

Abstract
Despite numerous reports citing the acute hepatotoxicity caused by MDMA (3,4-methylenedioxymethamphetamine, ecstasy), the underlying mechanism of organ damage is poorly understood. We hypothesized that key mitochondrial proteins are oxidatively-modified and inactivated in MDMA-exposed tissues. The aim of this study was to identify and investigate the mechanism of inactivation of oxidatively-modified mitochondrial proteins, prior to the extensive mitochondrial dysfunction and liver damage following MDMA exposure. MDMA-treated rats showed abnormal liver histology with significant elevation in plasma transaminases, nitric oxide synthase, and the level of hydrogen peroxide. Oxidatively-modified mitochondrial proteins in control and MDMA-exposed rats were labeled with biotin-N-maleimide (biotin-NM) as a sensitive probe for oxidized proteins, purified with streptavidin-agarose, and resolved using 2-DE. Comparative 2-DE analysis of biotin-NM-labeled proteins revealed markedly increased levels of oxidatively-modified proteins following MDMA exposure. Mass spectrometric analysis identified oxidatively-modified mitochondrial proteins involved in energy supply, fat metabolism, antioxidant defense, and chaperone activities. Among these, the activities of mitochondrial aldehyde dehydrogenase, 3-ketoacyl-CoA thiolases, and ATP synthase were significantly inhibited following MDMA exposure. Our data show for the first time that MDMA causes the oxidative inactivation of key mitochondrial enzymes which most likely contributes to mitochondrial dysfunction and subsequent liver damage in MDMA-exposed animals.
Keywords: MDMA, oxidative stress, liver damage, protein oxidation, mitochondria
1 Introduction
The abuse of 3,4-methylenedioxymethamphetamine (MDMA, Ecstasy)1 is a growing problem in the United States and represents a significant public health problem [1-3]. According to recent epidemiological data, the use of MDMA has reached epidemic proportions not only in the United States but also in many parts of the world [1-3]. Acute exposure to MDMA alone or in combination with other abused substances, including alcohol or cocaine can damage several organs such as the heart, liver, kidney, and brain. This damage can be fatal, as evidenced by multiple deaths related to MDMA abuse [4-7]. For instance, acute MDMA toxicity can lead to myocardial infarction often accompanied with tachycardia, hypertension, and hyperthermia, all of which usually precede disseminated intravascular coagulation, rhabdomyolysis, and multiple organ failure or death [8]. The life threatening clinical manifestations of MDMA toxicity also include acute hepatic damage [8-10], hyponatremia, and rhabdomyolysis-induced renal failure [9]. Despite the well-established toxicities associated with its abuse, MDMA is the second most common cause of liver injury in people under the age of 25 years [9], suggesting a lack of public knowledge of or disregard for its adverse health affects. Various factors may contribute to MDMA-induced tissue injury. For instance, MDMA metabolism [11], the increased efflux of neurotransmitters [11,12], hyperthermia [13], and the oxidation of catecholamines [14,15] are suggested to be involved in MDMA-related neuronal injury. Although many reports have demonstrated MDMA-induced liver damage [8-10], the underlying mechanism accounting for hepatic toxicity is poorly understood. One proposed mechanism suggests that reactive metabolites of MDMA are responsible for causing the MDMA-mediated hepatotoxicity [11,16]. The metabolism of MDMA involves N-demethylation to 3,4-methylenedioxyamphetamine while both MDMA and 3,4-methylenedioxyamphetamine are demethylated to catecholamines [N-methyl-α-methyldopamine and α-methyldopamine, respectively] that can undergo oxidation to corresponding quinones. Newly produced quinones are highly redox-active molecules that can undergo the following pathways: (a) a redox cycle producing semiquinones radicals, leading to the generation of reactive oxygen species (ROS) [17,18]; (b) irreversible 1,4-intramolecular cyclization with subsequent formation of aminochromes [14]; (c) conjugation with reduced glutathione (GSH) to form a glutathionyl adduct that can further react with GSH and protein thiols, leading to GSH depletion [16,19]; and d) formation of protein adducts, leading to inactivation of the target proteins [20]. Taken together, these results indicate that MDMA metabolism with increased production of ROS and/or toxic oxidation products with GSH depletion may be responsible for liver damage. Since antioxidant defense systems become severely impaired upon MDMA exposure, administration of small molecule antioxidants such as N-acetylcysteine and ascorbic acid or over-expression of antioxidant enzymes like superoxide dismutase, can inhibit the toxic effects of MDMA [10,16,21]. Depletion of GSH levels following MDMA exposure correlates with increased lipid peroxidation and cell damage [16,18]. Furthermore, it was shown that nitric oxide and reactive nitrogen species (RNS), including peroxynitrite, are also involved in MDMA-mediated toxicity [22].
Despite the well-established role of increased oxidative/nitrosative stress in MDMA-induced tissue damage [16-18,22], the underlying mechanism by which increased oxidative stress causes organ damage is still poorly understood. Mitochondria, which play a critical role in energy supply, fat degradation, anti-oxidant defense and apoptosis, are known to be a major target of increased oxidative/nitrosative stress upon exposure to toxic compounds and/or in pathological conditions [23,24]. It is possible that MDMA and/or its reactive metabolites [16-18,20] inhibit the mitochondrial function by directly interacting with mitochondrial proteins, as recently demonstrated [20]. In addition, MDMA and metabolites can indirectly cause mitochondrial dysfunction through increased oxidative/nitrosative stress, as similar to the pathological states or the conditions after exposure to potentially toxic drugs [23,24]. Therefore, we hypothesized that MDMA-mediated oxidative/nitrosative stress causes the inactivation of certain key mitochondrial proteins, leading to mitochondrial dysfunction and ultimately tissue damage. To our knowledge, there has not been a systematic proteomic analysis on MDMA-exposed tissues. Instead of investigating the overall pattern of protein changes, we specifically focused on the mitochondrial proteins that are oxidatively-modified following MDMA treatment. Characterization of the oxidatively-modified proteins is expected to increase our understanding of the molecular mechanisms of mitochondrial dysfunction and liver injury associated with MDMA. In this study, we identified oxidatively-modified mitochondrial proteins in MDMA-exposed rat livers by using the cysteinyl residue (Cys)-targeted proteomic method that was recently developed in this laboratory [25-27]. In addition, we determined the enzymatic activities of select oxidatively-modified mitochondrial proteins. The observed oxidative modification and enzymatic inactivations are likely to account for MDMA-induced mitochondrial dysfunction prior to full-blown hepatic injury.
2 Materials and Methods
2.1 Chemicals and other materials
MDMA, biotin-conjugated N-maleimide (biotin-NM), anti-β-ATP synthase antibody, propionyl aldehyde, pyrazole, dithiothreitol (DTT), and sodium dithionite were purchased from Sigma Chemical (St. Louis, MO, USA) in the highest purity available. Anti-3-nitrotyrosine (3-NT) antibody was purchased from Upstate Biotechnologies (Waltham, MA, USA). Specific antibody to horse radish peroxidase (HRP)-conjugated MAb-biotin was purchased from Cell Signaling (Beverly, MA, USA). Heparinized vacutainer tubes were purchased from Becton Dickinson (Franklin Lakes, NJ, USA). The specific anti-3-ketoacyl-CoA thiolase antibody was kindly provided by Dr. Nancy Braverman, Institute of Genomic Medicine, Johns Hopkins University Medical School, Baltimore, MD, USA.
2.2 Animal maintenance and MDMA treatment
Male Sprague Dawley rats (n≥6 per treatment group; 225-250 grams body weight) were purchased from Harlan Laboratories (Indianapolis, IN, USA). Animals were maintained on a 12:12 light:dark cycle in a temperature and humidity controlled environment and given ad libitum access to food and water. The protocol for the animal studies was approved by the Institutional Animal Care and Use Committee of the University of Maryland, School of Pharmacy. MDMA (10 mg/kg), dissolved in water, was administered per orally (p.o.) on Day 1 and 2 in a volume of 4 ml/kg. Control rats received water p.o. on Day 1 and 2 in a similar volume. On Day 2, rats were euthanized by carbon dioxide asphyxiation, 12 h after the last dose of MDMA or water treatment. Blood was collected by cardiac puncture using heparinized syringes, centrifuged for 10 min at 5000 × g, and plasma was stored at -80 °C until analyzed. Liver tissue was immediately excised, blotted dry and stored at -80 °C until analyzed.
