C); 200 μm (D and E); and 20 μm (F). G, Southern blotting confirmed 8.3-kb genomic DNA fragment isolated from hearts of flox/flox/Cre mice without tamoxifen injection (lane 1) and 4.3 kb after tamoxifen injection (lane 2). Cre-mediated recombination was absent after tamoxifen injection in homozygous wild type carrying the Cre-ER™ transgene (lane 3). H, Northern blotting demonstrated that Nkx2-5 mRNA remained unchanged without tamoxifen injections, but was below detection level with tamoxifen injection (lane 2 vs 4). I, Experimental time course study of Nkx2-5 perinatal knockout. J, Northern blotting confirmed near complete loss of Nkx2-5 mRNA from PD2 to PD7 in flox/flox/Cre mice (lanes 2, 5, and 8). HD indicates homeodomain; fl, flox; w, wild; RA, right atrium; LA, left atrium; RV, right ventricle; LV, left ventricle; TV, tricuspid valve; MV, mitral valve; AV, atrioventricular.
We analyzed cardiac β-galactosidase expression in R26R reporter mice and found it throughout the heart, including atria, ventricles, valves, and AV node, which expresses acetylcholine esterase in the adjacent tissue section 4 days after a single injection of tamoxifen into pregnant mice on gestational day 19 (Figure 1B through 1E and Figure 1 in the online data supplement). In ventricles, ubiquitously expressed lacZ staining was detected (Figure 1F), consistent with a previous report.24
Using this system, Nkx2-5 genes were effectively deleted in the heart, as demonstrated by Southern (Figure 1G), and Northern (Figure 1H) blotting. To delete floxed-Nkx2-5 alleles, both Cre-ER™ transgene and tamoxifen injections were required (Figure 1G and 1H). Time course studies and Nkx2-5 mRNA expression at postnatal day (PD)2 to PD7 are shown in Figure 1I and 1J. Of note, this knockout mouse model is not heart-specific for deletion of Nkx2-5 genes. Although during the fetal period, Nkx2-5 is detected in cardiac and extracardiac tissues including, spleen, stomach, liver, tongue, and anterior larynx, after birth, Nkx2-5 expression in extracardiac tissues is markedly downregulated and is essentially limited to cardiomyocytes.12-14 Nkx2-5 expression appeared to be slightly higher in flox/flox mice compared to wild/wild/Cre mice. This could be attributable to the modification of the Nkx2-5 alleles during insertion of loxP sites in intronic and 3′ noncoding sequences, leading to increased transcription of Nkx2-5, by modifying transcription factor binding or alternating microRNA regulation in these regions.25
Rapid Progressive Conduction Defects and Heart Failure in Perinatal Nkx2-5 Knockout Mice
Surface ECG recordings at PD4 demonstrate that Nkx2-5 knockout mice exhibited prolonged PR interval and QRS duration (Figure 2A, top traces, and the Table), which was further prolonged and accompanied by occasional 2° to 3° AV blocks at PD12 (Figure 2A, bottom traces, and the Table). Heart weight/body weight was also increased significantly at PD4 and was further increased at PD7 and PD12 (Figure 2B). Enlarged hearts in Nkx2-5 knockout mice at PD12 with distended ventricular cavities (Figure 2C), accompanied by reduced cardiac contraction using 55 MHz ultrasound imaging, are shown (Figure 2D). Survival studies revealed that approximately 50% of Nkx2-5 knockout mice died before 3 weeks of age (9 of 16 mice). All Nkx2-5 knockout mice surviving at 4 weeks of age demonstrated conduction defects and enlarged hearts (supplemental Figure II).
Figure 2.
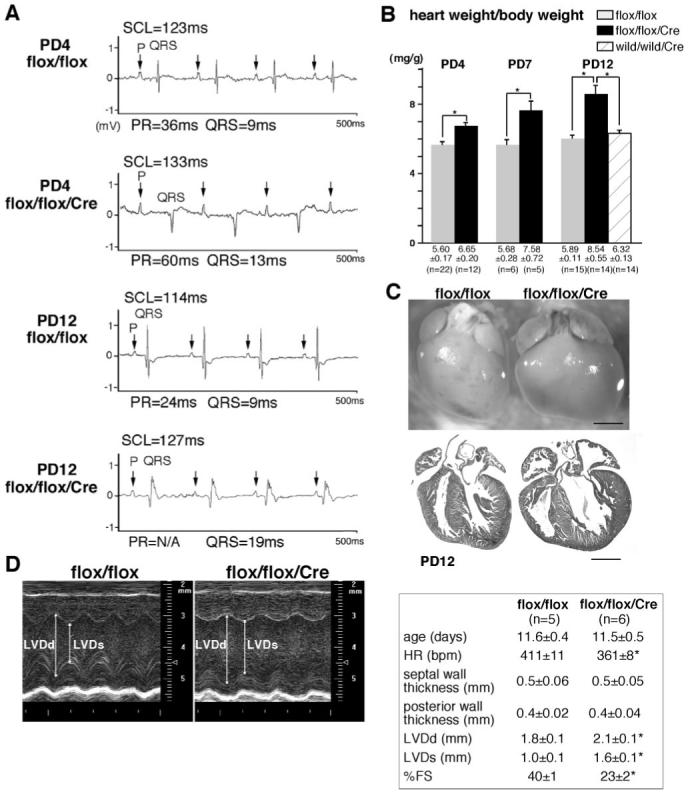
Progressive AV block, prolongation of QRS duration, and heart enlargement in Nkx2-5 knockout mice. A, Surface ECG recordings (lead I) from PD4 and PD12 flox/flox mice showed normal sinus rhythm, but PD4 flox/flox/Cre mice showed PR-prolongation and left bundle branch block, and flox/flox/Cre mice at PD12 showed independent P and QRS waves (complete AV block) with wide QRS complex (see the Table). B, Heart weight/body weight (mg/g) of flox/flox, flox/flox/Cre and wild/wild/Cre mice with tamoxifen injections (mean±SE). C, Hearts dissected from PD12 flox/flox and flox/flox/Cre mice demonstrate dilation of atria and ventricles in Nkx2-5 knockout mice. Hematoxylin/eosin-stained heart sections are shown. Scale bars=1 mm. D, Representative imaging of M-mode ultrasound biomicroscope. Echocardiographic indices of control and Nkx2-5 knockout mice around PD12 (mean±SE) are shown. SCL indicates sinus cycle length (beat-to-beat heart rate); LVDd, left ventricular diastolic dimension; LVDs, left ventricular systolic dimension; HR, heart rate; %FS, percentage of left ventricular fractional shortening.
