

Abstract
The peptide ligase subtiligase, derived from subtilisin, has been employed in the identification of protein N-termini in complex mixtures. Here, the peptide ester substrates for the ligation reaction were optimized with respect to solubility, resulting in greater incorporation of the N-terminal tags. Additionally, the quantitation of the incorporated tags was explored, and a “click” chemistry-based derivatization provided the ability to quantitate the tag to low nanomolar concentrations by sandwich ELISA. These new tags should expand the utility of subtiligase for the proteomic study of N-termini.
The study of the proteome is aided by tools that can separate specific subsets of proteins. For example, in studying post-translational modifications (PTM), methods for enriching phosphoproteins and phosphopeptides have been developed.1 One common PTM in biology is the production of new N-termini by proteolysis; another is the modification of N-termini through acetylation3 or myristoylation.4 Several labs have described indirect means for isolation or enrichment of N-terminal peptides. These techniques include strong cation exchange chromatography of tryptic peptides,5 and the chemical labeling of amine-containing functionality in the proteome6 followed by either diagonal chromatography7 or affinity purification.8 Alternatively, our laboratory has developed a direct enzymatic method to affinity tag and enrich for the subset of proteins with free N-termini in order to characterize cellular substrates of proteolysis, or, more generally, characterize the ensemble of native N-termini. A recent study using this method identified nearly three hundred caspase substrates and their precise cleavage sites from apoptotic Jurkat cells.9
This method utilizes subtiligase, a variant of the protease subtilisin BPN' that contains a cysteine residue in place of the active site serine-221 and an alanine residue replacing proline-225.10 Unlike the wild-type enzyme, the resulting double mutant has negligible protease activity, yet it can efficiently acylate free N-termini of peptides or proteins using ester substrates. The workflow for the application of subtiligase to the study of the proteome is outlined in Scheme 1. A complex mixture of proteins is N-terminally labeled with subtiligase using a peptide ester containing a biotin affinity tag and a TEV protease11,12 cleavage site. Tagged proteins are separated from the unincorporated tag and digested with trypsin. Tagged N-terminal peptides are captured on avidin beads, cleaved off the beads by TEV protease treatment and analyzed by LC-MS/MS. While approximately 80% of N-termini are blocked in mammalian cells, mostly due to acetylation,13,14 certain populations of proteins are believed to have a greater fraction of free N-termini. These include signal-sequence-dependent proteins transported across membranes, such as nuclear-encoded mitochondrial proteins.15,8