2.3 Identification of oxidized proteins using mass spectrometry
Mitochondrial fractions were prepared from pooled rat livers (n≥6 per group) obtained from each treatment group using a recently described method [25,27]. Labeling of oxidized proteins with biotin-NM was performed as described [25-27]. Purified biotin-NM labeled proteins bound to the streptavidin-agarose beads were washed twice prior to their separation using two-dimensional polyacrylamide gel electrophoresis (2-DE). The gels were then silver-stained, scanned, and analyzed. In-gel digestion of protein gel spots, nanoflow reversed-phase liquid chromatography—tandem mass spectro-metry and bioinformatic analyses were performed as recently described [25-27]. Detailed experimental design and protein identification methods were performed according to the recommended guidelines [28] and described in Supplementary Table 1.
2.4 Immunoprecipitation and immunoblot analyses
A separate aliquot of mitochondrial proteins was incubated with 5 μg of anti-β-ATP synthase for 2 h with constant agitation followed by addition of protein G-agarose for an additional 1 h [29]. Proteins bound to the protein G-agarose were washed three times with phosphate buffered saline containing 1% 3-[(3-cholamidopropyl)-1-dimethylammonio]-propanesulfonic acid (CHAPS), to remove non-specifically bound proteins. After centrifugation, bound proteins were dissolved in Laemmli buffer for immunoblot analysis using specific antibodies against each target protein [29].
2.5 Determination of transaminases, hydrogen peroxide, GSH/GSSG ratio, lipid peroxidation, triglyceride and cholesterol concentrations
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were measured in plasma samples using a clinical chemistry analysis system (PROCHEM-V; DREW Scientific, Oxford, CT, USA). Triglycerides and cholesterol levels were determined in the supernatant fraction of liver homogenates (normalized for protein concentration) using a QVET® kit as per manufacturer’s instructions (DREW Scientific) and normalized for the protein concentration. The level of hydrogen peroxide produced in isolated mitochondria was determined using the Amplex® Red Hydrogen Peroxide assay kit (Molecular Probes, Eugene, OR) in the presence of pyruvate (5 mM) and malate (2 mM) [30]. The GSH/GSSG ratio was measured in the supernatant fraction of liver homogenates by using a kit for colorimetric determination of reduced and oxidized glutathione (OXIS International, Inc, Foster City, CA, USA). Malondialdehyde (MDA) levels were measured using Lipid Peroxidation Assay Kit (Calbiochem, San Diego, CA, USA).
2.6 Histological analysis of liver samples
Liver samples were fixed in 10% buffered formalin. After paraffin embedding and cutting 5 μm slices, all sections were stained with hematoxylin and eosin (H&E). Histological evaluation was performed in a blinded manner.
2.7 Activity measurements of various mitochondrial enzymes
Nitric oxide synthase (NOS) activity was measured using 0.5 mg of protein with a fluorescence indicator 4-amino-5-methyl-amino-2′, 7′-difluorofluorescene diacetate, which has excitation and emission wavelengths of 490 and 510 nm, respectively [27]. ALDH2 activity was measured by increased production of NADH, as described [27]. One unit of ALDH2 activity represents a reduction of 1 μmol NAD+/min/mg protein. Activity of 3-ketoacyl-CoA thiolase was determined via an absorbance change at 303 nm following disappearance of the Mg2+—enolate complex of acetoacetyl-CoA (ε303 = 16.5 mM-1 cm-1) [27]. One unit of thiolase was defined as the amount of 3-ketoacyl-CoA thiolase that catalyzes the cleavage of 1 nmol acetoacetyl-CoA to acetyl-CoA/min/mg-protein at ambient temperature. ATP synthase activity was measured using an ATP bioluminescence assay kit (Roche, Mannheim, Germany) following the manufacturer’s protocol. One unit of ATP synthesis activity represents 1 nM ATP produced/min/mg protein at ambient temperature. Activity of mitochondrial complex I (NADH-ubiquinone oxidoreductase) was determined by measuring OD at 340 nm and subtracting the OD values in the presence of rotenone, an inhibitor of NADH-ubiquinone oxidoreductase [31]. One unit represents a reduction of 1 μmol NADH/min/mg protein.
2.8 Data processing and statistical analysis
All data in this report represent results from at least three separate experiments, unless stated otherwise. Statistical analyses were performed using the Student’s t test and p<0.05 was considered statistically significant.
3 Results
3.1 Selection of MDMA dosage
As a first step, we determined the appropriate MDMA dosage for our study. Since damaged liver cells leak cellular enzymes into the plasma, determination of plasma concentrations of the liver enzymes (ALT, AST) is a frequently used and reliable marker for detecting liver damage. Twelve hours after the administration of MDMA at a single oral administration of 10 mg/kg, we observed trends toward liver damage with a mild but statistically insignificant increase in plasma ALT and AST levels (data not shown). However, a significant increase in plasma ALT and AST levels was observed 12 h after the second administration of two consecutive MDMA doses (24 hours apart with 10 mg/kg, oral administration) (Fig. 1A). Accordingly, this MDMA dosing regimen was selected for our study. Our conditions are different from those known to cause severe liver damage (necrosis) observed with a higher dose (20 mg/kg, ip) and multiple exposures [10] but were specifically chosen to identify early changes in oxidative modification of mitochondrial proteins that lead to mitochondrial dysfunction and ultimately liver damage.
Figure 1.
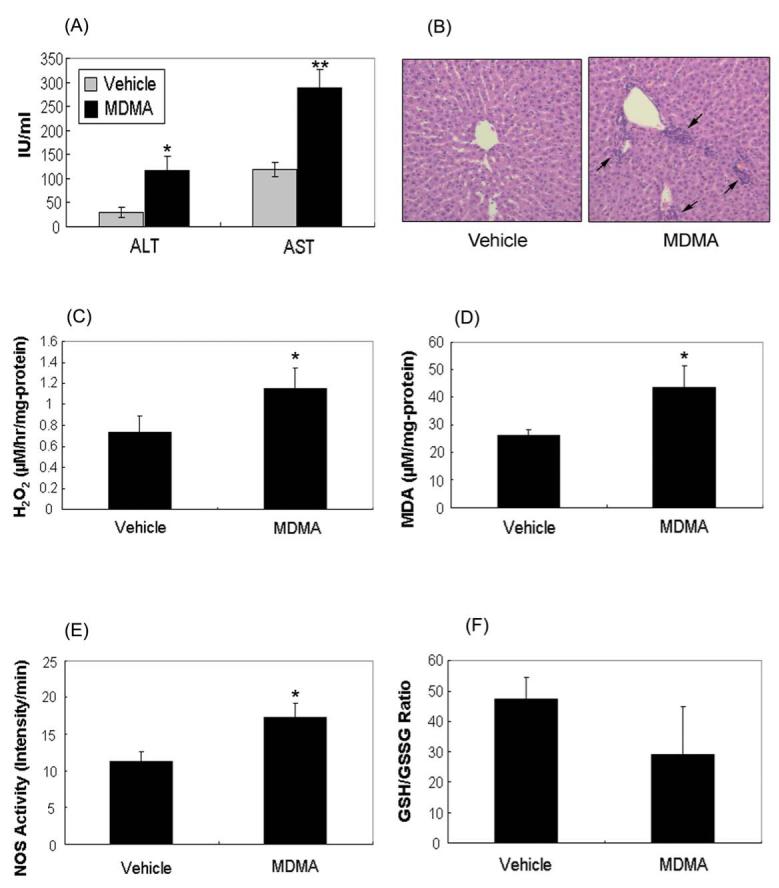
Hepatic damage and oxidative/nitrosative stress are altered following MDMA administration. (A) Plasma ALT and AST levels, (B) hematoxylin and eosin (H&E) staining (magnification × 200), with infiltrating immune cells marked with arrows, (C) mitochondrial hydrogen peroxide production, (D) liver MDA level, (E) NOS activity, and (F) GSH/GSSG ratio were measured in rat livers exposed with vehicle or MDMA. *significantly different from the vehicle-treated control (* p< 0.05 and ** p< 0.01).
3.2 Increased levels of liver damage and oxidative stress after MDMA exposure
To assess hepatocellular damage in exposed rat livers, liver histopathology were evaluated at 12 h after the second dose of MDMA. Under our conditions, vehicle control rats showed normal hepatic histology by H&E staining, while increased sinusoidal congestion and inflammatory cells were observed in MDMA-exposed rat livers (Fig. 1B). To determine the degree of oxidative stress in MDMA-exposed rat liver, the levels of mitochondrial hydrogen peroxide, lipid peroxidation, reduced and oxidized glutathione (GSH/GSSG) ratio, and NOS activity were measured. The level of mitochondrial hydrogen peroxide, an indicator of mitochondrial ROS production, was increased 58% in MDMA-exposed rats compared to vehicle control rats (Fig. 1C). Liver cytosolic level of MDA, an indicator of lipid peroxidation, was significantly elevated from 26.1 ± 2.0 to 43.3 ± 8.1 μM per mg protein after MDMA administration (Fig. 1D). In parallel, the activity of NOS, a source of nitrosative stress, was increased approximately 60% in MDMA-exposed rats compared to vehicle controls (Fig. 1E). Consistent with this result, the mitochondrial nitrite level in MDMA-exposed rats increased by 76% to 2.61 ± 0.48 from 1.48 ± 0.07 μM within control rats (p=0.079). Furthermore, the GSH/GSSG ratio, an indicator of oxidative stress, decreased from 47.3 ± 7.1 to 29.1 ± 15.6 following MDMA treatment (Fig. 1F). These results confirm that under our experimental conditions, MDMA increased oxidative/nitrosative stress and liver damage as seen by others [17,18,22].