Table. Electrocardiographic data of perinatal knockout of Nkx2-5 (surface ECG).
| SCL, ms | HR, bpm | P, ms | PR, ms | QRS, ms | QTc, ms | |
|---|---|---|---|---|---|---|
| PD4 | ||||||
| flox/flox (n = 9) | 106 ± 9 | 551 ± 46 | 8 ± 0 | 37 ± 3 | 10 ± 2 | N/A |
| flox/flox/Cre (n = 5) | 105 ± 13 | 575 ± 76 | 8 ± 0 | 46 ± 6* | 14 ± 2* | N/A |
| PD12 | ||||||
| flox/flox (n = 6) | 112 ± 11 | 536 ± 50 | 8 ± 0 | 31 ± 4 | 12 ± 2 | 50 ± 4 |
| flox/flox/Cre (n = 7) | 107 ± 12 | 564 ± 61 | 8 ± 1 | 36 ± 5* | 17 ± 4* | 50 ± 9 |
| wild/wild/Cre (n = 7) | 108 ± 16 | 567 ± 89 | 8 ± 0 | 28 ± 2 | 10 ± 1 | 57 ± 12 |
Values are expressed as mean±SE *P<0.05. SCL, sinus cycle length; HR, heart rate (beat per minute); QTc, rate-corrected QT interval.
These results indicate that perinatal deletion of Nkx2-5 results in early conduction and contraction defects with approximately 50% lethality before 3 weeks of age. Notably, these defects appear sooner and progress more rapidly than those seen in mice with embryonic ventricular-restricted deletion of Nkx2-5, likely attributable to loss of Nkx2-5 throughout the heart without compensation during embryonic cardiac development.17
Reduced Expression of Nkx2-5 Protein in Perinatal Nkx2-5 Knockout Hearts
Immunostaining confirmed decreased Nkx2-5 protein expression in Nkx2-5 knockout ventricles at PD4 (Figure 3A, left images). Despite heart enlargement and reduced contraction in PD12 Nkx2-5 knockout mice, apparent interstitial fibrosis was not detected in ventricles (Figure 3A, right images). In adjacent tissue sections including acetylcholine esterase positive AV nodes, positive Nkx2-5 staining was demonstrated in control but not in Nkx2-5 knockout hearts (Figure 3B). Although formation of the AV node is complete before birth,26 whole-mount acetylcholine esterase staining demonstrated that AV nodal surface area size was smaller in Nkx2-5 knockout heart at PD12 (Figure 3C and 3D), in agreement with a previous study using ventricular-specific deletion of the Nkx2-5 genes at the embryonic stage.17 In contrast to a previous study, perinatal Nkx2-5 knockout mice did not show apparent fibrosis in the AV node (Figure 3E).
Figure 3.
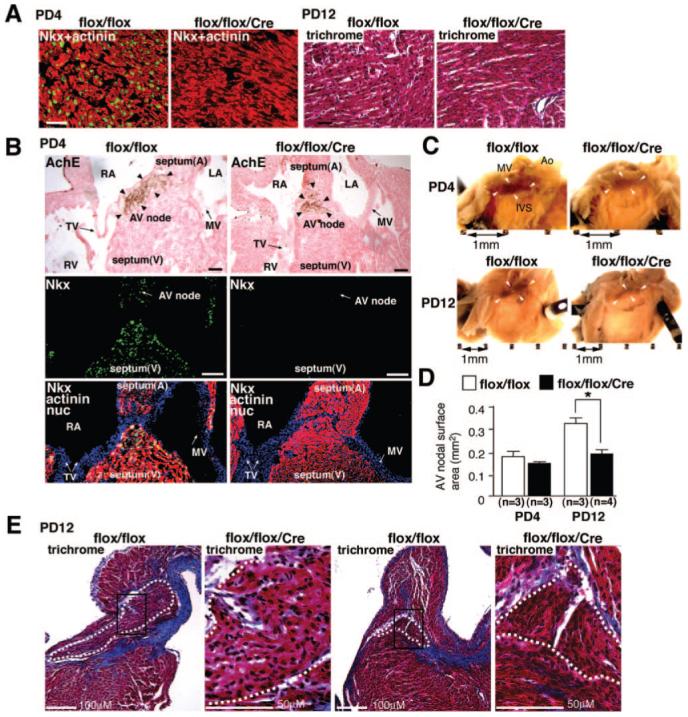
Nkx2-5 protein expression in contractile myocardium and AV node. A, Coimmunostaining of Nkx2-5 (green) and sarcomeric actinin (red) demonstrate reduced expression of Nkx2-5 proteins in ventricles dissected from Nkx2-5 knockout mice at PD4 (left images). Heart sections from PD12 mice stained with Masson’s trichrome show no apparent interstitial fibrosis in Nkx2-5 knockout mice (right images). B, Tissue sections of PD4 hearts demonstrate AV nodes positive for acetylcholine esterase in flox/flox and flox/flox/Cre mice (top images, arrowheads). Adjacent tissue sections including AV node are positive for Nkx2-5 (green) in flox/flox mice but not in flox/flox/Cre mice (middle images). Merged images of Nkx2-5 (green), sarcomeric actinin (red), and nuclear stainings (blue) are shown (bottom images). Scale bars=100 μm/L. C, Whole-mount acetylcholine esterase staining demonstrates AV nodes (brown) both in flox/flox and flox/flox/Cre mice at PD4 and PD12. Scale bars=1 mm. D, Surface area positive for acetylcholine esterase compared between flox/flox and flox/flox/Cre mice at PD4 and PD12 (mean±SE). *P<0.01. E, No fibrosis was observed in Nkx2-5 knockout AV node by Masson’s trichrome staining at PD12. AV nodal area is marked with white dots. Scales are indicated. A indicates atrium; V, ventricle; RA, right atrium; LA, left atrium; RV, right ventricle; TV, tricuspid valve; MV, mitral valve; Ao, aorta; IVS, interventricular septum.
