

Abstract
Multiple sclerosis (MS) is characterized by a dynamic inflammatory process in which CNS lesions of distinct cellular composition coexist. In particular the formation of B cell plaques has been ascribed an important role as predictor of disease progression. Here we show that the novel MBP-PLP fusion protein (MP4)-induced experimental autoimmune encephalomyelitis (EAE) of C57BL/6 mice fulfils these criteria inducing differential cellular infiltration of B cells, T cells, macrophages and granulocytes and permitting the quantification and staging of the disease. On the contrary, both key features – dynamic CNS inflammation and B cell infiltration – were absent in the classical MOG:35–55-induced EAE of C57BL/6 mice, which was characterized by a static CD4+ T cell and macrophage-mediated CNS immunopathology throughout the disease. MP4-induced EAE may thus provide a unique opportunity for studying immune-pathomechanims of the disease that have been previously neglected due to experimental shortcomings in murine EAE.
Keywords: Autoimmunity, C57BL/6 mice, CNS inflammation, Disease staging, EAE, Infiltrate composition, MS, Neuroimmunology
Introduction
Multiple sclerosis (MS) is thought to be a chronic autoimmune disease of the central nervous system (CNS). The disease is primarily characterized by multifocal inflammation and demyelination leading to severe deficits in motor, sensory, autonomic and neurocognitive function. There is persuasive evidence that MS does not represent a single and uniform disease entity, but that different subtypes of the disease could instead result from the involvement of different target antigens and distinct immune effector pathways [1–7].
Studies of the immune pathology of MS still largely depend on animal models, in particular variants of experimental autoimmune encephalomyelitis (EAE) (reviewed in [8–11]). Here, immunizations of susceptible animal strains with myelin antigens induce inflammatory CNS lesions that are similar to those seen in MS [8,9,12,13]. However, no single EAE model known so far can fully reproduce the spectrum of disease manifestations of MS. Instead hosts on different genetic backgrounds need to be immunized with different neuroantigens in order to mirror the different immuno-and histopathological aspects of the disease [5,14–24]. Yet, the number of variables in such models is remarkable making it very difficult to experimentally dissect and understand the respective contribution of genetic background, target antigen and immune mechanism to the disease pathogenesis. To eliminate the variable of the host’s genetic background it is desirable to study EAE in a single mouse strain, but such models are rare. For example, the C57BL/6 (B6) mouse was originally considered to be EAE-resistant since immunizations with the classic myelin basic protein (MBP) did not elicit the disease [25–28]. Also, immunizations with proteolipid protein (PLP) were found to be non-encephalitogenic [23,28,29]. Only the discovery that the myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 (MOGp) was able to trigger severe EAE in B6 mice made this strain join the EAE field [30]. Recently it has been reported that PLP peptide 178–191 (PLPp) was also capable of inducing EAE in the B6 mouse [31].
In previous work we set out to establish an alternative EAE model for B6 mice using the MBP-PLP fusion protein MP4, which has originally been generated as a drug candidate for MS [32]. MP4 proved to be capable of inducing aggressive EAE in B6 mice [33]. Our initial characterization of the MP4 model showed fundamental differences to MOGp-induced EAE. In particular, unlike the MOGp model, the MP4 model was dependent on CD8+ T cells and B cells. Moreover, the gross histopathological examination of the CNS showed that the two models differed in the kinetics and topology of lesions suggesting involvement of different immune mechanisms [34].
In the present study we set out to further investigate the histopathological differences between the two models, in particular by comparing the dynamics of central nervous system (CNS) lesion composition and development. While MOGp-induced EAE was characterized by a uniform prevalence of CD4+ T cells, macrophages and granulocytes within the lesions throughout the disease, in the MP4 model the lesion composition was dynamic shifting from CD4+ T cell, macrophage and granulocyte predominance in acute EAE to additional involvement of CD8+ T cells, dendritic cells and substantial B cell infiltration as the disease progressed.
Therefore, MOGp- versus MP4-induced EAE of B6 mice may be well-suited for modelling different immune pathologies and manifestations of MS providing insights which a single model would not cover. Finally, a major strength of working with these different EAE models on the B6 background resides in the fact that this strain is ideally suited for mechanism-oriented experimentation since gene-modified mice are typically created on this background.
Materials and Methods
Mice
60 female wild-type (WT) C57BL/6 mice (6–8 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained at the animal facilities of Case Western Reserve University, Cleveland, OH. All treatments complied with the institutional guidelines.
Induction and clinical assessment of EAE
MP4/Apogen was obtained from Alexion Pharmaceuticals (Cheshire, CT). MOG:35–55 (MEVGWYRSPFSRVVHLYRNGK) and hen egg lysozyme (HEL) protein were purchased from Princeton Biomolecules (Langhorne, PA). Incomplete Freund’s adjuvant (IFA) was prepared as a mixture of Mannide Monooleate (Sigma-Aldrich, St. Louis, MO) and Paraffin Oil (EMScience, Gibbstown, NJ), complete Freund’s adjuvant (CFA) was obtained by mixing Mycobacterium tuberculosis H37RA (Difco Laboratories, Franklin Lakes, NJ) at 5 mg/ml into IFA. For disease induction, mice were immunized subcutaneously in both sides of the flank with either 150 μg MP4 or 100 μg MOG:35–55 in CFA. Mice immunized with 200 μg HEL protein in CFA in addition to unimmunized mice served as negative controls. 200 ng pertussis toxin (PTX; List Biological Laboratories, Hornby, ONT, Canada) in 500 μl sterile phosphate-buffered saline (PBS) were given on the day of immunization and 48 h later. Clinical assessment of EAE was performed daily according to the following criteria: (0), no disease; (1), floppy tail; (2), hind limb weakness; (3), full hind limb paralysis; (4), quadriplegia; (5), death. Mice that were in between the clear-cut gradations of clinical signs were scored intermediate in increments of 0.5.
Tissue preparation
On the first day mice showed clinical symptoms of the disease (acute EAE) and on day 60 after EAE induction when clinical scores had reached a stable plateau (chronic EAE), mice were euthanized under deep anaesthesia. Brain, spinal cord and cerebellum/brainstem were removed. Brains were placed into a mouse brain matrix (World Precision Instruments, Berlin, Germany) and dissected. As a rostral border, we took the midpoint of the olfactory bulbs (bregma 2.7 mm) and as dorsal border, the beginning of the vermis cerebelli (bregma − 4.2 mm). All specimens were snap-frozen in liquid nitrogen and kept at − 80°C until further analysis.
Consecutively, 7 μm thick sections were cut on a Reichert-Jung Cryocut CM1850 cryostat (Leica, Deerfield, IL), mounted on SUPERFROST/Plus slides (Carl Roth, Karlsruhe, Germany) and kept at − 80°C until staining. For each animal, coronal brain, longitudinal spinal cord and coronal cerebellum/brainstem sections were obtained from the entire tissue. Sections from CNS areas that showed infiltration were then used for immunohistochemistry.