3.3 Increased levels of oxidized protein in rat liver mitochondria following MDMA exposure
Since an increase in oxidative/nitrosative stress was observed, we next examined whether the number of oxidatively-modified mitochondrial proteins increased in the MDMA-exposed rat livers compared to vehicle controls. Coomassie blue staining (Fig. 2A, left panel) shows that equal amounts of total proteins were analyzed for the two groups. The number and intensity of biotin-NM labeled proteins detected by immunoblot analysis using the HRP-conjugated-anti-biotin-antibody were markedly elevated in MDMA-exposed rats (Fig. 2A, right panel, lane 2) compared to the controls (lane 1). The levels of the immunoreactive proteins as determined by densitometry were significantly elevated following MDMA treatment (Fig. 2B).
Figure 2.
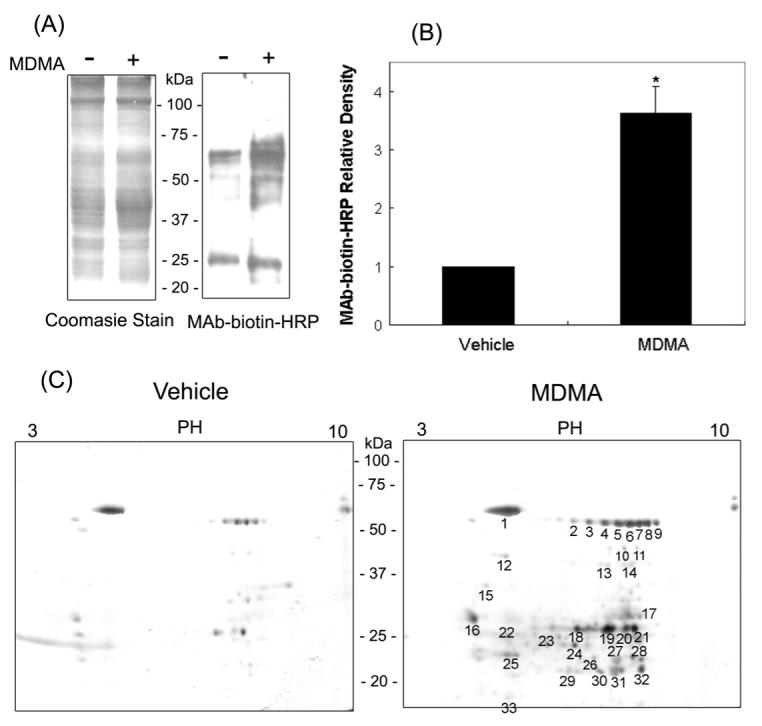
Detection and identification of oxidatively-modified proteins in vehicle control and MDMA-exposed rat livers. (A) Increased levels of oxidized mitochondrial proteins in rat liver following MDMA treatment. Rat liver mitochondria from vehicle control or MDMA-exposed rats were labeled with biotin-NM. Biotin-NM labeled mitochondrial proteins (20 μg/well) were then separated on 12 % SDS-PAGE and stained with Coomassie (left) or subjected to immunoblot analysis (right) using HRP-conjugated monoclonal antibody against biotin (MAb-biotin-HRP). This figure represents a typical result from three different experiments. (B) The density of the MAb-biotin-recognized bands in each lane was determined by using a gel digitizing software (UN-SCAN-IT™, Orem, Utah, USA). The relative density of MDMA-exposed sample was then calculated by comparing the density of the vehicle lane. *significantly different from the vehicle-treated control (* p< 0.025). (C) Comparison of oxidized mitochondrial proteins by 2-DE in vehicle and MDMA-exposed rat livers. Oxidized mitochondrial proteins (10 mg/sample) from vehicle and MDMA-treated rat liver were labeled with biotin-NM and then purified with streptavidin-agarose. Purified biotin-NM labeled proteins (0.25 mg/sample) were resolved by 2-DE, and silver stained. Individual protein spots (spots 1-33) with differential intensities were marked with different numbers, excised out of this particular gel (pH range 3-10), and subjected to MS analysis following in-gel trypsin digestion for protein identification by mass spectrometry. This figure represents a typical result from three independent experiments.
3.4 Identification of oxidatively-modified mitochondrial proteins
We identified the biotin-NM-labeled mitochondrial proteins using tandem mass spectrometry (MS/MS) following 2-DE fractionation [25-27]. The number and intensity of biotin-NM labeled oxidatively-modified protein spots were significantly increased in rat livers obtained from the MDMA-treated group (Fig. 2C, right panel with the statistical analysis of 33 spots shown in Supplementary Fig. 1) compared to livers obtained from the control group (Fig. 2C, left panel). Identities of thirty three spots, representing oxidatively-modified mitochondrial proteins with increased spot intensity in MDMA-exposed samples (right panel), are summarized in Table 1. The proteins identified as being oxidatively-modified after MDMA administration include those involved in: energy supply and oxidative phosphorylation (α- and β-ATP synthase); anti-oxidative defense system [mitochondrial aldehyde dehydrogenase 2 (ALDH2) and methylmalonate-semialdehyde dehydrogenase (ALDH6), ALDH7, ALDHX]; fatty acid and intermediary metabolism [3-ketoacyl-CoA thiolase, 3-hydroxyacy-CoA dehydrogenase 2, enoyl-CoA hydratase, acetyl-CoA acetyltransferase, acyl-CoA dehydrogenase, acetyl-CoA dehydrogenase, glutamate dehydrogenase (GDH), and UDP-glucose pyrophosphorylase]; molecular chaperon proteins [heat shock protein (Hsp) 60, Hsp86, Hsp75, protein disulfide isomerase, and glucose regulated protein (GRP) 75]; mitochondrial electron transport and voltage-dependent calcium protein.
Table 1.