Reduced Expression of Ion Channels in Nkx2-5 Knockout Hearts
In addition to the intriguing morphological changes in the AV node, within 4 days after tamoxifen injection, we found a reduction in levels of several ion channels in Nkx2-5 knockout hearts using gene expression profiling in whole hearts. The 6 highest reduced gene products are T-type Ca2+ channels α1H, KcneI/minK, T-type Ca2+ channels α1G, Nav1.5-α subunit, RyR2, and gap junction protein α 5 (connexin40) from a total of 191 (supplemental Tables I and II). We confirmed reduction of T-type Ca2+ channels (both α1G and α1H), Nav1.5-α subunit, and RyR2 (Figure 4A), and KcneI/minK (Figure 4B) from PD2 to PD7. Expression of the validated genes were decreased progressively, comparable to a known direct target of Nkx2-5, nuclear protein HOP/HOD (homeodomain only protein) (Figure 4B).27,28 Considering different turnover of individual mRNA after loss of Nkx2-5, the validated genes would be directly regulated by Nkx2-5. Thus, in agreement with previous studies, reduction of KcneI/minK and HOP/HOD,17 as well as connexin40,17,29,30 was demonstrated in perinatal Nkx2-5 knockout hearts. A reduction of Nav1.5(α), T-type Ca2+ channels (both α1G and α1H), and RyR2 is newly identified in this study.
Figure 4.
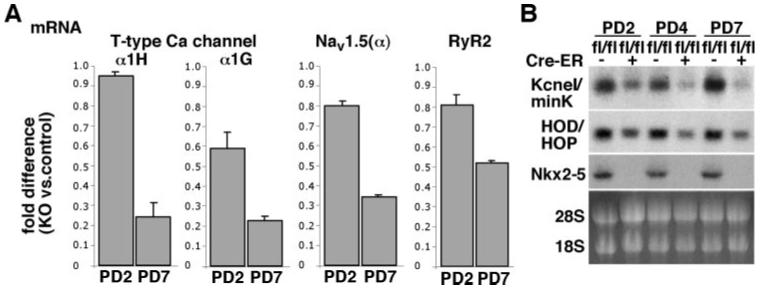
Reduced expression of ion channels in Nkx2-5 knockout hearts. A, Real-time RT-PCR shows fold difference (Nkx2-5 vs control) of mRNA of T-type Ca channels (α1G and α1H), Nav1.5(α), and RyR2 at PD2 and 7. B, Northern blotting reveals KcneI/minK and HOP/HOD mRNA reduction in Nkx2-5 knockout hearts. Of note, HOP/HOD is reported as a direct target of Nkx2-5.27,28 HOP/HOD indicates homeodomain only protein. See supplemental Tables I and II.
Reduced Expression of Nav1.5(α) in Ventricles but Not in Atria of Nkx2-5 Knockout Mice
Na+ channels are responsible for rapid depolarization in contractile myocardium, His bundle, and Purkinje fibers. Nav1.5 is the primary cardiac isoform of Na+ channel, which is composed of a pore-forming α subunit and auxiliary β subunits. Thus, reduction of Nav1.5(α) may be substantial determinant for conduction defects seen in Nkx2-5 knockout mice. We further investigated reduction of Nav1.5(α) mRNA from PD2 to PD12 (Figure 5A) and protein at PD4 and PD12 (Figure 5B) using whole hearts, as well as in the Na+ channel density in ventricular cardiomyocytes (Figure 5C through 5E) in perinatal Nkx2-5 knockout mice.
Figure 5.