Immunohistochemistry (IHC)
Immunohistochemistry was performed according to the following protocol. Designated sections were air-dried for 2 h at room temperature. Consecutively, a hydrophobic barrier was drawn around each section. The sections were rehydrated with 0.1 M PBS (pH 7.4) for 4 min and fixed with 3.7 % paraformaldehyde in the dark for 15 min followed by four washes (3 min each) with 0.1 M tris-buffered saline (TBS) at pH 7.6 and another wash with PBS for 3 min. Sections were then incubated with 5% goat serum (Sigma) in PBS for 60 min at room temperature, washed with PBS for 1 min before addition of the primary antibodies (all diluted in 0.8% BSA) for 48 h incubation in a humid chamber at 4°C. Rat anti-mouse GK1.5 was used at 1:500 dilution for the detection of CD4+ T cells (eBioscience, San Diego, CA), rat anti-mouse 53–6.7 at 1:500 for CD8+ T cells (eBioscience), rat anti-mouse RA3–6B2 at 1:500 for B cells (eBioscience), rat anti-mouse NLDC-145 at 1:25 for dendritic cells (Serotec, Duesseldorf, Germany), rat anti-mouse BM8 at 1:500 for macrophages (eBioscience) and rat anti-mouse RB6–8C5 at 1:200 for granulocytes (Molecular Probes/Invitrogen, Karlsruhe, Germany). Sections were then washed four times with PBS for 3 min followed by 60 min of incubation at 4°C with goat anti-rat Alexa Fluor 546-conjugated secondary antibodies (Molecular Probes) diluted at 1:1000 in 0.8% BSA. Sections were washed with PBS and Hoechst stain solution (bisbenzimide H; Sigma), diluted at 1:3000 in 0.8% BSA was added for 15 min of incubation at room temperature. Finally sections were washed with PBS as above, followed by two washes in deionized water for 10 min before air-drying and coverslipping with Fluoromount-G (EMS, Fort Washington, USA). Specificity controls included the omission of primary and/or secondary antibodies. The potency and reactivity of each monoclonal antibody were regularly checked by testing on sections of mouse spleen tissue.
Fluorescence microscopy
Sections were observed with a Zeiss Axioskop 50 epifluorescence microscope through the “rhodamine” filter (Nr. 15 of Carl Zeiss, excitation BP 546/12, beamsplitter FT 580, emission LP 590), the “ultraviolet” filter (Nr. 1 of Carl Zeiss, excitation BP 365/12, Emission LP 397) and through the “fluorescein” filter (Nr. 9 of Carl Zeiss, excitation BP 450–490, beamsplitter FT510, emission LP 520). Digital images of CNS infiltrates for each desired cell type were acquired using a slow scan CCD camera (SPOT RT, Diagnostic Instruments, Sterling Heights, USA) and software. The acquisition software ImagePro Plus (Media Cybernetics, Silver Spring, USA) was used for consecutive image analysis (measurements of infiltrate sizes, counting of cells).
Counting of cells
In each mouse, six brain, cerebellum and spinal cord sections were stained for each primary antibody and analyzed by counting all marker positive cells with a visible cell nucleus (stained with Hoechst stain solution). All counts were independently performed by two observers who had no information about the respective treatment of the mice and the antibodies used. This allowed us to evaluate the mean number of marker positive cells per mm2 infiltrate as well as the percentage of each particular cell type constituting the infiltrate in quantitative terms.
Statistical analysis
SigmaStat software (Version 7.0; SPSS, Chicago, IL) was used to assess the strength of correlation between clinical scores and the number of marker positive cells within CNS lesions in MP4- and MOG:35–55-induced EAE. The coefficients of determination (r2-values) were calculated to evaluate the percent of data closest to the line of best fit and thereby to show how well the regression model described the data. Statistical significance of the differences in lesion composition between two groups was assessed by Student’s t-test. Statistical significance was set at P ≤ 0.05.
Results
Our previous studies in which haematoxylin/eosin staining was performed only suggested that the MP4 and MOGp model fundamentally differed in the topology and kinetics of inflammatory lesions [34]. In order to further elucidate the immune reaction underlying these differences between the models, in the present study we evaluated the cellular composition of CNS lesions in acute and chronic EAE. A total of 30 mice was studied in each of the two EAE models. 15 mice were sacrificed when displaying acute EAE and 15 in chronic EAE. The entire CNS of each mouse was cut into 7 μm thick sections, resulting in 250 coronal brain sections, 50 longitudinal spinal cord and 80 coronal cerebellar sections. According to the fractionator selection strategy, Nissl staining was performed on every 25th brain, 5th spinal cord and 8th cerebellar section to screen for the presence of infiltrates. Positive areas were then subjected to detailed immune histological analysis, staining for CD4+ and CD8+ T cells, CD205+ dendritic cells, B220+ B cells, F4/80+ macrophages and Gr-1+ granulocytes. The major findings of this systematic study are outlined in the following.
During acute EAE the overall cellular composition of CNS infiltrating cells is similar in both MP4- and MOGp-induced EAE
Acute EAE (MP4- or MOGp-induced) was defined as the first day of paralytic symptoms reaching a severity of at least a score of 1. Since the MP4 model showed a slightly earlier onset (Fig. 1) sections for this model were obtained between days 6–16. Sections for the MOGp model were obtained on days 10–16. Regions that scored positive for cellular infiltration via Nissl staining were subjected to immune histological characterization. The mean number of infiltrating cells per mm2 (irrespective of cell type, DAPI staining) was counted by ImagePro software.
Figure 1.
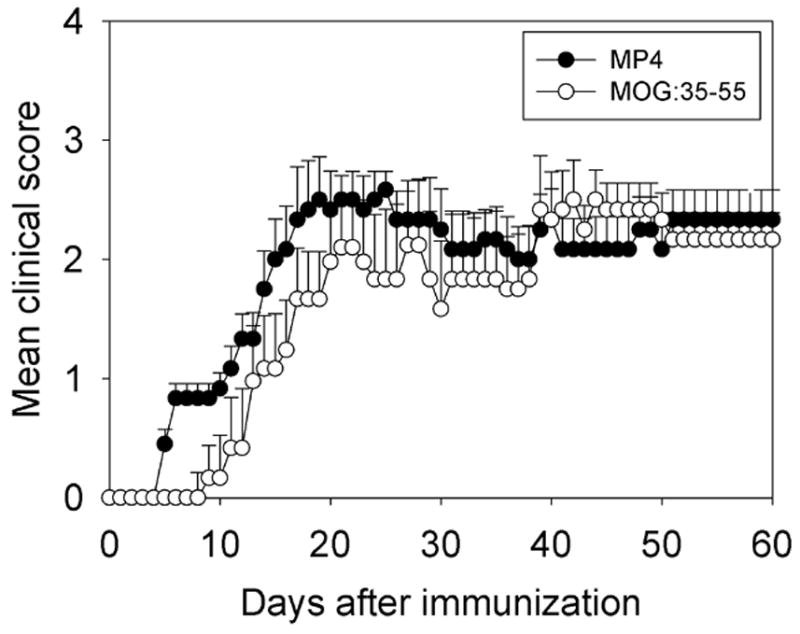
The clinical course of disease in MP4- and MOG:35–55-induced EAE of B6 mice. B6 mice were immunized with either MP4 (solid circles) or MOG:35–55 (open circles) in CFA. PTX was injected on days 0 and 2 after immunization, as specified in the Materials and Methods. The mean course of disease, assessed daily, is shown for eight individual mice in each group. Mean ± SD are given for each group and time point. Scoring of disease followed the 1–5 standard scale (see Materials and Methods). The data are shown for one representative experiment out of three performed.
The results for all lesions analyzed (also irrespective of their location in brain, cerebellum and spinal cord) are shown in Fig. 2A. This figure summarizes the results of 216 sections stained in each model and delineates that the overall density of cellular infiltration was significantly higher in acute MP4-induced lesions than in the MOGp model (P = 0.024).
Figure 2.