Summary of LC-MS/MS peptide sequence analyses for oxidized proteins in MDMA treated rat liver mitochondria
| Spot No |
Protein Identified | Swiss-Prot Accession |
Detect No |
|---|---|---|---|
| 1 | 60-kDa Heat shock protein (HSP 60) | P63039 | 79 |
| Stress-70 protein (GRP 75) | P48721 | 11 | |
| 78-kDa Glucose-regulated protein (GRP 78) | P06761 | 5 | |
| 2 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 8 |
| Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P11884 | 3 | |
| Acyl-CoA oxidase 2 | P97562 | 2 | |
| Catalase | P04762 | 2 | |
| Heat shock protein HSP 90-alpha (HSP 86) | P82995 | 2 | |
| 3 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 16 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 9 | |
| Aldehyde dehydrogenase 2, mitochondrial (ALDH2) | P11884 | 2 | |
| 4 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 16 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 14 | |
| UDP-glucose pyrophosphorylase 2 | Q4V8I9 | 5 | |
| Aldehyde dehydrogenase family 7 member A1 (ALDH7) | Q64057 | 3 | |
| Aldehyde dehydrogenase X (ALDH X) | Q66HF8 | 3 | |
| Voltage-dependent T-type calcium channel subunit α-1G (VSCC) | O54898 | 2 | |
| ATP synthase subunit alpha (α-ATP) | P15999 | 2 | |
| 5 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 21 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 12 | |
| UDP-glucose pyrophosphorylase 2 | Q4V8I9 | 8 | |
| ATP synthase subunit alpha (α-ATP) | P15999 | 3 | |
| Aldehyde dehydrogenase family 7 member A1 (ALDH7) | Q64057 | 2 | |
| Aldehyde dehydrogenase X (ALDH X) | Q66HF8 | 2 | |
| Cytochrome P450 11B1 | P15393 | 2 | |
| 6 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 16 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 15 | |
| Cytochrome P450 11B1 | P15393 | 2 | |
| UDP-glucose pyrophosphorylase 2 | Q4V8I9 | 2 | |
| 7 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 24 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 12 | |
| UDP-glucose pyrophosphorylase 2 | Q4V8I9 | 5 | |
| ATP synthase subunit alpha (α-ATP) | P15999 | 3 | |
| Fumarate hydratase | P14408 | 2 | |
| 8 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 16 |
| Cytochrome P450 11B1 | P15393 | 3 | |
| Fumarate hydratase | P14408 | 3 | |
| Cytochrome P450 19A1 | P22443 | 2 | |
| 9 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 17 |
| Cytochrome P450 11B1 | P15393 | 2 | |
| 10 | 3-Ketoacyl-CoA thiolase | P13437 | 7 |
| Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 6 | |
| Acetyl-CoA dehydrogenase, short chain | Q6IMX3 | 3 | |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 2 | |
| Long-chain specific acyl-CoA dehydrogenase | P15650 | 2 | |
| 11 | 3-Ketoacyl-CoA thiolase | P13437 | 12 |
| Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 8 | |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 4 | |
| Fumarate hydratase | P14408 | 3 | |
| Acetyl-CoA acetyltransferase | P17764 | 2 | |
| Medium-chain specific acyl-CoA dehydrogenase | P08503 | 2 | |
| 12 | 3-Ketoacyl-CoA thiolase | P13437 | 1 |
| 13 | Probable oxidoreductase | Q68FT3 | 4 |
| 3-Ketoacyl-CoA thiolase | P13437 | 3 | |
| Acetyl-CoA dehydrogenase, short chain | Q6IMX3 | 3 | |
| Protein disulfide-isomerase A6 precursor | Q63081 | 3 | |
| 3-Ketoacyl-CoA thiolase A, peroxisomal | P21775 | 2 | |
| Peroxisomal delta3, delta2-enoyl-CoA isomerase | Q5XIC0 | 2 | |
| 14 | Acetyl-CoA dehydrogenase, short chain | Q6IMX3 | 4 |
| 3-Ketoacyl-CoA thiolase | P13437 | 3 | |
| Peroxisomal delta3, delta2-enoyl-CoA isomerase | Q5XIC0 | 3 | |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 2 | |
| Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 2 | |
| 15 | 60-kDa Heat shock protein (HSP 60) | P63039 | 4 |
| 16 | 60-kDa Heat shock protein (HSP 60) | P63039 | 10 |
| ATP synthase subunit beta (β-ATP) | P10719 | 2 | |
| 17 | Glutamate dehydrogenase 1 (GDH) | P10860 | 4 |
| Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 2 | |
| 18 | Enoyl-CoA hydratase | P14604 | 6 |
| 19 | Enoyl-CoA hydratase | P14604 | 9 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 2 | |
| 20 | Electron transfer flavoprotein subunit beta (β-ETF) | Q68FU3 | 9 |
| 3-Hydroxyacyl-CoA dehydrogenase 2 | O70351 | 7 | |
| Enoyl-CoA hydratase | P14604 | 6 | |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 5 | |
| 21 | Enoyl-CoA hydratase | P14604 | 4 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 2 | |
| 22 | Enoyl-CoA hydratase | P14604 | 5 |
| Heat shock protein 75 kDa (HSP 75) | Q5XHZ0 | 4 | |
| 3-Hydroxyacyl-CoA dehydrogenase 2 | O70351 | 3 | |
| 23 | Enoyl-CoA hydratase | P14604 | 5 |
| 24 | Enoyl-CoA hydratase | P14604 | 5 |
| 25 | Enoyl-CoA hydratase | P14604 | 2 |
| 26 | ATP synthase subunit alpha (α-ATP) | P15999 | 14 |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 2 | |
| 27 | ATP synthase subunit alpha (α-ATP) | P15999 | 8 |
| Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 2 | |
| 28 | ATP synthase subunit alpha (α-ATP) | P15999 | 9 |
| 29 | ATP synthase subunit alpha (α-ATP) | P15999 | 6 |
| ATP synthase D chain | P31399 | 2 | |
| 30 | ATP synthase subunit alpha (α-ATP) | P15999 | 7 |
| 31 | Glutamate dehydrogenase 1 (GDH) | P10860 | 3 |
| Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 3 | |
| 32 | Methylmalonate-semialdehyde dehydrogenase (ALDH6) | Q02253 | 2 |
| 33 | 60-kDa Heat shock protein (HSP 60) | P63039 | 21 |
| Cytochrome b5 | P00173 | 5 | |
| Glutamate dehydrogenase 1 (GDH) | P10860 | 2 | |
| 10-kDa Heat shock protein (HSP 10) | P26772 | 2 | |
| DNA polymerase subunit gamma 1 (Mitochondrial DNA polymerase catalytic subunit) |
Q9QYV8 | 2 |
Biotin-NM labeled oxidized proteins were isolated with streptavidin-agarose, washed, resolved on 2-D gels, and stained with silver. Each protein spot as indicated was picked up with a razor blade and subjected to protein identification using mass spectrometric analysis, as described in the Experimental Procedures and Supplementary Table 1.
Many proteins were identified from spots with apparent molecular weights (e.g. spots 26-30 for α—ATP synthase) lower than that based on their full-length sequence. These spots likely represent fragments of their full-length parent proteins (i.e. 58 kDa for α-ATP synthase). Increased degradation products of proteins were also observed for GDH, HSP60, and ALDH6. Detection of smaller protein fragments may be related to increased susceptibility of oxidized mitochondrial proteins to elevated proteasomal degradation reported under conditions of oxidative stress [32,33] or alternatively, spontaneous fragmentation of oxidized proteins [34].
3.5. Inactivation of Oxidatively-Modified Mitochondrial Enzymes in MDMA-Exposed Rat Livers
Certain amino acids such as Cys, Met, Trp, His and Tyr can undergo various oxidative modifications [34]. The oxidative modifications of multiple mitochondrial proteins observed in this study (Fig. 2) led us to test whether the biological functions of some of these proteins could be inhibited through oxidative modifications of critical Cys and Tyr residues. In fact, several mitochondrial proteins are known to contain Cys residue(s) within their catalytic sites. For instance, ALDH isozymes, including ALDH2, ALDH5, ALDH6, and ALDH7, contain a highly conserved Cys in their catalytic site [35] while the catalytic site of 3-ketoacyl-CoA thiolase contains two Cys residues [36]. GDH also contains a critical Cys residue near the catalytic site [37]. Therefore, we evaluated whether the catalytic activities of ALDH2 and 3-ketoacyl-CoA thiolase were inhibited following MDMA treatment. The ALDH2 activity was 0.16 ± 0.12 units after MDMA exposure. This value represents an approximately 72% reduction in the ALDH2 activity compared to vehicle controls (Fig. 3A). This suppressed ALDH2 activity was completely recovered by the pre-incubation of the MDMA-exposed sample with 12 mM DTT, suggesting that the Cys residues within ALDH2 could be oxidized to form disulfides or sulfenic acid. Alternatively, Cys residues can be S-nitrosylated or S-glutathionylated after MDMA exposure [27,35]. To further demonstrate that the inactivation of ALDH2 enzyme activity was due to its oxidative modification, biotin-NM labeled mitochondrial proteins were analyzed before and after purification with streptavidin-agarose. Immunoblot analysis using a specific anti-ALDH2 antibody detected similar amounts of ALDH2 protein in all treatment groups before purification (Fig. 3B, left panel). After affinity purification using streptavidin-beads, a single band was recognized with the anti-ALDH2 antibody in the MDMA-administrated sample (Fig. 3B, right panel, lane 2) but not in vehicle control groups (lane 1). Addition of DTT to the MDMA-exposed samples prior to biotin-NM labeling causes the disappearance of immunoreactive ALDH2 in the MDMA-treated sample (lane 3). These immunoblot data clearly support that ALDH2 was reversibly oxidized and/or S-nitrosylated after MDMA administration and that this leads to its inactivation.
Figure 3.