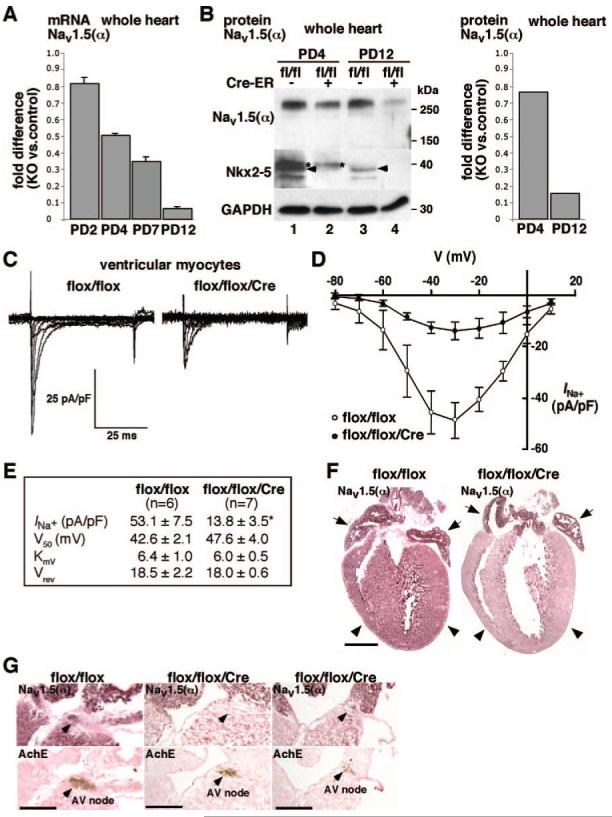
Reduced expression of Nav1.5 α-subunit and reduced INa in Nkx2-5 knockout ventricles. A, Real-time RT-PCR demonstrates progressive reduction of Nav1.5(α) mRNA at PD2, PD4, PD7, and PD12. B, Western blotting demonstrates progressive reduction of Nav1.5(α) protein at PD4 and 12 (lanes 1 vs 2 and 3 vs 4). Fold difference compared to control hearts normalized to GAPDH is shown in right image. Nkx2-5 proteins (arrowheads) are below the level of detection in flox/flox/Cre mice at PD4 and PD12 (arrowheads). C, Representative INa trace recordings from cardiac myocytes obtained from flox/flox (left) and flox/flox/Cre mice (right) using 10 mmol/L Na+ as charge carrier. Myocytes were maintained at -110 mV holding potential with stimulation to -80 to +10 mV at 10-mV steps for 100 ms at 0.1 Hz. D, Current-voltage relation plot for INa in flox/flox (n=6) and flox/flox/Cre (n=7) myocytes, total 7 to 8 days after tamoxifen injections. INa are normalized by cell capacitance and presented as mean±SE. E, Table of kinetic values with maximal current density obtained at -30-mV stimulation. V50, obtained from steady-state activation analysis is the voltage potential determined to elicit the half-maximal INa; KmV is the slope factor; and Vrev is the reversal potential for INa. F, Immunostaining of Nav1.5(α) demonstrates reduced Nav1.5(α) protein in ventricles from Nkx2-5 knockout mice (right images, arrowheads) but preserved expression in atria at PD12 (right images, arrows). Scale bar=1 mm. G, Immunostaining of Nav1.5(α) protein (top) and acetylcholine esterase staining in adjacent tissue sections (bottom). In flox/flox mice, part of AV node is positive for Nav1.5(α) (left images). In flox/flox/Cre mice, serial sections including AV node demonstrate Nav1.5(α) staining absent in the section shown in middle images but is present at a limited part of AV node in the section shown in right images. Scale bars=500 μm. *P<0.01.
Immunohistochemistry in control flox/flox hearts demonstrates Nav1.5(α) staining in both atria and ventricles, with slightly higher intensity in atria in agreement with a recent study (Figure 5F, flox/flox, left).31 Consistently, ventricular Nav1.5(α) staining was markedly reduced in Nkx2-5 knockout hearts; however, atrial Nav1.5(α) staining was similar to control hearts despite the loss of Nkx2-5 (Figure 5F, flox/flox/Cre, right; see Figure 3B and supplemental Figures 3 through 5). In adjacent tissue sections including acetylcholine esterase positive AV node, Nav1.5(α) staining was detected in a limited area of AV node in control hearts and was further limited in Nkx2-5 knockout hearts (Figure 5G). Thus, despite global knockout of Nkx2-5 in the heart, Nav1.5(α) expression is reduced in ventricles, and potentially in AV node, whereas its expression is maintained in atria, and potentially in His-Purkinje fibers (see supplemental Figure IV).
Expression of other Na+ channels in the heart was compared between control and Nkx2-5 knockout heart at PD12 using quantitative real-time PCR; Nav1.4(α), 1.53±0.09 (fold difference of knockout versus control, average±SE, n=2); Nav1.7(α), 2.44±0.04; Nav2.3(α), 0.33±0.00; Nav1(β), 1.90±0.06. Expression of Nav1.1(α), Nav1.3(α), and Nav1.6(α) was below the level of detection. Thus, Nkx2-5 knockout hearts demonstrated increases in Nav1.4(α), Nav1.7(α), and Nav1(β) and reduction in Nav2.3(α).
Scn5a Proximal Promoter Region Contains Nkx2-5 Responsive Elements
Relatively rapid and progressive reduction of Nav1.5(α) expression in Nkx2-5 knockout hearts suggests that transcription of Scn5a may be directly regulated by Nkx2-5 in most ventricles. Scn5a is a large gene approximately 144 kb, including 95 kb for 28 exons, and 30 kb of 5′ and 19 kb of 3′ noncoding sequence. In this study, we focused on identification of Nkx2-5-responsive elements in upstream regulatory sequences conserved among mouse, rat and human.
Proximal promoter regions (-736 to +119 bp) were examined in reporter assays in the absence or presence of Nkx2-5 in 10T1/2 cells (Figure 6A). Nkx2-5 transactivated the reporter constructs Scn5a(-736 to +119) by 1.8-fold relative to the promoter-less reporter construct defined as 1. Two Nkx2-5 core binding sequences (AAG) are found at -628 bp and -590 bp. Mutations at 2 sites (AAG to GGG) abolished Nkx2-5 responsiveness similar to baseline luciferase reporter activity.
Figure 6.