Scheme 1.
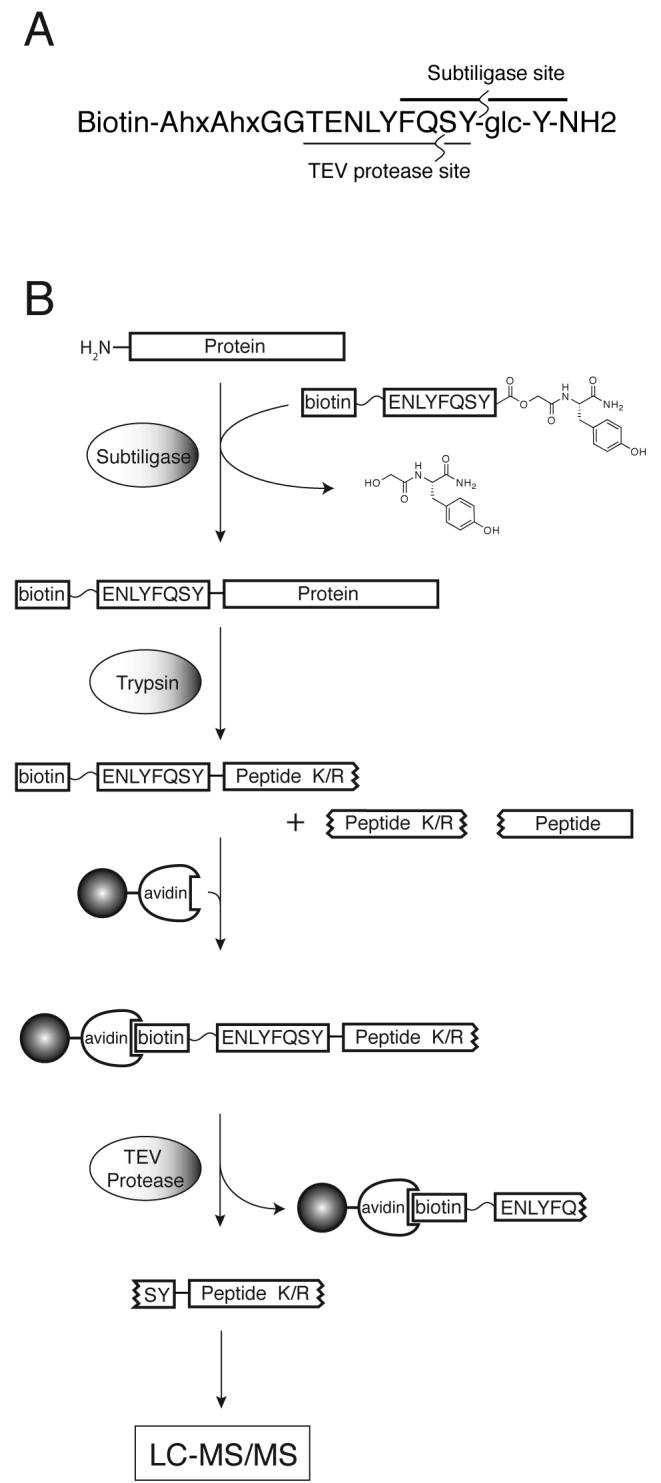
A. Structure of peptide ester 1 with ligation and TEV protease cleavage sites annotated. B. Proteomics workflow for N-terminal labeling using subtiligase.
While this approach is a powerful and sensitive tool for identifying free protein N-termini and the precise sites of proteolysis, it has limitations. Subtiligase will hydrolyze the ester substrate and ligation yields vary depending on the accessibility and, to some extent, on the nature of the N-terminal residues.16 In principle, increasing amount of peptide ester will increase the extent of the ligation reaction by providing more ester for ligation before it is consumed by hydrolysis. TEVest2 (1, Table 1), the prototypical peptide ester, is somewhat insoluble in aqueous solution and cannot be used at concentrations higher than 1 mM.
Table 1.
Peptide ester structures and solubilities
| Ester | Sequence | Solubility (mM) |
|---|---|---|
| 1 | Biotin-AhxAhxGGTENLYFQSY-glc-Y-NH2 | ∼1-2 |
| 2 | Biotin-AhxAhx-EEEE-GGTENLYFQSY-glc-Y-NH2 | ∼10 |
| 3 | Biotin-Linker-dRdRdRdR-prG-TENLYFQSY-glc-R-NH2 | >40 |
| 4 | Biotin-Linker-dRdRdRdR-GTENLYFQS-prG-glc-R-NH2 | >50 |
| 5 | Biotin-Linker-dRdRdR-prG-TENLYFQSY-glc-RR-NH2 | >40 |
![]()
Ahx = 6-aminohexanoyl
prG = L-propargylglycinyl
glc = glycoloyl
Here, we explored whether it would be possible to improve the yield of the tagging reaction and increase proteomic coverage by producing more soluble peptide esters. The general strategy in these designs was to add basic or acidic residues such that the net charge of the ester would be strongly positive or negative at neutral pH. Some of the esters conjugated to biotin using a PEG-type linker instead of the two aminohexanoic acid residues in 1. In addition, to facilitate the routine quantitation of the tagged proteins in complex mixtures by sandwich ELISA, the incorporation of 3-nitrotyrosine and propargylglycine residues into the esters was also investigated. The primary structures of the peptide esters and their solubilities are listed in Table 1.
Ester 2, a derivative of TEVest2 (1) that contained an additional four glutamic acid residues at the N-terminus of the peptide portion, showed improved solubility. However, a much greater increase in solubility was observed with analogues that instead contained additional arginine residues (3-5). We chose to incorporate D-arginine in order to preserve the integrity of the tag during the trypsin digest step of the proteomic workflow.