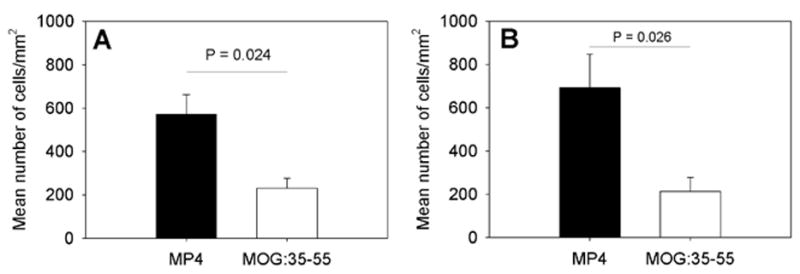
The density of cellular infiltrates in the CNS of MP4- and MOGp-immunized mice in acute and chronic EAE. WT B6 mice were immunized with MP4 (solid bars) or MOGp (open bars) as above. The CNS tissue was removed on the first day on which mice showed clinical symptoms of the disease and DAPI staining was performed on 7 μm thick cryostat sections. The mean total number of cells/mm2 ± SD infiltrating the CNS is given in panel (A) for acute EAE and for chronic EAE in panel (B). Results are shown for a total of 15 mice and 216 sections in each model and for each time point.
The cellular composition of the infiltrates in brain, cerebellum and spinal cord is given in Fig. 3A, B and C, respectively. In these panels, the mean number of marker positive cells per mm2 (the density of these cells) is shown. As can be seen, the density of marker positive cells showed model- and organ-specific differences. In the cerebellum, infiltrating cells were present only in the MOGp model and these consisted of CD4+ T cells, granulocytes and macrophages. These data are in line with our previous observation that the cerebellum is targeted in MP4-induced EAE at a late stage of the disease only (Kuerten et al., 2007). In the brain, macrophages occurred in high and comparable densities in both models. In contrast, the number of CD4+ T cells (P = 0.006), B cells (P < 0.001) and granulocytes (P < 0.001) was higher in the MP4 model. In the spinal cord, the numbers of B cells and macrophages were comparable; however the densities of CD4+ T cells (P < 0.001) and granulocytes (P < 0.001) were significantly higher in the MP4 model. CD8+ T cell and dendritic cell densities were low in all CNS regions.
Figure 3.
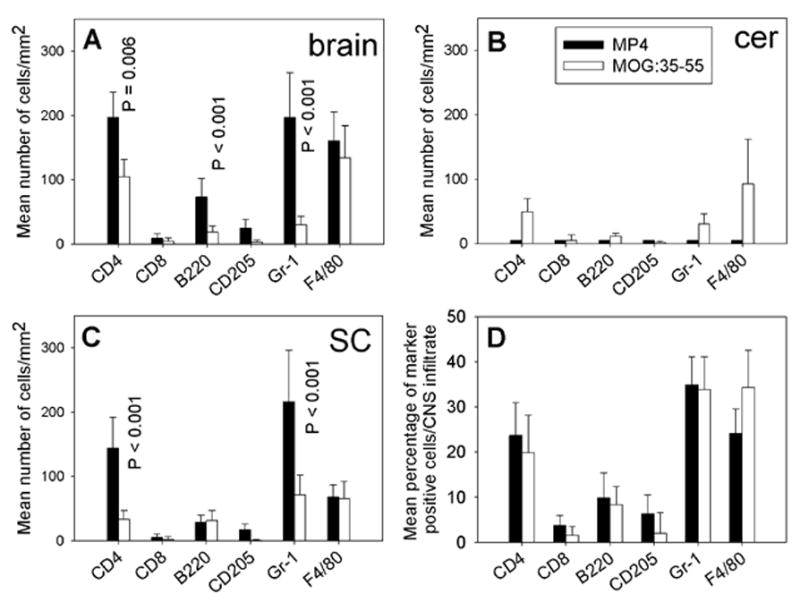
The cellular composition of CNS infiltrates in acute MP4- and MOGp-induced EAE. The CNS tissue was removed on the first day on which mice showed clinical symptoms of the disease and IHC was conducted on 7 μm thick cryostat sections to detect CD4+ and CD8+ T cells, B220+ B cells, CD205+ dendritic cells, Gr-1+ granulocytes and F4/80+ macrophages within the lesions. Solid bars refer to the MP4 model, open bars to MOGp-induced EAE. The mean total numbers of marker positive cells/mm2 ± SD infiltrating the brain are given in panel (A), for the cerebellum in panel (B) and for the spinal cord in panel (C). The mean percentage of infiltrating cells ± SD in all acute lesions analyzed independent of the CNS region are given in panel (D). Results are shown for a total of 15 mice analyzed in each group.
Because we noted that the overall numbers of infiltrating cells differed between the two models (Fig. 3A to C) we re-evaluated the density-based figures, now accounting for the mean percentage of marker positive cells in each CNS infiltrate (Fig. 3D). While for brain, spinal cord and cerebellum minor differences were seen comparing cellular densities, for all three CNS regions analyzed the relative cellular composition of infiltrates was comparable for acute EAE in both models.
In summary, the CNS histology showed a higher density of infiltration in acute MP4-induced EAE that paralleled the more rapid onset of disease in this model [33]. No early targeting of the cerebellum was seen in MP4-immunized mice unlike in the MOGp model. In the brain, but not in the other tissues, there was a tendency for increased B cell numbers already early on (which will be further accentuated in chronic EAE), but was not prevalent in spinal cord and cerebellum yet. Overall, the cellular composition of infiltrates, however, was comparable in both models with CD4+ T cells, granulocytes and macrophages each constituting 20–40% of the infiltrates whereas CD8+ T cells, B cells and dendritic cells each accounted for less than 10% of the infiltrating cells.
During chronic EAE B cells become codominant in MP4-, but not in MOGp-induced EAE
Chronic MP4- and MOGp-induced EAE were evaluated on day 60 after immunization. As shown in Fig. 1 after the initial delay in onset, MOGp-induced EAE reached similar severity and an overall comparable course as the MP4-induced disease. Animals in both categories displayed a mean disease score of 1.85 when sacrificed. As described above for acute disease the density and cellular composition of CNS infiltrates was assessed in brain, spinal cord and cerebellum. The significantly higher density of infiltrating cells that was seen in the acute stage of MP4-induced EAE (Fig. 2A) was maintained in the chronic stage (P = 0.026; Fig. 2B). As above the numbers reflect a total of 216 sections studied in each model. The immune histological analysis of the lesions showed a major shift towards the prevalence of B cells in the MP4 model, however (Fig. 4A to C). In the brain and spinal cord, B cells outnumbered both CD4+ T cells and macrophages. In the cerebellum, the density of infiltration was now significantly higher for all cell types in MP4-induced EAE compared to the MOGp model (P < 0.001 for all cell types). In this CNS region, the numbers of B cells also closely approximated those of CD4+ T cells (Fig. 4B). In all three regions, the densities of dendritic cells and CD8+ T cells were significantly higher in MP4-induced EAE than in the MOGp model (P < 0.001). When these results were expressed as mean percentage of each cell type within the infiltrate (normalizing for the different densities of infiltration) and after pooling the data for brain, spinal cord and cerebellum (Fig. 4D) the patterns of infiltration stayed unchanged in chronic versus acute MOGp-induced disease - the only major change being the decrease in the numbers of infiltrating granulocytes (P < 0.001). CD4+ T cells and macrophages each accounted for 25–40% of infiltrating cells with CD8+ T cells, B cells and dendritic cells representing less than 10% each. In contrast, while for the MP4 disease CD4+ T cells and macrophages still contributed more than 20% to the infiltrating cells each, a major participation of other cell types was also evident in the aggregate numbers. In these mice the numbers of B cells, macrophages and dendritic cells constituted about 46% of the infiltrates.
Figure 4.