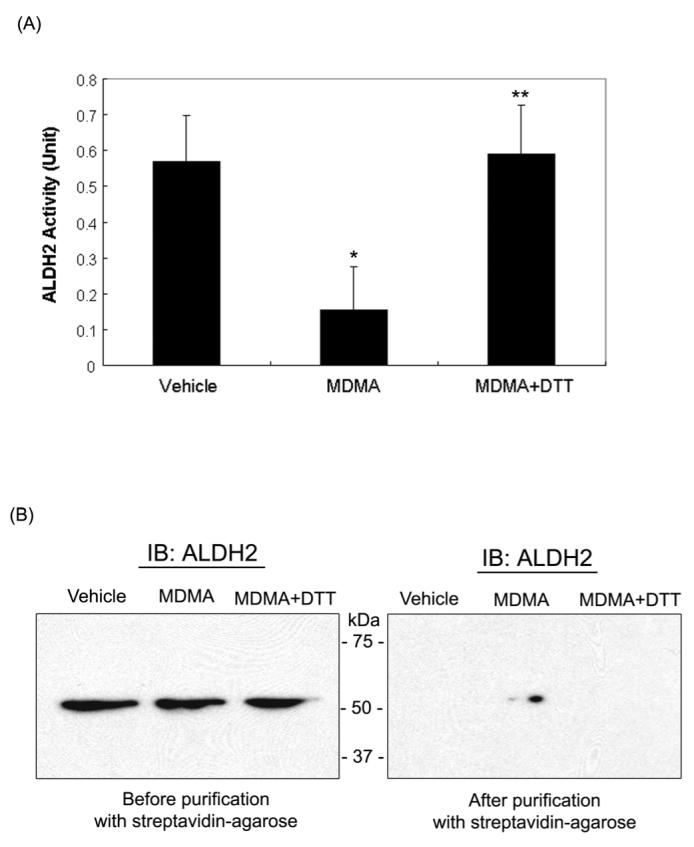
Reversible inactivation and oxidation of mitochondrial ALDH2 in MDMA-exposed rat livers. (A) Mitochondrial ALDH2 activities in vehicle and MDMA-exposed rat livers in the absence or presence of DTT are presented. Before determining the ALDH2 enzyme activity, 12 mM DTT was added to MDMA-exposed group for 30 min at room temperature (third lane). *significantly different from the vehicle treated control (*p<0.005); **significantly different from MDMA-treated group (**p < 0.005). (B) Presence of ALDH2 protein in oxidized mitochondrial proteins was confirmed by immunoblot analysis. Another part of MDMA-administration sample was incubated with 12 mM DTT at room temperature for 30 min (lane 3 on left and right panels) before being subjected to biotin-NM labeling. Biotin-NM-labeled mitochondrial proteins before (left) or after (right) purification with streptavidin-agarose were analyzed by 1D SDS-PAGE, and then subjected to immunoblot analysis (IB) using the specific anti-ALDH2 antibody. This figure represents a typical result from at least two different experiments.
Two Cys residues (Cys92 and Cys382), which are essential for the activity of 3-ketoacyl-CoA thiolase, are likely targets of oxidative modification leading to inactivation of the enzyme [36]. Indeed, mitochondrial 3-ketoacyl-CoA thiolase activity was inhibited approximately 89.1 % by MDMA treatment, and its activity was restored by 83.3% after the addition of 12 mM DTT to the MDMA-exposed groups (Fig. 4A). To further demonstrate that oxidized 3-ketoacyl-CoA thiolase was inactive, biotin-NM labeled mitochondrial proteins were analyzed before and after purification with streptavidin-agarose. Immunoblot analysis using the specific anti-3-ketoacyl-CoA thiolase antibody revealed that similar amounts of 3-ketoacyl-CoA thiolase protein were detected in all treatment groups prior to purification (Fig. 4B, left panel). After streptavidin-agarose affinity purification, a single band was recognized in the MDMA-exposed group (Fig. 4B, right panel, lane 2) but not in vehicle control groups (lane 1) or DTT-treated MDMA-exposed group prior to biotin-NM labeling (lane 3). These immunoblot data indicate that 3-ketoacyl-CoA thiolase becomes reversibly oxidized after MDMA administration, leading to its inactivation. The reduction in the activity of 3-ketoacyl-CoA thiolase is likely to lead to increased free fatty acids in the liver since this enzyme plays a key role in the β-oxidative breakdown of fatty acids in the mitochondria and peroxisomes [38]. Since free fatty acids are used in hepatic triglyceride synthesis, hepatic triglyceride and cholesterol levels were measured to further evaluate functional implication of MDMA-mediated inhibition of this enzyme. The hepatic triglyceride and cholesterol were increased by 26% (p=0.16) and 45% (p=0.11), respectively, in the MDMA-treated samples compared to those of vehicle controls (Fig. 4C). The relatively small increases in the fat levels may result from the moderate MDMA dosage and different exposure time. Although statistically insignificant, the increases in triglycerides and cholesterol levels observed in this study are consistent with an earlier report [10] showing MDMA administration results in elevated levels of triglyceride and cholesterol.
Figure 4.
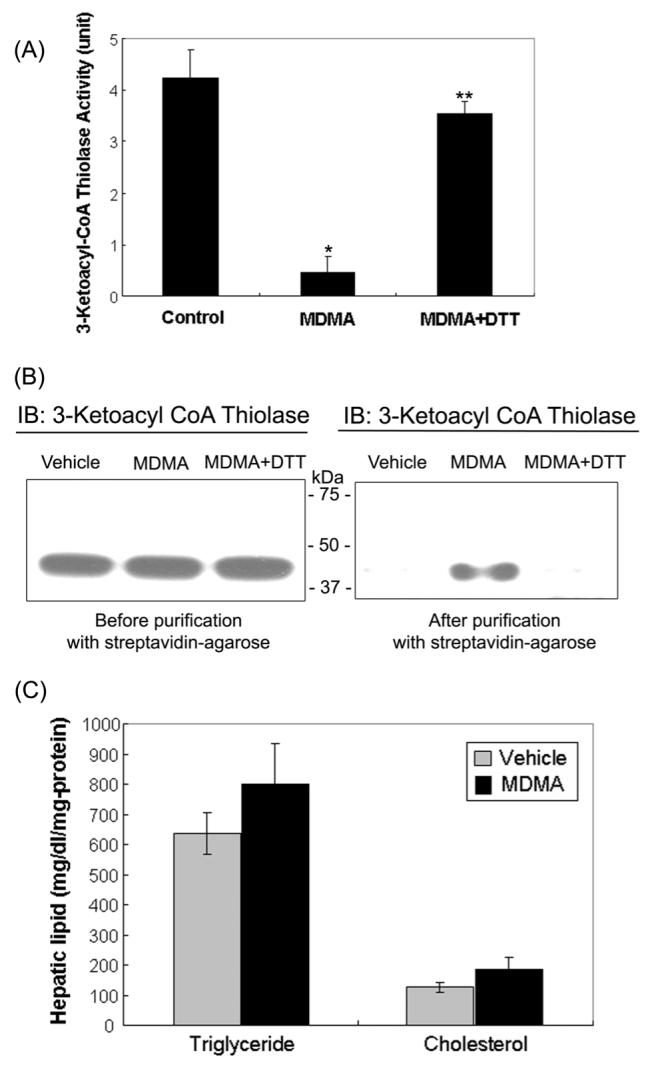
Oxidative inactivation of 3-ketoacyl-CoA thiolase in MDMA-exposed rat liver mitochondria and fat accumulation following MDMA exposure. (A) Mitochondrial 3-ketoacyl-CoA thiolase activities in vehicle and MDMA-treated rat livers were determined before or after preincubation of mitochondrial proteins with and without DTT. To reverse the suppressed enzyme activity, 12 mM DTT was added to the MDMA-treated sample for 30 min at room temperature (third lane) prior to actual assay. *significantly different from the vehicle samples (*p< 0.005); **significantly different from the MDMA samples (**p< 0.0005). (B) Presence of 3-ketoacyl-CoA thiolase protein in oxidized mitochondrial proteins was confirmed by immunoblot analysis. To reverse the oxidized enzyme, another part of MDMA-exposed sample was incubated with 12 mM DTT at room temperature for 30 min (lane 3 on left and right panels) before being subjected to biotin-NM labeling. Biotin-NM-labeled mitochondrial proteins before (left) or after (right) purification with streptavidin-agarose were analyzed by 1D SDS-PAGE, and then subjected to immunoblot analysis (IB) using the specific anti-3-ketoacyl-CoA thiolase antibody. This figure represents a typical result from at least two different experiments. (C) Hepatic levels of total triglyceride and cholesterol in vehicle and MDMA-induced rat livers are presented.
In addition to Cys, other amino acid residues such as Tyr, Trp, His, and Met are also known to undergo oxidative modification [34]. Therefore, we evaluated whether critical Tyr residues in the active site of ATP synthase [39] may undergo nitration by elevated levels of nitrite and/or peroxynitrite in MDMA induced rat livers, leading to its inactivation. Mitochondrial ATP synthase activity was significantly inhibited in MDMA-exposed rats. The hepatic ATP synthase activity was 147.9 ± 13.9 units in vehicle controls, while it was 92.0 ± 16.5 units in MDMA-treated rats (Fig. 5A). To elucidate the mechanism for MDMA-mediated inhibition of ATP synthase activity, β-ATP synthase protein was immunoprecipitated from the mitochondria fractions of vehicle control and MDMA-treated samples. Immunoblot analysis with the specific anti-β-ATP synthase antibody showed that similar levels of β-ATP synthase exist in all conditions (Fig. 5B, left panel). However, the specific anti-3-NT antibody detected an immunoreactive protein in the samples from the MDMA-exposed rat livers (right panel, lane 2) but not from the vehicle control (lane 1). The disappearance of the 3-NT band in the presence of dithionite (lane 3) suggested its conversion to 3-aminoTyr. Furthermore, mitochondrial complex I activity, which could be inactivated by peroxynitrite through the modification of its Tyr residues [40], was significantly inhibited by 69% in MDMA-treated samples (3.90 ± 0.43 units) compared to that of controls (1.19 ± 0.15 units). These results of Tyr modifications of ATP synthase and possibly of the mitochondrial complex I suggest an elevated level of peroxynitrite, although it could not be accurately determined due to an extremely short half-life of peroxynitrite, in MDMA-treated samples compared to controls.