We also wished to produce peptide analogs that could be used to introduce functionality for quantitation and labeling post-TEV protease cleavage. Quantitation would be an important new feature allowing users to analyze how much labeling one has in the sample prior to any further fractionation and LC-MS/MS. We chose to test incorporating 3-nitrotyrosine since this single residue can be used in very quantitative and sensitive ELISA assays.17 Peptides containing this residue, however, were significantly less soluble. Alternatively, we found that propargylglycine could be incorporated without loss in solubility. The terminal alkyne side chain of propargylglycine provides a “click”-able handle for derivatization with a variety of functional groups post-subtiligase labeling using the copper-catalyzed alkyne-azide cyclization reaction18,19 with an azide-bearing chemical tag. Ester 4 was thus designed to combine some of the features of a highly soluble peptide ester and contain a propargylglycine residue immediately N-terminal to the ester group. Since non-β-branched hydrophobic residues at this position are good substrates for subtilisin20,21 it was anticipated that the propargyl side chain would be well tolerated. At this position, the propargylglycine residue can function in two roles. First, it can function as a tag for derivatization and quantitation, in principle along the entire workflow and not only prior to the TEV protease cleavage step as with 3. Second, as a non-natural amino acid, it will produce unique a2 and b2 ions when tagged peptides are fragmented during tandem mass spectrometry. Abundant a2 and b2 ions, corresponding to the residual serine and tyrosine residues of the tag, are observed with a QqTOF instrument when analyzing proteomic samples labeled using ester 1 and are a hallmark of tagged N-terminal peptides. The incorporation of propargylglycine instead could provide an even more distinct signature.
The performance of the new esters was assessed by small-scale ligation reactions with Jurkat cell lysates, which were then analyzed by SDS-PAGE and blotted with streptavidin to detect the incorporated tag (Figure 1). At 1 mM, all the esters show comparable or modest improvements in labeling compared to 1. However at higher concentrations the more soluble penta-Arg containing esters (3, 4 and 5) showed significantly higher levels of labeling. Redistribution of some of the Arg residues from the N- to the C-terminal side of the ester (compare 3 vs. 5) did not significantly alter their performance. Ester 4, which introduces a propargylglycine residue directly adjacent to the ligated protein's N-terminus, does not appear to perform as well with respect to labeling as 3 or 5 but is still significantly better than 1. We note that esters 3, 4 and 5 caused some protein precipitation in cell lysates. It is known that proteins containing N-terminal polyarginine tags are less thermostable,22 but we could prevent precipitation by the addition of sodium chloride, urea or guanidinium chloride.
Figure 1.
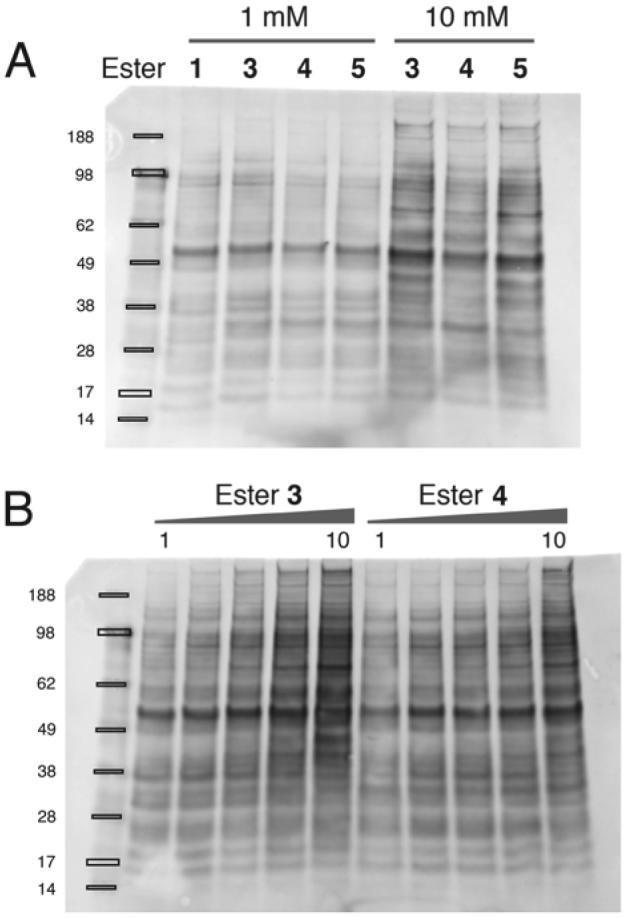
Avidin blots of ligation reactions in Jurkat cell lysates. A. Comparison of peptide esters at 1 mM and 10 mM. The limited solubility of 1 precludes its use at 10 mM. B. Lysates incubated with increasing concentrations (1, 2, 4, 7 & 10 mM) of peptide ester in the presence of 10 μM subtiligase.