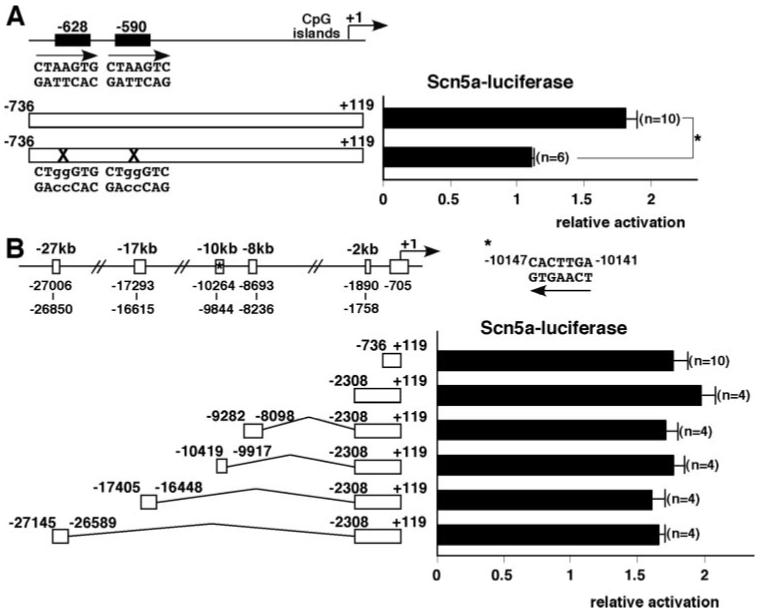
Proximal promoter of Scn5a gene contains Nkx2-5 responsive elements. A, Schematics of 2 Nkx2-5 consensus binding sites in the Scn5a promoter at -628 and -590 bp. Scn5a promoter has CpG islands, but not a TATA box. Scn5a(-736 to +119) (top) and mutated Scn5a(-736 to +119) (bottom), and corresponding relative luciferase reporter activities with pcDNA3-Nkx2-5 cotransfection normalized to β-galactosidase activity and pcDNA empty vector with the value in promoter-less luciferase reporter defined as 1 (means ± SE). ANOVA demonstrated significant difference between 2 groups (F=22.1, P=0.0003). *P=0.0003 by Bonferroni’s post hoc test. B, Schematics of conserved genomic regions among mouse, rat, and human Scn5a gene approximately -27, -17, -10, -8, and -2 kb upstream of the transcriptional start site. Six luciferase reporter constructs and corresponding relative luciferase reporter activities with pcDNA3-Nkx2-5 cotransfection normalized to β-galactosidase activity and pcDNA empty vector with the value in promoter-less luciferase reporter defined as 1 (means±SE). ANOVA demonstrated no significant difference among groups (F=1.08, P=0.3966).
Next, 5 upstream conserved genomic regions were examined for potential enhancer activities (Figure 6B). The -10 147-bp site contains the conserved perfect Nkx2-5 binding sequence uniquely identified in the entire Scn5a genome using rVista 2.0 program (http://rvista.dcode.org). However, inclusion of a -2-kb fragment (-2308 to +119), as well as addition of 4 conserved regions did not enhance luciferase activities compared to the proximal promoter construct (-736 to +119). Therefore, Nkx2-5 responsiveness was demonstrated only in the proximal promoter region in reporter assays.
Reduced Contractility Accompanied by Ca2+-Handling Defects in Cardiomyocytes Isolated From Nkx2-5 Knockout Heart
Because cardiac conduction and contraction are closely coupled, reduction of Nav1.5(α) alone likely causes contraction defects as in human patients with SCN5A mutations.32,33 In addition, gene expression profiling demonstrated reduction of RyR2, a cardiac isoform responsible for Ca2+ release from SR in Nkx2-5 knockout hearts (supplemental Table I), which was confirmed by real-time RT-PCR (Figures 4A and 7A) and Western blotting (Figure 7B). Expression of other Ca2+-handling proteins responsible for restoration of Ca2+ into SR, including sarcoendoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) and its regulatory protein phospholamban was modestly reduced in Nkx2-5 knockout hearts (Figure 7B). Consistently, Nkx2-5 knockout cardiomyocytes showed reduced contractile and Ca2+-handling parameters (Figures 7C and 7D). Therefore, altered expression of Ca2+-handling proteins, including RyR2, SERCA2a, and phospholamban, is also responsible for the depressed contractility of Nkx2-5 knockout hearts.
Figure 7.