Figure 5.
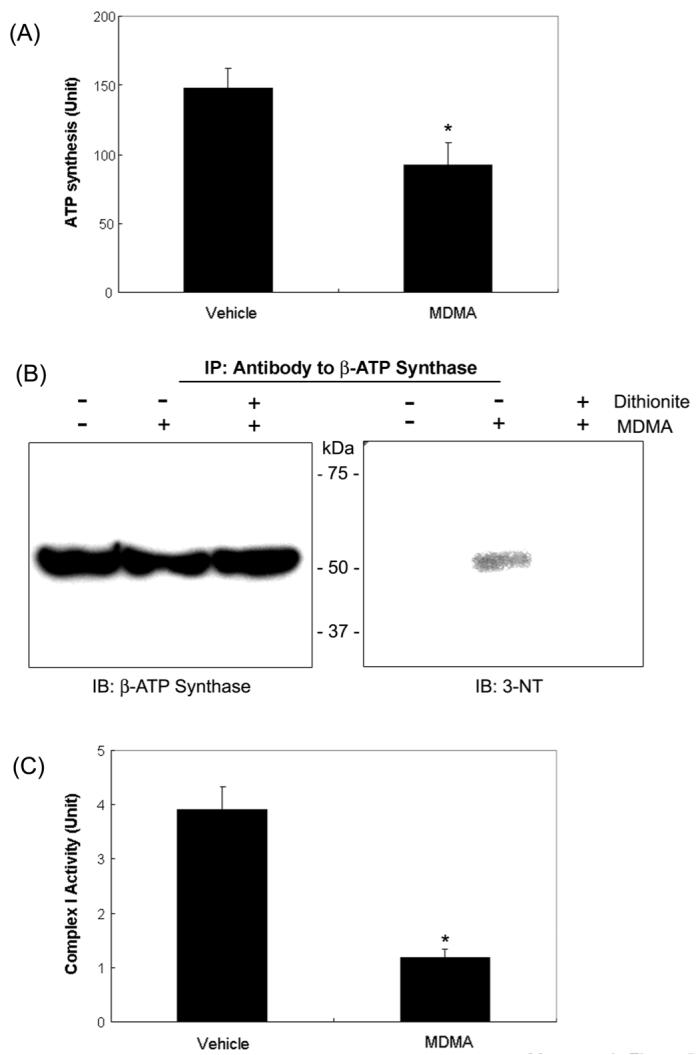
Inactivation of ATP synthase and mitochondrial complex I activities in MDMA-exposed rat liver mitochondria and immunoblot analysis. (A) Mitochondrial ATP synthase activities in vehicle and MDMA-treated rat livers are presented. *significantly different from the vehicle control (*p< 0.005). (B) Mitochondrial β-ATP synthase from the vehicle and MDMA-exposed rat livers in the absence or presence of 0.5 M sodium dithionite were immunoprecipitated with the specific antibody against β-ATP synthase, separated on 12 % SDS-PAGE, and subjected to immunoblot analysis using the anti-β-ATP synthase antibody (left) or the anti-3-NT antibody (right). This figure represents a typical result from three independent experiments. (C) NADH-ubiquinone oxidoreductase (complex I) activity was measured as described in Materials and Methods. *significantly different from the vehicle control (*p< 0.005).
4. Discussion
Over the past decade, the use of MDMA has grown beyond the boundaries of the rave scene and has spread to schools and colleges [2,3]. Unfortunately, acute exposure to MDMA alone or in combination with alcohol or other abused substances can damage various organs such as liver, heart, brain, and kidney [4-10]. MDMA organ toxicities are known to depend on route, time, dosage, and metabolism of MDMA as well as genetic polymorphism of cytochrome P450 isozymes within individuals. Despite extensive clinical descriptions of MDMA-mediated organ injury, the molecular mechanisms for these effects are not well understood. Although research is being actively carried out to understand the mechanism of MDMA-mediated neurotoxicity [15,17-19], little is known about the mechanisms of MDMA-induced hepatotoxicity. Recent studies suggest that increased oxidative and nitrosative stress plays an important role in MDMA-mediated tissue damage [18,22]. We hypothesized that MDMA and/or its reactive metabolites can inhibit the normal function of mitochondria (i.e. mitochondrial dysfunction) through increased oxidative/nitrosative stress, although these compounds can directly inhibit the mitochondrial function, as recently reported [20]. To test this hypothesis, we focused on studying the mechanism by which increased oxidative/nitrosative stress causes mitochondrial dysfunction after MDMA exposure. To identify early changes and evaluate functional implications of oxidatively-modified mitochondrial proteins, we selected a relatively low dose of MDMA (10 mg/kg, twice, orally). Under this mild condition significant, but not full-blown, hepatic damage with elevated plasma transaminases and increased immune cells infiltrated into the liver was observed. Subsequently we used this MDMA dosing regimen for our study to identify oxidatively-modified mitochondrial proteins.
Reactive metabolites of MDMA, produced via cytochrome P450-catalyzed metabolism, are critically important in MDMA-related organ damage, since direct administration of MDMA to brain areas did not cause tissue damage [41,42]. These reactive metabolites, especially quinone metabolites may produce oxidative and nitrosative stress by inhibiting the mitochondrial respiratory chain [18,20,43], although the effect of specific MDMA metabolites on the respiratory chain remains to be determined. In addition, the reactive metabolites of MDMA significantly decrease the level of reduced GSH, which protects against many types of free radical metabolites of toxic compounds (including MDMA-quinone related metabolites) [16,17,20,44]. The decreased ratio of GSH/GSSG, along with increased ROS/RNS production in MDMA-exposed rats is expected to increase oxidative and nitrosative stress. Presence of both reactive oxygen and nitrogen species can produce a more potent oxidant peroxynitrite (ONOO-), which can cause irreversible cell and tissue damage [45]. Although peroxynitrite could not be accurately measured in MDMA-exposed rat livers, the presence of peroxynitrite was indirectly shown by nitration of ATP synthase and its disappearance in the presence of dithionite. In addition, the inhibition of the mitochondrial complex I activity provides further support of peroxynitrite production (Fig. 5). Based on these results, we conclude that MDMA treatment increased the levels of ROS/RNS, resulting in the oxidative modification and inactivation of numerous mitochondrial proteins. These results not only support the important role of oxidative/nitrosative stress [17,18,22] but also provide an underlying mechanism for MDMA-mediated liver damage through oxidative inactivation of many mitochondrial proteins and subsequent mitochondrial dysfunction.
Recent studies from this laboratory and others showed that many hepatic mitochondrial proteins are oxidatively-modified and inactivated in alcohol-exposed rats [27,46]. In addition, many mitochondrial proteins are nitrated and their activities inactivated in diabetic mice, leading to mitochondrial dysfunction [47]. Interestingly, there is a significant overlap in the type of proteins that are oxidatively-modified in these different groups suggesting that similarities exist in the downstream actions of ROS/RNS produced in these conditions. However, in the current study, additional mitochondrial proteins such as acetyl-CoA dehydrogenase, UDP-glucose pyrophosphorylase 2, Hsp86, Hsp 75, and cytochrome P450 11B1 (CYP11B1), that were not identified in previous studies, were found. Although our study shows that CYP11B1 and CYP19A1 were oxidized after MDMA exposure, we did not find oxidation of cytochromes P450 such as CYP2D6, CYP1A2, and CYP3A4 that are involved in the MDMA metabolism [48], possibly due to their relatively low expression in the mitochondria. The oxidative modification of mitochondrial aconitase was reported by some investigators [49] but not by others [25,27,46]. In our current study, we did not detect the oxidative modification of aconitase in MDMA-exposed tissues. The reason for the apparent discrepancy among these studies is currently unclear. All these results suggest that some differences exist in the oxidative modifications of protein among different states. Although we have not tested functional alterations of all the oxidized enzymes that we have identified in this study, we believe that many of the enzymes and proteins that are oxidatively-modified following MDMA exposure could be inactivated, as demonstrated (Figs. 3-5). In addition to oxidative modifications, it is likely that MDMA or its reactive quinone metabolites can directly interact with or bind many mitochondrial proteins, resulting in their decreased functions, as recently demonstrated with cytochrome c [20]. Thus, these different modifications of mitochondrial proteins most likely contribute to mitochondrial dysfunction following MDMA exposure.