As a model to test the performance of a quantitation strategy involving derivatizaton of a propargylglycine residue (Figure 2a), tripeptides consisting of (Ac)GlyTyr(NO2)Ala, a PEG linker and an azide group were prepared. The derivatization of the propargylglycine residue of 3 pre-bound to neutravidin in 96-well plates proceeded efficiently under typical conditions (Figure 2b). The large excess (250 equivalents or greater) of the azide-bearing nitrotyrosine peptide appears to ensure the complete reaction of the avidin-immobilized 3, as standard curves using peptides containing nitrotyrosine residues and pre-derivatized peptides showed similar sensitivity compared to standard curves prepared by on-plate derivatization. Using this assay, 3 can be reliably detected at 30 nM, a level of sensitivity that allows quantitation of ligated N-termini to an estimated lower limit of 0.2% of free N-termini in a eukaryotic cell lysate. In practice, the material analyzed would be a complex mixture of proteins, with some fraction N-terminally labeled and any residual unreacted tag removed by gel filtration.
Figure 2.
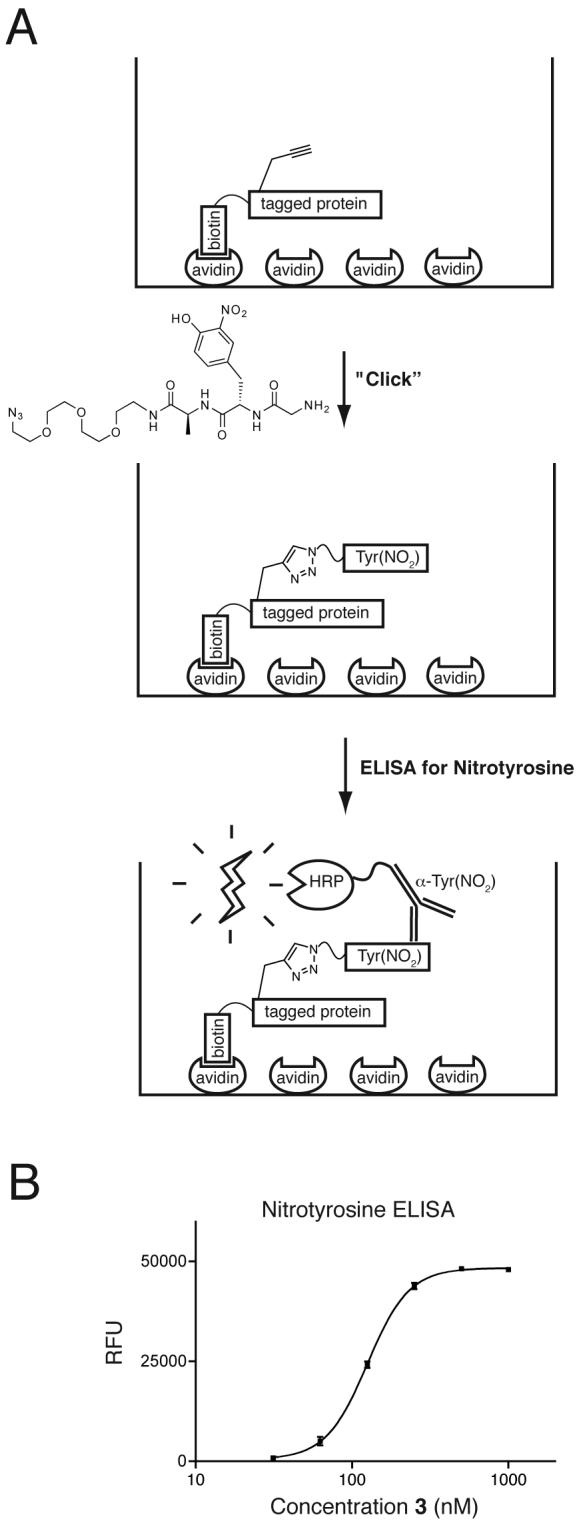
Quantitation of tagging by derivatization and ELISA. A. Tagged proteins are captured on an analytical scale in neutravidin-coated 96-well plates. The propargylglycine residue is derivatized using a copper(I)-catalyzed azide-alkyne cyclization reaction and the incorporated 3-nitrotyrosine is quantitated using an antibody conjugated to horseradish peroxidase (HRP). B. ELISA using peptide ester 3.
In summary, we have developed new peptide esters as subtiligase substrates for use in the proteomic labeling of free N-termini. By incorporating several additional arginine residues, the solubility of the peptides is markedly improved, and the ligation reaction can be driven further by the addition of more ester. Adding propargylglycine residues into the peptide esters allows the quantitiation of the labeled N-termini by sandwich ELISA following derivatization with a nitrotyrosine and azide containing peptide. These features should to expand the utility of subtiligase for the study of proteolysis and other PTMs of protein N-termini.