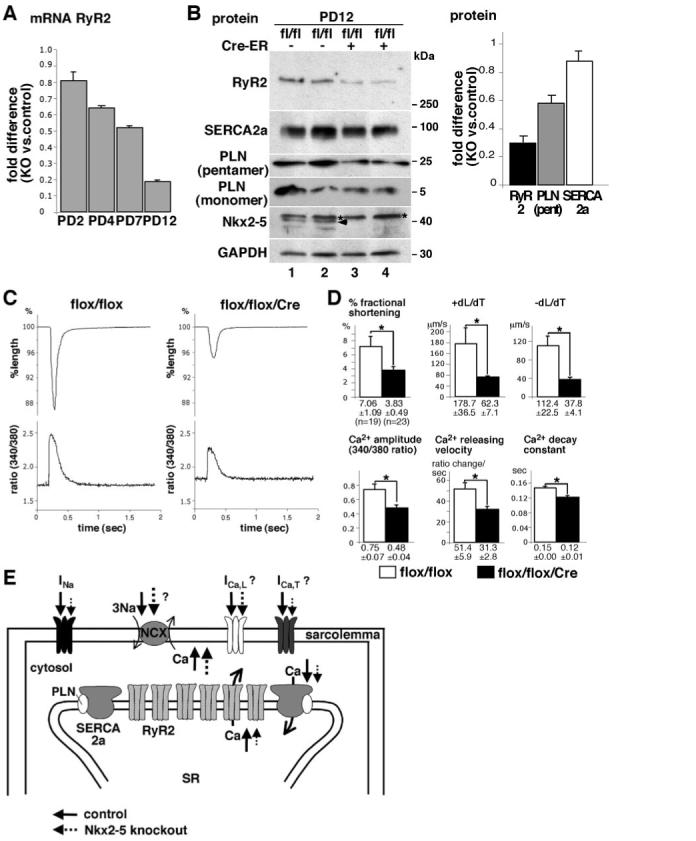
Reduced contractility accompanied by Ca2+-handling defects in cardiomyocytes isolated from Nkx2-5 knockout heart. A, Real-time RT-PCR demonstrates progressive reduction of RyR2 mRNA at PD2, PD4, PD7, and PD12. B, Western blotting demonstrates marked reduction of RyR2 proteins and modest reduction of SERCA2a and phospholamban (PLN) (pentamer and monomer) in heart lysates obtained from Nkx2-5 knockout mice compared to control mice at PD12. Fold difference compared to control hearts normalized with GAPDH is shown at right (mean±SE, n=2). Note, because of different molecular sizes of RyR2 (≈560 kDa), phospholamban monomer (≈5 kDa) and GAPDH (≈30 kDa), equal amounts of protein were loaded on separate gels. Signal intensities in Western blotting were normalized to those of GAPDH. C, Representative tracings of a single contraction (top) and simultaneous Ca2+ transients (bottom) in an isolated cardiomyocyte from control flox/flox (left) or Nkx2-5 knockout (flox/flox/Cre) mice (right). D, Summarized data (mean±SE) obtained from multiple cardiomyocytes demonstrate that Nkx2-5 knockout cardiomyocytes show decreased percentage fractional shortening, +dL/dT (speed of contraction), and -dL/dT (speed of relaxation), Ca2+ amplitude, Ca2+-releasing velocity, and Ca2+ decay constant. *P<0.01. E, Simplified Na+ and Ca2+ transport through sarcolemma and SR in cardiomyocytes (modified from a previously published figure41). Model for the altered excitation-contraction coupling in Nkx2-5 knockout cardiomyocytes; reduced Na+ inward currents through Nav1.5, reduced Ca2+ inward currents possibly through T-type Ca2+ channels and reverse mode of sodium-calcium exchanger (increased Na+ entry and Ca2+ exit) may reduce Ca2+ inflow into cytosol (dotted lines). In addition, Ca2+ release from SR to cytosol, as well as Ca2+ uptake to SR, is reduced by reduction of RyR2 and SERCA2a in Nkx2-5 knockout hearts (dotted lines). Overall, altered cytosolic Ca2+ handling would decrease cardiac contraction.
Of note, cardiomyocytes isolated from surviving mice were examined at 3 weeks of age for contraction because of technical difficulties in isolation when using younger mice.
Discussion
Nkx2-5 function is not limited to embryonic heart development; it is critical for both conduction and contraction in the perinatal heart, within 4 days after tamoxifen injection, as demonstrated in this study. Initiation of action potential in rapid conduction cardiomyocytes relies on Na+ inward currents, which were markedly reduced in the ventricles of Nkx2-5 knockout mice, because of reduced expression of pore-forming subunit Nav1.5(α). In addition, Ca2+ release from SR was diminished, accompanied by substantial reduction in RyR2 expression. Because excitation and contraction are tightly coupled, reduced handling of Na+ and Ca2+ demonstrated in Nkx2-5 knockout cardiomyocytes may mutually lead to rapid conduction and contraction defects.
Mutations in the human SCN5A gene, which encodes Nav1.5(α), lead to Brugada syndrome and long-QT syndrome, resulting from loss-of-function or gain-of-function mechanisms.2,4,5,34 Homozygous knockout of Scn5a resulted in embryonic lethality around ED10.5,35 and heterozygous knockout of Scn5a, in which Na+ current density was reduced by approximately 43% (21 pA/pF in Scn5a+/- versus 37 pA/pF in wild type) with virtually identical channel-gating characteristics, led to prolonged P wave, AV block, and intraventricular conduction defects.35 Na+ current density in ventricular cardiomyocytes was reduced by approximately 75% in Nkx2-5 knockout mice compared to wild type (13.8 versus 53.1 pA/pF; Figure 5E) at equivalent PD7 to PD8, likely resulting in more profound ventricular conduction defects than those found in Scn5a+/- mice.
Nkx2-5 knockout mice did not demonstrate prolongation of P wave (Table). Consistently, expression of Nav1.5(α) in atria of Nkx2-5 knockout mice was not reduced despite marked reduction of Nkx2-5. This indicates that Nkx2-5 is critical for transcription of Scn5a in the majority of ventricular cardiomyocytes but not atrial cardiomyocytes.
Despite a number of studies on functional regulation of Nav1.5,4,5 transcriptional regulation of Scn5a gene is not well understood. It is repressed by the zinc finger protein Snail.5,36 It is unlikely that upregulation of Snail is the mechanism in Nkx2-5 knockout hearts, based on gene expression profiling data (fold difference flox/flox/Cre versus flox/flox is 0.98±0.04 in Snail1; 1.04±0.03 in Snail2; mean±SE; n=4). We examined 5′ upstream genomic regions of Scn5a and found that 2 Nkx2-5 binding sites in the proximal promoter separated by 38 bp are responsive for Nkx2-5dependent transcription, whereas 5 potential enhancer elements did not further increase transcription in reporter assays.