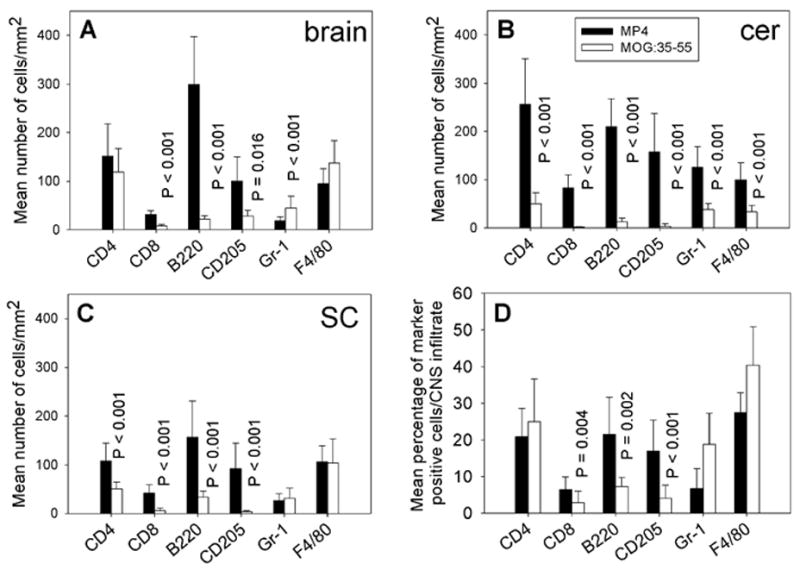
The cellular composition of CNS infiltrates in chronic MP4- and MOGp-induced EAE. The CNS tissue was removed on day 60 after immunization and IHC was conducted on 7 μm thick cryostat sections to detect CD4+ and CD8+ T cells, B220+ B cells, CD205+ dendritic cells, Gr-1+ granulocytes and F4/80+ macrophages within the lesions. Solid bars refer to the MP4 model, open bars to MOGp-induced EAE. The mean total numbers of marker positive cells/mm2 ± SD infiltrating the brain are given in panel (A), for the cerebellum in panel (B) and for the spinal cord in panel (C). The mean percentage of infiltrating cells ± SD in all chronic lesions analyzed independent of the CNS region are given in panel (D). Results are shown for a total of 15 mice analyzed in each group.
B cells are organized in a lymphoid tissue-like manner in CNS infiltrates of chronic MP4-induced EAE
A striking difference between chronic MP4- and MOGp-induced disease resided in the fact that in the former B cells occurred in cellular aggregates – reminiscent of lymphoid follicules – while in the latter they showed random distribution within the infiltrate. This lymph-follicule like organization of B cells was seen in 14/15 of the MP4-immunized mice and in 0/15 of the MOGp mice. The lymph follicle-like structures in MP4 mice occurred in the spinal cord, cerebellum and brain (Fig. 5A, C and D). In the brain the infiltration in MP4-induced disease occurred in a single extended plaque located in the transition from the lateral to the third ventricle. In the cerebellum the infiltrates frequently reached close to perfectly round shapes and many times consisted close to entirely of B cells that were organized in a follicule-like architecture (Fig. 5C) with the remaining cells representing mostly CD4+ T cells, dendritic cells and granulocytes (see Fig. 4B). In the spinal cord infiltrating B cells were predominantly found in the submeningeal regions representing cuffs that occasionally extended into the parenchyma. The rest of infiltrates in all CNS regions analyzed consisted primarily of macrophages and CD4+ T cells. Dendritic cells frequently showed colocalization with B cells (data not shown).
Figure 5.
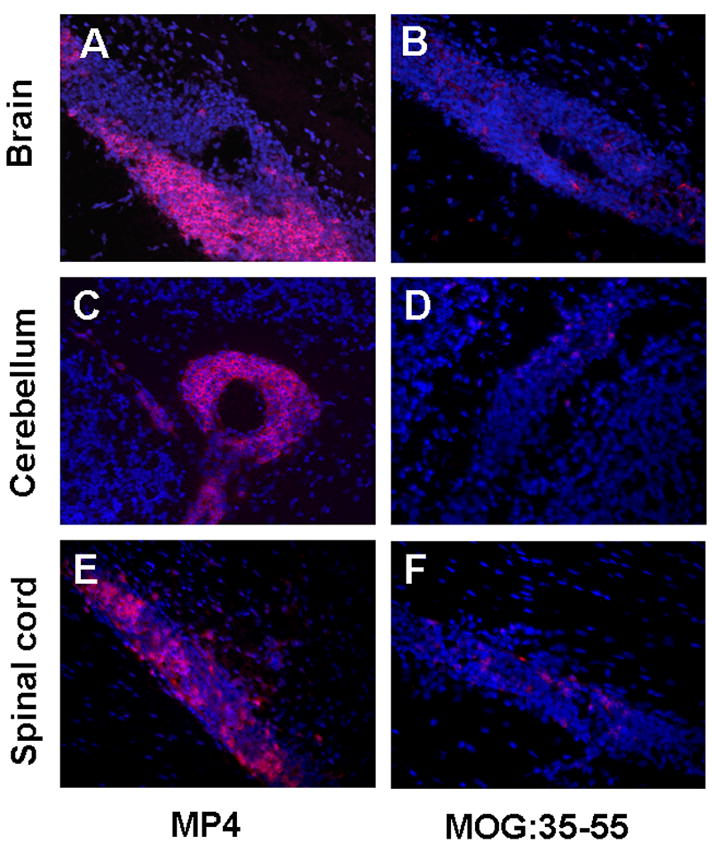
B cells are organized in a lymphoid tissue-like manner in CNS infiltrates of MP4-immunized mice. Representative images depict how B cells are organized in lymph follicle-like structures in lesions of the brain (A), cerebellum (C) and spinal cord (E) in MP4-immunized mice. In the MOGp model B cells are scattered throughout the infiltrate in brain (B), cerebellum (D) and spinal cord (F). Rat anti-mouse B220 served as primary antibody, goat anti-rat IgG (H+L) conjugated to Alexa Fluor 546 as secondary antibody in both models. Thus, marker positive cells are shown in red. DAPI staining is shown in blue. IHC. CNS tissue. 400 × magnification.
Overall, the distribution and organization of cells suggests that in the case of MP4-induced disease the infiltrating cells are organized in the form of actual lymphoid tissue with germinal centers that may support ongoing immune T cell activation, proliferation and differentiation. On the contrary, in the MOGp-induced disease this follicule-like structure of CNS infiltrations was absent (Fig. 5B, D and F) being more consistent with plain effector functions asserted by the T cells.
The cellular composition of infiltrates in MP4-, but not in MOGp-induced EAE allows the staging of the disease into an acute and chronic phase
One of the primary criticisms on EAE models in general and the MOGp model in particular has been that histological manifestations of the disease do not reflect the stage of the disease as it progresses [35]. We studied whether in MP4-induced EAE changes in the cellular composition of infiltrates would provide such information. Therefore, the density of marker positive cells was compared in the acute and chronic stage of the disease. Fig. 6 shows that for MOGp-induced disease neither CD4+ T cells (A), CD8+ T cells (B), B cells (C) nor dendritic cells (D) showed a significant difference in CNS lesion densities between the acute and chronic stage of the disease. Only the densities of granulocytes (P < 0.001) and macrophages (P = 0.001) in the spinal cord reached significance (Fig 6E and F, respectively). On the contrary, for MP4-induced EAE the numbers of CD4+ T cells in the cerebellum (Fig. 7A), and of all other cell types in all CNS regions studied (Fig. 7B to F) provided a significant difference between the acute and chronic disease with P ≤ 0.01 in all cases.
Figure 6.