Our results represent a deeper understanding of the mechanism involved in MDMA-mediated liver damage. For instance, the inhibition of ALDH2 by MDMA is expected to cause accumulation of toxic acetaldehyde and lipid aldehydes, contributing to cell death. In fact, our current data showing the increased malondialdehyde level are in agreement with the earlier results in MDMA-exposed tissues [16,21,50]. In addition, the significant inhibition of the 3-ketoacyl-CoA thiolase activity involved in the mitochondrial β-oxidation pathway most likely elevates the level of free fatty acids, which are likely to indirectly elevate triglyceride levels (Fig. 4). Our current results thus explain the earlier results of fat accumulation following MDMA exposure [10]. Furthermore, MDMA-related inactivation of 3-ketoacyl-CoA thiolase may lead to an insufficient supply of ATP produced from the fat degradation pathway, especially during fasting and states of insufficient glucose utilization [38]. This putative decrease in ATP level may be further exacerbated by the decreased mitochondrial ATP synthase activity upon MDMA treatment. Because of the markedly decreased ATP level resulting from both blockade of the fat degradation pathway and direct inhibition of ATP synthase, hepatocytes may not properly carry out many cellular functions and may thus eventually undergo the necrotic cell death process. All these results provide evidence for MDMA-mediated mitochondrial dysfunction, which leads to liver damage. Our results indicate that increased oxidative/nitrosative stress following MDMA treatment promotes oxidative-modifications of many mitochondrial proteins, resulting in suppression of their normal functions. Functional assays for several other modified proteins identified upon MDMA treatment are underway.
In conclusion, we have investigated the mechanism of MDMA-induced mitochondrial dysfunction. Numerous oxidatively-modified mitochondrial proteins from rat livers after acute MDMA exposure have been identified. We have demonstrated that the oxidative modification of some of these proteins results in their inactivation. These data suggest that the increased oxidative/nitrosative stress induced by MDMA promotes oxidation and nitration of various mitochondrial proteins, leading to mitochondrial dysfunction and hepatic injury, as summarized in Fig. 6. To our knowledge, our results represent the first report which identifies specific proteins which are oxidatively-modified and inactivated upon MDMA exposure. These results enhance our understanding of the molecular mechanisms by which MDMA causes liver damage. Although we have only studied the mechanism of liver damage in this study, the molecular mechanism of MDMA-mediated toxicity to other tissues such as brain and kidney may be very similar to that observed in the liver.
Figure 6.

Schematic diagram of MDMA-mediated liver damage. Hepatic P450 enzymes are responsible for the metabolism of MDMA, generating reactive metabolites of MDMA and ROS/RNS, which can lead to an increased level of peroxynitrite. All these reactive metabolites and oxygen/nitrogen species can decrease the level of GSH while they can increase the level of lipid peroxides and the oxidative-modification of mitochondrial proteins. The inactivation of key mitochondrial enzymes following MDMA exposure can lead to mitochondrial dysfunction, ultimately contributing to acute hepatotoxicity.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of National Institute on Alcohol Abuse and Alcoholism. This project has been also funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the United States Government. We thank Drs. Norman Salem Jr. and Nancy Braverman for the support and providing the specific antibody to 3-ketoacyl-CoA thiolase, respectively.
Footnotes
- MDMA
- 3,4-methylenedioxymethamphetamine
- ROS
- reactive oxygen species
- RNS
- reactive nitrogen species
- MDA
- malondialdehyde
- iNOS
- inducible nitric oxide synthase
- H&E
- hematoxylin and eosin
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- Biotin-NM
- biotin-N-maleimide
- 2-DE
- two-dimensional polyacrylamide gel electrophoresis
- MS/MS
- tandem mass spectrometry
- ALDH2
- mitochondrial aldehyde dehydrogenase 2
- α-ATP synthase
- ATP synthase α-subunit
- β-ATP synthase
- ATP synthase β-subunit
- HRP
- horse radish peroxidase
- HSP
- heat shock protein
- GRP
- glucose-regulated protein
- GDH
- glutamate dehydrogenase
- MAb
- monoclonal antibody
- 3-NT
- 3-nitro-tyrosine
- GSH
- reduced glutathione
- GSSG
- oxidized glutathione
- Complex I
- NADH-ubiquinone oxidoreductase
- CYP11B1
- cytochrome P450 11B1
REFERENCES
- [1].Landry MJ. MDMA: a review of epidemiologic data. J. Psychoactive Drugs. 2002;34:163–169. doi: 10.1080/02791072.2002.10399950. [DOI] [PubMed] [Google Scholar]
- [2].Schifano F, Corkery J, Deluca P, Oyefeso A, Ghodse AH. Ecstasy (MDMA, MDA, MDEA, MBDB) consumption, seizures, related offences, prices, dosage levels and deaths in the UK (1994-2003) J. Psychopharmacol. 2006;20:456–463. doi: 10.1177/0269881106060147. [DOI] [PubMed] [Google Scholar]
- [3].Reid LW, Elifson KW, Sterk CE. Ecstasy and gateway drugs: initiating the use of ecstasy and other drugs. Ann. Epidemiol. 2007;17:74–80. doi: 10.1016/j.annepidem.2006.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Henry JA, Hill IR. Fatal interaction between ritonavir and MDMA. Lancet. 1998;352:1751–1752. doi: 10.1016/s0140-6736(05)79824-x. [DOI] [PubMed] [Google Scholar]
- [5].Harrington RD, Woodward JA, Hooton TM, Horn JR. Life-threatening interactions between HIV-1 protease inhibitors and the illicit drugs MDMA and gamma-hydroxybutyrate. Arch. Intern. Med. 1999;159:2221–2224. doi: 10.1001/archinte.159.18.2221. [DOI] [PubMed] [Google Scholar]
- [6].Nutt DJ. A tale of two Es. J. Psychopharmacol. 2006;20:315–317. doi: 10.1177/0269881106064592. [DOI] [PubMed] [Google Scholar]
- [7].Kuypers KP, Samyn N, Ramaekers JG. MDMA and alcohol effects, combined and alone, on objective and subjective measures of actual driving performance and psychomotor function. Psychopharmacology. 2006;187:467–475. doi: 10.1007/s00213-006-0434-z. [DOI] [PubMed] [Google Scholar]
- [8].Ellis AJ, Wendon JA, Portmann B, Williams R. Acute liver damage and ecstasy ingestion. Gut. 1996;38:454–458. doi: 10.1136/gut.38.3.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Andreu V, Mas A, Bruguera M, Salmeron JM, et al. Ecstasy: a common cause of severe acute hepatotoxicity. J. Hepatol. 1998;29:394–397. doi: 10.1016/s0168-8278(98)80056-1. [DOI] [PubMed] [Google Scholar]
- [10].Beitia G, Cobreros A, Sainz L, Cenarruzabeitia E. Ecstasy-induced toxicity in rat liver. Liver. 2000;20:8–15. doi: 10.1034/j.1600-0676.2000.020001008.x. [DOI] [PubMed] [Google Scholar]
- [11].de la Torre R, Farre M, Roset PN, Pizarro N, et al. Human pharmacology of MDMA: pharmacokinetics, metabolism, and disposition. Ther. Drug Monit. 2004;26:137–144. doi: 10.1097/00007691-200404000-00009. [DOI] [PubMed] [Google Scholar]
- [12].O’Shea E, Escobedo I, Orio L, Sanchez V, et al. Elevation of ambient room temperature has differential effects on MDMA-induced 5-HT and dopamine release in striatum and nucleus accumbens of rats. Neuropsychopharmacology. 2005;30:1312–1323. doi: 10.1038/sj.npp.1300673. [DOI] [PubMed] [Google Scholar]
- [13].Green AR, O’Shea E, Colado MI. A review of the mechanisms involved in the acute MDMA (ecstasy)-induced hyperthermic response. Eur. J. Pharmacol. 2004;500:3–13. doi: 10.1016/j.ejphar.2004.07.006. [DOI] [PubMed] [Google Scholar]
- [14].Bindoli A, Rigobello MP, Deeble DJ. Biochemical and toxicological properties of the oxidation products of catecholamines. Free Radic. Biol. Med. 1992;13:391–405. doi: 10.1016/0891-5849(92)90182-g. [DOI] [PubMed] [Google Scholar]