Acknowledgements
We wish to thank Pete Wildes, Nick Agard and Emily Crawford for helpful discussions. Financial support was provided by the Sandler Foundation, NIH (RO1 GM081051), and the Hartwell Foundation. HAIY was supported in part by a Stewart Trust Cancer Research Award from the UCSF Cancer Center.
References
- 1.Reinders J, Sickmann A. Proteomics. 2005;5:4052. doi: 10.1002/pmic.200401289. [DOI] [PubMed] [Google Scholar]
- 2.Bradshaw RA, Brickey WW, Walker KW. Trends Biochem. Sci. 1998;23:263. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 3.Polevoda B, Sherman F. J. Biol. Chem. 2000;275:36479. doi: 10.1074/jbc.R000023200. [DOI] [PubMed] [Google Scholar]
- 4.Towler DA, Gordon JI, Adams SP, Glaser L. Annu. Rev. Biochem. 1988;57:69. doi: 10.1146/annurev.bi.57.070188.000441. [DOI] [PubMed] [Google Scholar]
- 5.Dormeyer W, Mohammed S, Breukelen B, Krijgsveld J, Heck AJ. J. Proteome Res. 2007;6:4634. doi: 10.1021/pr070375k. [DOI] [PubMed] [Google Scholar]
- 6.Enoksson M, Li J, Ivancic MM, Timmer JC, Wildfang E, Eroshkin A, Salvesen GS, Tao WA. J. Proteome Res. 2007;6:2850. doi: 10.1021/pr0701052. [DOI] [PubMed] [Google Scholar]
- 7.Gevaert K, Goethals M, Martens L, Van Damme J, Staes A, Thomas GR, Vandekerckhove J. Nat. Biotechnol. 2003;21:566. doi: 10.1038/nbt810. [DOI] [PubMed] [Google Scholar]
- 8.Timmer JC, Enoksson M, Wildfang E, Zhu W, Igarashi Y, Denault JB, Ma Y, Dummitt B, Chang YH, Mast AE, Eroshkin A, Smith JW, Tao WA, Salvesen GS. Biochem. J. 2007;407:41. doi: 10.1042/BJ20070775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL, Wells JA. Cell. 2008;134:866. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrahmsen L, Tom J, Burnier J, Butcher KA, Kossiakoff A, Wells JA. Biochemistry. 1991;30:4151. doi: 10.1021/bi00231a007. [DOI] [PubMed] [Google Scholar]
- 11.Carrington JC, Dougherty WG. Proc. Natl. Acad. Sci. U. S. A. 1988;85:3391. doi: 10.1073/pnas.85.10.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speers AE, Cravatt BF. J. Am. Chem. Soc. 2005;127:10018. doi: 10.1021/ja0532842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown JL. Biochim. Biophys. Acta. 1970;221:480. doi: 10.1016/0005-2795(70)90218-7. [DOI] [PubMed] [Google Scholar]
- 14.Brown JL, Roberts WK. J. Biol. Chem. 1976;251:1009. [PubMed] [Google Scholar]
- 15.Neupert W. Annu. Rev. Biochem. 1997;66:863. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- 16.Chang TK, Jackson DY, Burnier JP, Wells JA. Proc. Natl. Acad. Sci. U. S. A. 1994;91:12544. doi: 10.1073/pnas.91.26.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye YZ, Strong M, Huang ZQ, Beckman JS. Methods Enzymol. 1996;269:201. doi: 10.1016/s0076-6879(96)69022-3. [DOI] [PubMed] [Google Scholar]
- 18.Speers AE, Cravatt BF. Chem Biol. 2004;11:535. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J. Am. Chem. Soc. 2003;125:3192. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 20.Gron H, Meldal M, Breddam K. Biochemistry. 1992;31:6011. doi: 10.1021/bi00141a008. [DOI] [PubMed] [Google Scholar]
- 21.Wells JA, Cunningham BC, Graycar TP, Estell DA. Proc. Natl. Acad. Sci. U. S. A. 1987;84:5167. doi: 10.1073/pnas.84.15.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuchs SM, Raines RT. Protein Sci. 2005;14:1538. doi: 10.1110/ps.051393805. [DOI] [PMC free article] [PubMed] [Google Scholar]