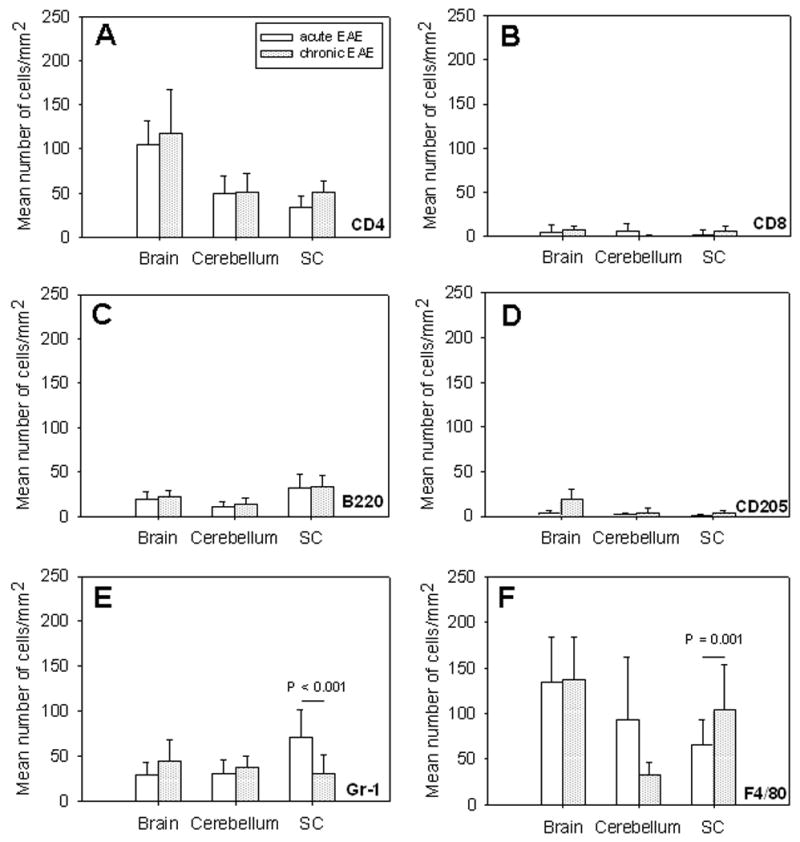
Similar cellular composition of CNS infiltrates in acute versus chronic MOGp-induced EAE. WT B6 mice were immunized with MOGp in CFA. Brain, spinal cord and cerebellum were removed during acute (open bars) and chronic (dotted bars) EAE as defined in the text. IHC was performed staining 7 μm thick cryostat sections for the presence of CD4+ T cells (A), CD8+ T cells (B), B220+ B cells (C), CD205+ dendritic cells (D), Gr-1+ granulocytes (E) and F4/80+ macrophages (F) within CNS lesions as described above. Results are depicted as mean number of positive cells/mm2 ± SD in CNS lesions of a total of 15 mice per group. P-values denote whether the differences between the acute and chronic stage of the disease were significant.
Figure 7.
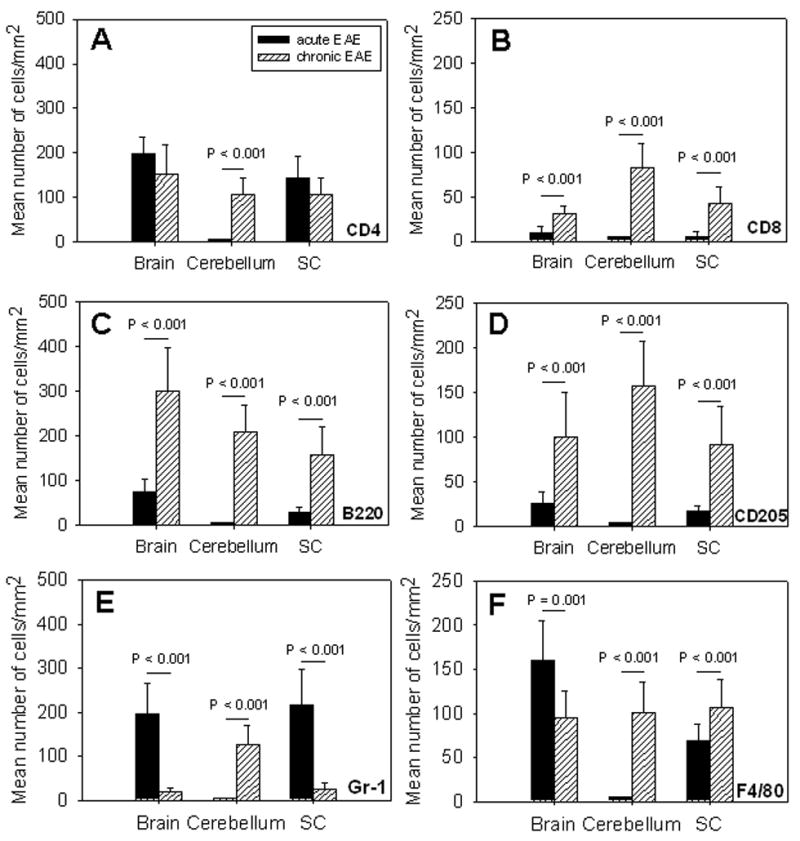
Numbers of infiltrating cells permit the differentiation between acute and chronic EAE in MP4-induced EAE. WT B6 mice were immunized with MP4 in CFA. Brain, spinal cord and cerebellum were removed during acute (solid bars) and chronic (hatched bars) EAE as defined in the text. IHC was performed staining 7 μm thick cryostat sections for the presence of CD4+ T cells (A), CD8+ T cells (B), B220+ B cells (C), CD205+ dendritic cells (D), Gr-1+ granulocytes (E) and F4/80+ macrophages (F) within CNS lesions as described above. Results are depicted as mean number of positive cells/mm2 ± SD in CNS lesions of a total of 15 mice per group. P-values denote whether the differences between the acute and chronic stage of the disease were significant.
The cellular composition of CNS infiltrates correlates with the clinical score in MP4-induced EAE, but not in the MOGp model
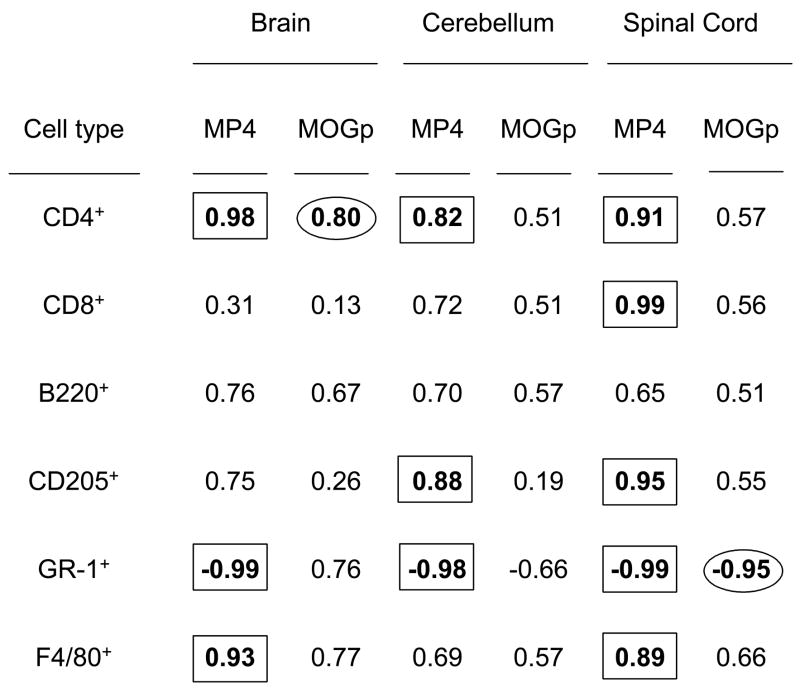
Another alleged weakness of the MOGp and other EAE models is that it does not show a correlation between functional damage (i.e. the clinical score) and structural changes in the CNS (i.e. size, density and composition of lesions) [34,35]. With this systematic histological material at hand, we set out to study whether such differences could be seen in the MP4 model. For both models and for all three CNS regions analyzed we correlated the density of each particular cell type in each mouse with the clinical scores on the day of obtaining the CNS material. The data are summarized in Table 1.