Cardiomyocytes isolated from Nkx2-5 knockout mice demonstrated substantial Ca2+-handling defects, particularly in Ca2+ release, with approximately a 70% reduction of RyR2 protein at PD12. Intracellular Ca2+ homeostasis is well balanced in normal heart but is altered in a number of human and rodent heart failure models, and it is primarily responsible for the depressed contractility of failing hearts.37-39 Perhaps crosstalk among the Ca2+-handling proteins is important, as has been demonstrated in SERCA2a+/- mice, in which its negative regulator phospholamban is also decreased, maintaining the constant ratio of SERCA2a and phospholamban.40 Thus, reduction of Ca2+-handling proteins is not specific for Nkx2-5 knockout hearts, except for profound defects in Ca2+ release accompanied by substantial reduction of RyR2 expression. In addition, elevation of the Na+ inward current through Na+ channels is usually observed in failing hearts compensatory to increase intracellular Ca2+ through the sodium calcium exchanger (Ca2+ influx and Na+ efflux),41 although this was not likely the case in Nkx2-5 knockout hearts.
Shortly after Nkx2-5 knockout, in addition to reduction of Nav1.5(α) and RyR2 expression, expression of other ion channels, including T-type Ca2+ channels (α1G and α1H), was reduced. Because T-type Ca2+ channels also play a small role in the increase of intracellular Ca2+ for contraction, and a substantial role in depolarization in slow conduction tissues,1,3,42 reduction in mRNA of T-type Ca2+ channels in Nkx2-5 knockout hearts would also contribute to the phenotype.
We propose a model for the potential mechanisms of cardiac dysfunction following loss of Nkx2-5 (Figure 7E). Reduced Na+ entry into cardiomyocytes may lead to compensatory increases in reverse mode of the sodium-calcium exchanger (increased Na+ entry and Ca2+ exit) and a further decrease in intracellular Ca2+.33 A reduction of cardiacspecific myosin light chain kinase and phosphorylation of myosin light chain 2v was demonstrated in perinatal Nkx2-5 knockout mice,43 which likely also contributes to contractile defects.
Cre-ER™ transgenic mice,24 widely used for Cre-mediated gene deletion in different tissues, demonstrated near 100% recombination frequency in hearts from R26R-reporter mice; therefore, it is suitable for functional analyses of individual cardiomyocytes. Expression of Nkx2-5 in postnatal stage is nearly limited to cardiomyocytes12-14; thus, global deletion of Nkx2-5 using Cre-ER™ mice does not likely affect cardiac phenotype. An alternative approach would be to use mice with cardiomyocyte-specific expression of tamoxifen-inducible Cre-recombinase through the use of αMHC-Cre transgenic mice.44 However, myocyte-specific recombination frequency was reported to be approximately 70% to 80%, and, to our knowledge, spatial recombination efficacy in the highly specialized AV nodal cardiomyocytes has not been reported in this mouse model.44
In summary, we demonstrate that Nkx2-5 is critical for perinatal conduction and contraction by regulating expression of several ion channel genes. Our findings may provide potential explanations for progressive conduction and contraction defects seen after birth in patients with congenital heart disease associated with NKX2-5 mutations.
Supplementary Material
Acknowledgments
We greatly appreciate C. Ketcham, E. Scott, and E. Chan for valuable suggestions and technical support.
Sources of Funding
This work was supported by an American Heart Association National Scientific Development Grant and NIH grant HL081577 (to H.K.).
Footnotes
Disclosures
None.
References
- 1.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 2.Marban E. Cardiac channelopathies. Nature. 2002;415:213–218. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- 3.Mangoni ME, Couette B, Marger L, Bourinet E, Striessnig J, Nargeot J. Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog Biophys Mol Biol. 2006;90:38–63. doi: 10.1016/j.pbiomolbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Haufe V, Chamberland C, Dumaine R. The promiscuous nature of the cardiac sodium current. J Mol Cell Cardiol. 2007;42:469–477. doi: 10.1016/j.yjmcc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Abriel H. Roles and regulation of the cardiac sodium channel Na(v) 1.5: recent insights from experimental studies. Cardiovasc Res. 2007;76:381–389. doi: 10.1016/j.cardiores.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 6.Benndorf K, Boldt W, Nilius B. Sodium current in single myocardial mouse cells. Pflugers Arch. 1985;404:190–196. doi: 10.1007/BF00585418. [DOI] [PubMed] [Google Scholar]
- 7.Nuss HB, Marban E. Electrophysiological properties of neonatal mouse cardiac myocytes in primary culture. J Physiol. 1994;479(pt 2):265–279. doi: 10.1113/jphysiol.1994.sp020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrell MD, Harbi S, Hoffman JF, Zavadil J, Coetzee WA. Large-scale analysis of ion channel gene expression in the mouse heart during perinatal development. Physiol Genomics. 2007;28:273–283. doi: 10.1152/physiolgenomics.00163.2006. [DOI] [PubMed] [Google Scholar]
- 9.Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–729. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- 10.Harvey RP. NK-2 homeobox genes and heart development. Dev Biol. 1996;178:203–216. doi: 10.1006/dbio.1996.0212. [DOI] [PubMed] [Google Scholar]
- 11.Harvey RP, Rosenthal N, editors. Heart Development. Academic Press; San Diego, Calif: 1999. [Google Scholar]
- 12.Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development. 1993;119:419–431. doi: 10.1242/dev.119.2.419. [DOI] [PubMed] [Google Scholar]