- [15].Milhazes N, Cunha-Oliveira T, Martins P, Garrido J, et al. Synthesis and cytotoxic profile of 3,4-methylenedioxymethamphetamine (“ecstasy”) and its metabolites on undifferentiated PC12 cells: A putative structure-toxicity relationship. Chem. Res. Toxicol. 2006;19:1294–1304. doi: 10.1021/tx060123i. [DOI] [PubMed] [Google Scholar]
- [16].Carvalho M, Remiao F, Milhazes N, Borges F, et al. The toxicity of N-methyl-alpha-methyldopamine to freshly isolated rat hepatocytes is prevented by ascorbic acid and N-acetylcysteine. Toxicology. 2004;200:193–203. doi: 10.1016/j.tox.2004.03.016. [DOI] [PubMed] [Google Scholar]
- [17].Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem. Res. Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- [18].Montiel-Duarte C, Ansorena E, Lopez-Zabalza MJ, Cenarruzabeitia E, Iraburu MJ, Cenarruzabeitia E, Iraburu MJ. Role of reactive oxygen species, glutathione and NF-kappaB in apoptosis induced by 3,4-methylenedioxy methamphetamine (“Ecstasy”) on hepatic stellate cells. Biochem. Pharmacol. 2004;67:1025–1033. doi: 10.1016/j.bcp.2003.10.020. [DOI] [PubMed] [Google Scholar]
- [19].Miller RT, Lau SS, Monks TJ. 2,5-Bis-(glutathion-S-yl)-alpha-methyldopamine, a putative metabolite of (+/-)-3,4-methylenedioxyamphetamine, decreases brain serotonin concentrations. Eur. J. Pharmacol. 1997;323:173–180. doi: 10.1016/s0014-2999(97)00044-7. [DOI] [PubMed] [Google Scholar]
- [20].Fisher AA, Labenski MT, Malladi S, Gokhale V, et al. Quinone electrophiles selectively adduct “electrophile binding motifs” within cytochrome c. Biochemistry. 2007;46:11090–11100. doi: 10.1021/bi700613w. [DOI] [PubMed] [Google Scholar]
- [21].Jayanthi S, Ladenheim B, Andrews AM, Cadet JL. Overexpression of human copper/zinc superoxide dismutase in transgenic mice attenuates oxidative stress caused by methylenedioxymethamphetamine (Ecstasy) Neuroscience. 1999;91:1379–1387. doi: 10.1016/s0306-4522(98)00698-8. [DOI] [PubMed] [Google Scholar]
- [22].Darvesh AS, Yamamoto BK, Gudelsky GA. Evidence for the involvement of nitric oxide in 3,4-methylenedioxymethamphetamine-induced serotonin depletion in the rat brain. J. Pharmacol. Exp. Ther. 2005;312:694–701. doi: 10.1124/jpet.104.074849. [DOI] [PubMed] [Google Scholar]
- [23].Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- [25].Suh SK, Hood BL, Kim BJ, Conrads TP, et al. Identification of oxidized mitochondrial proteins in alcohol-exposed human hepatoma cells and mouse liver. Proteomics. 2004;4:3401–3412. doi: 10.1002/pmic.200400971. [DOI] [PubMed] [Google Scholar]
- [26].Kim BJ, Hood BL, Aragon RA, Hardwick JP, et al. Increased oxidation and degradation of cytosolic proteins in alcohol-exposed mouse liver and hepatoma cells. Proteomics. 2006;6:1250–1260. doi: 10.1002/pmic.200500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Moon KH, Hood BL, Kim BJ, Hardwick JP, et al. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- [28].Wilkins MR, Appel RD, Van Eyk JE, Chung MCM, et al. Guidelines for the next 10 years of proteomics. Proteomics. 2006;6:4–8. doi: 10.1002/pmic.200500856. [DOI] [PubMed] [Google Scholar]
- [29].Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006;281:21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- [30].Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- [31].Cardoso SM, Pereira C, Oliveira R. Mitochondrial function is differentially affected upon oxidative stress. Free Radic. Biol. Med. 1999;26:3–13. doi: 10.1016/s0891-5849(98)00205-6. [DOI] [PubMed] [Google Scholar]
- [32].Widmer R, Kaiser B, Engels M, Jung T, Grune T. Hyperammonemia causes protein oxidation and enhanced proteasomal activity in response to mitochondria-mediated oxidative stress in rat primary astrocytes. Arch. Biochem. Biophys. 2007;464:1–11. doi: 10.1016/j.abb.2007.03.027. [DOI] [PubMed] [Google Scholar]
- [33].Venugopal SK, Chen J, Zhang Y, Clemens D, et al. Role of MAP kinase phosphatase-1 in sustained activation of C-JUN N-terminal kinase (JNK) during ethanol-induced apoptosis in hepatocyte-like VL-17A cells. J. Biol. Chem. 2007;282:31900–31908. doi: 10.1074/jbc.M703729200. [DOI] [PubMed] [Google Scholar]
- [34].Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- [35].Moon KH, Abdelmegeed MA, Song BJ. Inactivation of cytosolic aldehyde dehydrogenase via S-nitrosylation in ethanol-exposed rat liver. FEBS Lett. 2007;581:3967–3972. doi: 10.1016/j.febslet.2007.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zeng J, Li D. Expression and purification of His-tagged rat mitochondrial 3-ketoacyl-CoA thiolase wild-type and His352 mutant proteins. Protein Expr. Purif. 2004;35:320–326. doi: 10.1016/j.pep.2004.01.018. [DOI] [PubMed] [Google Scholar]
- [37].Yang SJ, Cho EH, Choi MM, Lee HJ, et al. Critical role of the cysteine 323 residue in the catalytic activity of human glutamate dehydrogenase isozymes. Mol Cells. 2005;19:97–103. [PubMed] [Google Scholar]
- [38].Eaton S, Bursby T, Middleton B, Pourfarzam M, et al. The mitochondrial trifunctional protein: centre of a beta-oxidation metabolon? Biochem. Soc. Trans. 2000;28:177–182. doi: 10.1042/bst0280177. [DOI] [PubMed] [Google Scholar]
- [39].Bianchet MA, Hullihen J, Pedersen PL, Amzel LM. The 2.8-A structure of rat liver F1-ATPase: configuration of a critical intermediate in ATP synthesis/hydrolysis. Proc. Natl. Acad. Sci. USA. 1998;95:11065–11070. doi: 10.1073/pnas.95.19.11065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA. Oxidative damage to mitochondrial complex I due to peroxynitrite. Identification of reactive tyrosines by mass spectrometry. J. Biol. Chem. 2003;278:37223–37230. doi: 10.1074/jbc.M305694200. [DOI] [PubMed] [Google Scholar]
- [41].Schmidt CJ, Taylor VL. Direct central effects of acute methylenedioxymethamphetamine on serotonergic neurons. Eur. J. Pharmacol. 1988;156:121–131. doi: 10.1016/0014-2999(88)90154-9. [DOI] [PubMed] [Google Scholar]
- [42].Paris JM, Cunningham KA. Lack of serotonin neurotoxicity after intraraphe microinjection of (+)-3,4-methylenedioxymethamphetamine (MDMA) Brain Res. Bull. 1992;28:115–119. doi: 10.1016/0361-9230(92)90237-r. [DOI] [PubMed] [Google Scholar]
- [43].Alves E, Summavielle T, Alves CJ, Gomes-da-Silva J, et al. Monoamine oxidase-B mediates ecstasy-induced neurotoxic effects to adolescent rat brain mitochondria. J. Neurosci. 2007;27:10203–10210. doi: 10.1523/JNEUROSCI.2645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jones DC, Duvauchelle C, Ikegami A, Olsen CM, et al. Serotonergic neurotoxic metabolites of ecstasy identified in rat brain. J. Pharmacol. Exp. Ther. 2005;313:422–431. doi: 10.1124/jpet.104.077628. [DOI] [PubMed] [Google Scholar]
- [45].Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Venkatraman A, Landar A, Davis AJ, Ulasova E, et al. Oxidative modification of hepatic mitochondria protein thiols: effect of chronic alcohol consumption. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;286:G521–G527. doi: 10.1152/ajpgi.00399.2003. [DOI] [PubMed] [Google Scholar]
- [47].Turko IV, Li L, Aulak KS, Stuehr DJ, et al. Protein tyrosine nitration in the mitochondria from diabetic mouse heart. J. Biol. Chem. 2003;278:33972–33977. doi: 10.1074/jbc.M303734200. [DOI] [PubMed] [Google Scholar]
- [48].Oesterheld JR, Armstrong SC, Cozza KL. Ecstasy: pharmacodynamic and pharmacokinetic interactions. Psychosomatics. 2004;45:84–87. doi: 10.1176/appi.psy.45.1.84. [DOI] [PubMed] [Google Scholar]
- [49].Eaton P, Byers HL, Leeds N, Ward MA, Shattock MJ. Detection, quantitation, purification, and identification of cardiac proteins S-thiolated during ischemia and reperfusion. J. Biol. Chem. 2002;277:9806–9811. doi: 10.1074/jbc.M111454200. [DOI] [PubMed] [Google Scholar]
- [50].Sprague JE, Nichols DE. The monoamine oxidase-B inhibitor L-deprenyl protects against 3,4-methylenedioxymethamphetamine-induced lipid peroxidation and long-term serotonergic deficits. J. Pharmacol. Exp. Ther. 1995;273:667–673. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.