Table 1.
Correlation between the cellular composition of CNS infiltrates and the clinical severity of disease in MP4- and MOGp-induced EAE
|
The severity of MP4-, but not of MOGp-induced EAE correlates with the cellular lesion composition. Mice were immunized with MP4 or MOGp as described in the Materials and Methods. In each animal and depending on the clinical score, we evaluated the mean number of CD4+ and CD8+ T cells, B220+ B cells, CD205+ dendritic cells, GR-1+ granulocytes and F4/80+ macrophages per mm2 infiltrate via IHC. The r2-values are shown for each cell type and CNS regions analyzed differentiating between MP4- and MOGp-induced EAE. The cut-off for a high to perfect correlation was set at r2 > 0.8.
According to regression analysis, the cut-off for a high to perfect correlation can be set at r2 > 0.8. Taking this criterium, for the MOGp model only in two cases, i.e. for CD4+ T cells in the brain and for granulocytes in the spinal cord a relevant correlation with the clinical score was achieved. For the MP4 model in contrast, 11 positive correlations were identified.
Discussion
There is increasing evidence that MS is not a single disease entity leading to the common features of CNS inflammation and demyelination, but that different immune mechanisms drive distinct and unique manifestations of the disease. Besides differences in the clinical presentation, histological MS subtypes can be defined in which lesions bear signs of either isolated T cell activity with downstream activation of microglia and macrophages, or antibody- and complement-mediated demyelination is evident [5,7]. In EAE of rats and mice it has also been shown that the very nature of the neuroantigen used for disease induction can result in differences in lesion topology and kinetics [1,34]. Overall, the different clinical and histological features of MS are more pronounced in one patient than in the other making it difficult to understand the pathogenesis of the disease and develop therapeutic strategies.
When trying to study MS by using EAE as a model many of these differential disease manifestations were reproduced by immunizing susceptible animals from different genetic backgrounds. However, in addition to the immune pathology associated with the particular neuroantigen itself and with the T cell differentiation pattern caused by the immunization these genetic differences introduced further variables hampering clear-cut and “simple” studies of the disease mechanisms. Moreover, when EAE models are compared in the literature, naturally different mouse colonies were used for studying the disease. However, variable environmental influences have been shown to profoundly affect the outcome of autoimmune processes [36–38]. A major quest in the field should therefore be to consolidate models in a single mouse strain (thereby avoiding the genetic variables) for comparative studies in a single laboratory (thereby avoiding the environmental influences). Yet, single mouse strains have been shown to be susceptible to disease induction with only one or few neuroantigens. This may be related to Ir gene effects or reasons of self-tolerance. For example, the B6 mouse is notoriously resistant to MBP-induced EAE. For yet poorly understood reasons the B6 mouse is also resistant to PLP protein-induced EAE. Working with PLP protein itself is hardly a viable model because of the difficulty of preparing the protein and its poor solubility. Strikingly however, this mouse is highly susceptible to the MBP-PLP fusion protein (MP4)-induced disease [33]. The reasons may reside in the fact that PLP is exon-shuffled excising the hydrophobic parts of the molecule and thus increasing solubility. In addition, the associated changes in primary sequence may result in unique antigenic determinants that are more immunogenic in the B6 mouse than native PLP, yet cross-reacting with antigenic PLP. As a consequence, a mouse strain that is resistant to MBP and PLP individually has become susceptible to disease induced by the fusion of both, thereby complimenting the classic MOGp-induced EAE on the B6 background. Only recently, a third model has been introduced for B6 mice using PLP:178–191 as neuroantigen [31]. According to the initial characterization of this model [31,33] the disease induced by this peptide may or may not represent a distinct pathogenic entity relative to the MOGp-induced disease – beyond its initial description the PLP model has not been studied in depth yet.
While it is important to establish MS models on a single background, among such backgrounds, the B6 mouse indeed holds a unique place. Knock-out and knock-in mice are typically generated on the 129 background and backcrossed to the B6 background and the use of gene-modified mice is becoming increasingly indispensable for studies of the underlying pathomechanisms of the disease. For example, depleting a cell type with an antibody is technically not “clean”. The apparent depletion of the cells could result from (temporary) downregulation of the molecule, from redistribution of the cells in the organism in addition to the depletion itself. The depletion may work in tissues into which the antibodies readily penetrate, but may not affect tissues into which the antibodies are not penetrating. Moreover, massive cell destruction and depletions can induce cytokine storms and other inflammatory reactions that fundamentally change the host’s immune biology beyond the actual cell depletion itself. Therefore, it is much more effective to study a cell knock-out mouse in such a situation with and without the adoptive transfer of that particular cell controlling for the actual contribution of that cell type. Our follow-up studies will also need to focus on such experiments and the B6 background will facilitate such progress.
Here we set out to provide a closer histopathological characterization of the MP4 versus the MOGp model. Our initial observations showed that these models differed in the relative contribution of B cells to the disease as shown by a less severe course of the disease and a later disease onset in B cell KO mice immunized with MP4. B cells are thought to be key players in the pathology of MS, however most EAE models including the MOGp peptide model do not seem to include this component [39,40]. Therefore, we aimed at further gaining evidence for B cell contribution in the MP4 model. Studying both, the MP4 and the MOGp model systematically early and late after immunization, we indeed found that B cell infiltration was a common feature of the MP4 model, but in MOGp-induced disease B cells were scarce within the infiltrates. Moreover, it was striking that in chronic MP4-induced disease B cells showed clustering reminiscent of follicular structures while in the MOGp-induced disease they were scattered throughout the lesions. Remarkably, dendritic cells co-clustered with the B cells in follicular-like structures in the MP4 model, but not in MOGp-induced EAE. Such lymphoid organization of inflamed tissue can occur primarily in the setting of chronic immune pathology. In one scenario infiltrating T cells release cytokines that attract and stimulate macrophages and other cells of the immune system into the inflammatory process and eventually die [41]. This scenario seems to apply for the MOGp-induced disease where many CD4+ T cells and macrophages, but only few dendritic cells and B cells in the absence of lymphoid organization were seen. In the second scenario, the target organ itself and infiltrates within the target organ are organized into so-called tertiary lymphoid tissues. In such settings, the germinal centers are thought to facilitate the active immunological processes of proliferation and differentiation converting the target organ into a primitive lymph node. Processes like determinant spreading, the recruitment of other naïve T cells into the effector cell repertoire and the differentiation of the effector cells into subclasses such as TH1/TH17 or TH2 are facilitated in such tissues. Recent evidence provided by our group and others suggests that the inflamed CNS is indeed the site where determinant spreading first takes place in contrast to the draining lymph nodes of that organ [42–46]. In MS patients it has been shown that meningeal B cell follicles in secondary progressive MS are associated with early onset of disease and severe cortical pathology [47]. Lymphoid tissue organization is also regularly seen in rheumatoid arthritis [48] and Graves’ disease [49], possibly representing sites of ongoing immune sensitization, perpetuation and differentiation. Our data suggest that indeed the MOGp and the MP4 model fundamentally differ in this component; the CNS being primarily an end-organ for the MOGp model and “germinal center” for ongoing autoimmunity in the MP4 model.