- 13.Komuro I, Izumo S. Csx: a murine homeobox-containing gene specifically expressed in the developing heart. Proc Natl Acad Sci U S A. 1993;90:8145–8149. doi: 10.1073/pnas.90.17.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasahara H, Bartunkova S, Schinke M, Tanaka M, Izumo S. Cardiac and extracardiac expression of Csx/Nkx2.5 homeodomain protein. Circ Res. 1998;82:936–946. doi: 10.1161/01.res.82.9.936. [DOI] [PubMed] [Google Scholar]
- 15.Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–1280. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- 17.Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, Ho SY, Benson DW, Silberbach M, Shou W, Chien KR. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117:373–386. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 18.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 19.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasahara H, Benson DW. Biochemical analyses of eight NKX2.5 homeodomain missense mutations causing atrioventricular block and cardiac anomalies. Cardiovasc Res. 2004;64:40–51. doi: 10.1016/j.cardiores.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Konig K, Will JC, Berger F, Muller D, Benson DW. Familial congenital heart disease, progressive atrioventricular block and the cardiac homeobox transcription factor gene NKX2.5: identification of a novel mutation. Clin Res Cardiol. 2006;95:499–503. doi: 10.1007/s00392-006-0412-9. [DOI] [PubMed] [Google Scholar]
- 22.Meysen S, Marger L, Hewett KW, Jarry-Guichard T, Agarkova I, Chauvin JP, Perriard JC, Izumo S, Gourdie RG, Mangoni ME, Nargeot J, Gros D, Miquerol L. Nkx2.5 cell-autonomous gene function is required for the postnatal formation of the peripheral ventricular conduction system. Dev Biol. 2007;303:740–753. doi: 10.1016/j.ydbio.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 23.Moskowitz IP, Kim JB, Moore ML, Wolf CM, Peterson MA, Shendure J, Nobrega MA, Yokota Y, Berul C, Izumo S, Seidman JG, Seidman CE. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell. 2007;129:1365–1376. doi: 10.1016/j.cell.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 25.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 26.Sedmera D, Reckova M, DeAlmeida A, Coppen SR, Kubalak SW, Gourdie RG, Thompson RP. Spatiotemporal pattern of commitment to slowed proliferation in the embryonic mouse heart indicates progressive differentiation of the cardiac conduction system. Anat Rec A Discov Mol Cell Evol Biol. 2003;274:773–777. doi: 10.1002/ar.a.10085. [DOI] [PubMed] [Google Scholar]
- 27.Chen F, Kook H, Milewski R, Gitler AD, Lu MM, Li J, Nazarian R, Schnepp R, Jen K, Biben C, Runke G, Mackay JP, Novotny J, Schwartz RJ, Harvey RP, Mullins MC, Epstein JA. Hop is an unusual homeobox gene that modulates cardiac development. Cell. 2002;110:713–723. doi: 10.1016/s0092-8674(02)00932-7. [DOI] [PubMed] [Google Scholar]
- 28.Shin CH, Liu ZP, Passier R, Zhang CL, Wang DZ, Harris TM, Yamagishi H, Richardson JA, Childs G, Olson EN. Modulation of cardiac growth and development by HOP, an unusual homeodomain protein. Cell. 2002;110:725–735. doi: 10.1016/s0092-8674(02)00933-9. [DOI] [PubMed] [Google Scholar]
- 29.Kasahara H, Wakimoto H, Liu M, Maguire CT, Converso KL, Shioi T, Huang WY, Manning WJ, Paul D, Lawitts J, Berul CI, Izumo S. Progressive atrioventricular conduction defects and heart failure in mice expressing a mutant Csx/Nkx2.5 homeoprotein. J Clin Invest. 2001;108:189–201. doi: 10.1172/JCI12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 31.Dominguez JN, de la Rosa A, Navarro F, Franco D, Aranega AE. Tissue distribution and subcellular localization of the cardiac sodium channel during mouse heart development. Cardiovasc Res. 2008;78:45–52. doi: 10.1093/cvr/cvm118. [DOI] [PubMed] [Google Scholar]
- 32.McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 33.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolf CM, Berul CI. Inherited conduction system abnormalities-one group of diseases, many genes. J Cardiovasc Electrophysiol. 2006;17:446–455. doi: 10.1111/j.1540-8167.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 35.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci U S A. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hesse M, Kondo CS, Clark RB, Su L, Allen FL, Geary-Joo CT, Kunnathu S, Severson DL, Nygren A, Giles WR, Cross JC. Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel Scn5a. Cardiovasc Res. 2007;75:498–509. doi: 10.1016/j.cardiores.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 37.Houser SR, Piacentino V, III, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 38.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 39.Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115:556–564. doi: 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Periasamy M, Huke S. SERCA pump level is a critical determinant of Ca(2+)homeostasis and cardiac contractility. J Mol Cell Cardiol. 2001;33:1053–1063. doi: 10.1006/jmcc.2001.1366. [DOI] [PubMed] [Google Scholar]
- 41.Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 42.Yasui K, Niwa N, Takemura H, Opthof T, Muto T, Horiba M, Shimizu A, Lee JK, Honjo H, Kamiya K, Kodama I. Pathophysiological significance of T-type Ca2+ channels: expression of T-type Ca2+ channels in fetal and diseased heart. J Pharmacol Sci. 2005;99:205–210. doi: 10.1254/jphs.fmj05002x3. [DOI] [PubMed] [Google Scholar]
- 43.Chan JY, Takeda M, Briggs LE, Graham ML, Lu JT, Horikoshi N, Weinberg EO, Aoki H, Sato N, Chien KR, Kasahara H. Identification of cardiac-specific myosin light chain kinase. Circ Res. 2008;102:571–580. doi: 10.1161/CIRCRESAHA.107.161687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissuespecific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.