While this notion triggers an endless number of testable hypotheses at this time point, there is evidence to support this notion. When we studied the immune repertoire induced by MP4 we noted that an extremely diverse spectrum of determinants was recognized, reminiscent of a repertoire induced by determinant spreading [33]. Immunizations with proteins such as MP4 – for reasons of antigen processing and presentation on MHC class II molecules only – are prone to induce CD4+ T cell responses only. Yet, we noted that the MP4 disease – unlike MOGp-induced disease – is also dependent on CD8+ T cells [4,33,50,51] since CD8 KO mice showed a late onset of disease with decreased severity. In our histological studies as described here, CD8+ T cell infiltration accounted for approximately 10% of the infiltrates in MP4-induced EAE (see Fig. 4). Moreover, in the MP4 model CD8+ T cells were frequently significantly larger and expresses ruffled membrane extrusions relative to the rounder and smaller CD8+ T cells seen in MOGp-induced disease (data not shown) suggesting that in the former, they were activated T cell blasts while in the latter they might be resting bystander cells.
One of the difficulties in working with MS and EAE model has been that it was very difficult to stage the disease, that is to conclude from histological lesions how long the disease has been ongoing. In humans, studies have been further hampered by the fact that CNS material cannot be obtained for longitudinal histological studies. In this way, differences seen between patients could either represent different subtypes of MS or changes seen in the course of the disease. In our case, MOGp-induced EAE appeared static with respect to localization and cellular infiltration. In contrast, in the MP4 model histological changes throughout the disease allowed its staging. In particular, early infiltrates did not include the cerebellum, late ones did. B cell infiltration was scarce in the early disease while being prevalent in the chronic disease. Similarly, numbers of CD8+ T cells and dendritic cells increased towards the progression of the disease. Therefore, these changes that are all consistent with the recruitment of lymphoid cells into organisation of a tertiary lymphoid tissue provide criteria by which acute and chronic disease can be distinguished.
In conclusion, the observations made in our systematic immune histological study suggest that the MP4 model is not only an alternative to inducing EAE in B6 mice with MOGp, but represents a distinct immune pathological entity, thereby enhancing the ability to model MS while avoiding genetic background differences in different mouse strains.
Acknowledgments
We would like to thank Susan Faas (Alexion Pharmaceuticals) for providing us with the MP4 protein as well as Lukas P. Frenzel and Tomo ari and for their valuable discussions. This work was supported by grants to D.N.A. from the Deutsche Forschungsgemeinschaft (DFG), to P.V.L from the National Institutes of Health (NS-39434) and to M.T.L (AI-47756). S.K. was supported by a fellowship of the Studienstiftung des Deutschen Volkes, by the Koeln Fortune Program (University of Cologne) and the Maria-Pesch-Stiftung.
Abbreviations
- B6
C57BL/6
- CFA
Complete Freund’s Adjuvant
- CNS
central nervous system
- EAE
experimental autoimmune encephalomyelitis
- HEL
hen egg lysozyme
- IFA
Incomplete Freund’s Adjuvant
- IHC
immunohistochemistry
- MBP
myelin basic protein
- MOGp
MOG peptide 35–55
- MP4
MBP-PLP fusion protein
- MS
multiple sclerosis
- PBS
phosphate-buffered saline
- PLP
proteolipid protein
- PLPp
PLP peptide 178–191
- PTX
pertussis toxin
- SCH
spinal cord homogenate
- TBS
tris-buffered saline
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berger T, Weerth S, Kojima K, Linington C, Wekerle H, Lassmann H. Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab Invest. 1997;76:355–364. [PubMed] [Google Scholar]
- 2.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 3.Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. 2001;7:115–121. doi: 10.1016/s1471-4914(00)01909-2. [DOI] [PubMed] [Google Scholar]
- 4.Sun D, Zhang Y, Wie B, Peiper SC, Shao H, Kaplan HJ. Encephalitogenic activity of truncated myelin oligodendrocyte glycoprotein (MOG) peptides and their recognition by CD8+ MOG-specific T cells on oligomeric MHC class I molecules. Int Immunol. 2003;15:261–268. doi: 10.1093/intimm/dxg023. [DOI] [PubMed] [Google Scholar]
- 5.Lassmann H, Ransohoff RM. The CD4-Th1 model for multiple sclerosis: a critical [correction of crucial] re-appraisal. Trends Immunol. 2004;25:132–137. doi: 10.1016/j.it.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Hafler DA. Multiple sclerosis. Clin Invest. 2004;113:788–94. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 8.Wekerle H, Kojima K, Lannes-Vieira J, Lassmann H, Linington C. Animal models. Ann Neurol Suppl. 1994;36:S47–S53. doi: 10.1002/ana.410360714. [DOI] [PubMed] [Google Scholar]
- 9.Martin R, McFarland HF. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit Rev Clin Lab Sci. 1995;32:121–182. doi: 10.3109/10408369509084683. [DOI] [PubMed] [Google Scholar]
- 10.Goverman J, Brabb T. Rodent models of experimental allergic encephalomyelitis applied to the study of multiple sclerosis. Lab Anim Sci. 1996;46:482–492. [PubMed] [Google Scholar]
- 11.Baxter AG. The origin and application of experimental autoimmune encephalomyelitis. Nat Rev Immunol. 2007;7:904–12. doi: 10.1038/nri2190. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt S. Candidate autoantigens in multiple sclerosis. Mult Scler. 1999;5:147–160. doi: 10.1177/135245859900500303. [DOI] [PubMed] [Google Scholar]
- 13.Steinman L. Assessment of animal models for MS and demyelinating disease in the design of rational therapy. Neuron. 1999;24:511–514. doi: 10.1016/s0896-6273(00)81107-1. [DOI] [PubMed] [Google Scholar]
- 14.Levine S, Sowinski R, Shaw CM, Alvord EC., Jr Do neurological signs occur in experimental allergic encephalomyelitis in the absence of inflammatory lesions of the central nervous system? J Neuropathol Exp Neurol. 1975;34:501–506. doi: 10.1097/00005072-197511000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Sensharma GC, Singh S. Neuroglial relationship in experimental central demyelination in rabbits. Acta Anat. 1975;93:534–542. doi: 10.1159/000144531. [DOI] [PubMed] [Google Scholar]
- 16.Snyder DH, Valsamis MP, Stone SH, Raine CS. Progressive demyelination and reparative phenomena in chronic experimental allergic encephalomyelitis. J Neuropathol Exp Neurol. 1975;34:209–221. doi: 10.1097/00005072-197505000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Mitsuzawa E, Yasuda T. Experimental allergic encephalitis (EAE) in mice: histological studies on EAE induced by myelin basic protein, and role of pertussis vaccine. Jpn J Exp Med. 1976;46:205–212. [PubMed] [Google Scholar]
- 18.Lassmann H, Wisniewski HM. Chronic relapsing experimental allergic encephalomyelitis: clinicopathological comparison with multiple sclerosis. Arch Neurol. 1979;3:490–497. doi: 10.1001/archneur.1979.00500440060011. [DOI] [PubMed] [Google Scholar]
- 19.Waxman FJ, Fritz RB, Hinrichs DJ. The presence of specific antigen-reactive cells during the induction, recovery, and resistance phases of experimental allergic encephalomyelitis. Cell Immunol. 1980;49:34–42. doi: 10.1016/0008-8749(80)90053-2. [DOI] [PubMed] [Google Scholar]
- 20.Sobel RA, Blanchette BW, Bhan AK, Colvin RB. The immunopathology of experimental allergic encephalomyelitis. I. Quantitative analysis of inflammatory cells in situ. J Immunol. 1984;132:2393–2401. [PubMed] [Google Scholar]
- 21.Sobel RA, van der Veen RC, Lees MB. The immunopathology of chronic experimental allergic encephalomyelitis induced in rabbits with bovine proteolipid protein. J Immunol. 1986;136:157–163. [PubMed] [Google Scholar]
- 22.Matsumoto Y, Hara N, Tanaka R, Fujiwara M. Immunohistochemical analysis of the rat central nervous system during experimental allergic encephalomyelitis, with special reference to Ia-positive cells with dendritic morphology. J Immunol. 1986;136:3668–3676. [PubMed] [Google Scholar]
- 23.Tuohy VK, Sobel RA, Lees MB. Myelin proteolipid protein-induced experimental allergic encephalomyelitis. Variations of disease expression in different strains of mice. J Immunol. 1988;140:1868–1873. [PubMed] [Google Scholar]
- 24.Hofstetter HH, Shive CL, Forsthuber TG. Pertussis toxin modulates the immune response to neuroantigens injected in incomplete Freund’s adjuvant: induction of Th1 cells and experimental autoimmune encephalomyelitis in the presence of high frequencies of Th2 cells. J Immunol. 2002;169:117–125. doi: 10.4049/jimmunol.169.1.117. [DOI] [PubMed] [Google Scholar]
- 25.Bernard CC. Experimental autoimmune encephalomyelitis in mice: genetic control of susceptibility. J Immunogenet. 1976;3:263–274. doi: 10.1111/j.1744-313x.1976.tb00583.x. [DOI] [PubMed] [Google Scholar]
- 26.Gasser DL, Goldner-Sauve A, Hickey WF. Genetic control of resistance to clinical EAE accompanied by histological symptoms. Immunogenetics. 1990;31:377–382. doi: 10.1007/BF02115013. [DOI] [PubMed] [Google Scholar]
- 27.Skundric DS, Huston K, Shaw M, Tse HY, Raine CS. Experimental allergic encephalomyelitis. T cell trafficking to the central nervous system in a resistant Thy-1 congenic mouse strain. Lab Invest. 1994;71:671–679. [PubMed] [Google Scholar]
- 28.Fritz RB, Zhao ML. Active and passive experimental autoimmune encephalomyelitis in strain 129/J (H-2b) mice. J Neurosci Res. 1996;45:471–474. doi: 10.1002/(SICI)1097-4547(19960815)45:4<471::AID-JNR17>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 29.Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med. 2000;6:56–61. doi: 10.1038/71540. [DOI] [PubMed] [Google Scholar]
- 30.Mendel I, de Rosbo NK, Ben Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- 31.Tompkins SM, Padilla J, Dal Canto MC, Ting JP, Van Kaer L, Miller SD. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. J Immunol. 2002;168:4173–4183. doi: 10.4049/jimmunol.168.8.4173. [DOI] [PubMed] [Google Scholar]
- 32.Elliott EA, McFarland HI, Nye SH, Cofiell R, Wilson TM, Wilkins JA, Squinto SP, Matis LA, Mueller JP. Treatment of experimental encephalomyelitis with a novel chimeric fusion protein of myelin basic protein and proteolipid protein. J Clin Invest. 1996;98:1602–1612. doi: 10.1172/JCI118954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuerten S, Lichtenegger FS, Faas S, Angelov DN, Tary-Lehmann M, Lehmann PV. MBP-PLP fusion protein-induced EAE in C57BL/6 mice. J Neuroimmunol. 2006;177:99–111. doi: 10.1016/j.jneuroim.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 34.Kuerten S, Kostova-Bales DA, Frenzel LP, Tigno JT, Tary-Lehmann M, Angelov DN, Lehmann PV. MP4- and MOG:35–55-induced EAE in C57BL/6 mice differentially targets brain, spinal cord and cerebellum. J Neuroimmunol. 2007;189:31–40. doi: 10.1016/j.jneuroim.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerschensteiner M, Stadelmann C, Buddeberg BS, Merkler D, Bareyre FM, Anthony DC, Linington C, Bruck W, Schwab ME. Targeting experimental autoimmune encephalomyelitis lesions to a predetermined axonal tract system allows for refined behavioral testing in an animal model of multiple sclerosis. Am J Pathol. 2004;164:1455–1469. doi: 10.1016/S0002-9440(10)63232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leiter EH, Serreze DV, Prochazka M. The genetics and epidemiology of diabetes in NOD mice. Immunol Today. 1990;11:147–149. doi: 10.1016/0167-5699(90)90057-g. [DOI] [PubMed] [Google Scholar]
- 37.Anderson AC, Nicholson AB, Legge KL, Turchin V, Zaghouani H, Kuchroo VK. High Frequency of Autoreactive Myelin Proteolipid Protein–specific T Cells in the Periphery of Naive Mice: Mechanisms of Selection of the Self-reactive Repertoire. J Exp Med. 2000;191:761–770. doi: 10.1084/jem.191.5.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Racke MK. UNIT 9.7 Experimental Autoimmune Encephalomyelitis (EAE) Curr Prot Neurosci Supplement. 2001;14 doi: 10.1002/0471142301.ns0907s14. [DOI] [PubMed] [Google Scholar]
- 39.Hjelmstrom P, Juedes AE, Fjell J, Ruddle NH. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol. 1998;161:4480–4483. [PubMed] [Google Scholar]
- 40.Oliver AR, Lyon GM, Ruddle NH. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J Immunol. 2003;171:462–468. doi: 10.4049/jimmunol.171.1.462. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann PV. The fate of T cells in the brain : veni, vidi, vici and veni, vidi, mori. Am J Pathol. 1998;153:677–680. doi: 10.1016/S0002-9440(10)65609-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann PV, Sercarz EE, Forsthuber T, Dayan CM, Gammon G. Determinant spreading and the dynamics of the autoimmune T-cell repertoire. Immunol Today. 1993;14:203–208. doi: 10.1016/0167-5699(93)90163-F. [DOI] [PubMed] [Google Scholar]
- 44.McRae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. 1995;182:75–85. doi: 10.1084/jem.182.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Targoni OS, Baus J, Hofstetter HH, Hesse MD, Karulin AY, Boehm BO, Forsthuber TG, Lehmann PV. Frequencies of neuroantigen-specific T cells in the central nervous system versus the immune periphery during the course of experimental allergic encephalomyelitis. J Immunol. 2001;166:4757–4764. doi: 10.4049/jimmunol.166.7.4757. [DOI] [PubMed] [Google Scholar]
- 46.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 47.Maggliozi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, Reynolds R, Aloisi F. Meningeal B-cell follicules in secondary progressive multiple sclerosis associate with early onset of the disease and severe cortical pathology. Brain. 2007;130:1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- 48.Benoist C, Mathis D. A revival of the B cell paradigm for rheumatoid arthritis pathogenesis? Arthritis Res. 2000;2:90–94. doi: 10.1186/ar73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Misaki T, Konishi J, Nakashima T, Iida Y, Kasagi K, Endo K, Uchiyama T, Kuma K, Torizuka K. Immunohistological phenotyping of thyroid infiltrating lymphocytes in Graves’ disease and Hashimoto’s thyroiditis. Clin Exp Immunol. 1985;60:104–110. [PMC free article] [PubMed] [Google Scholar]
- 50.Koh DR, Fung-Leung WP, Ho A, Gray D, Acha-Orbea H, Mak TW. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8−/− mice. Science. 1992;256:1210–1213. doi: 10.1126/science.256.5060.1210. [DOI] [PubMed] [Google Scholar]
- 51.Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]