

Abstract
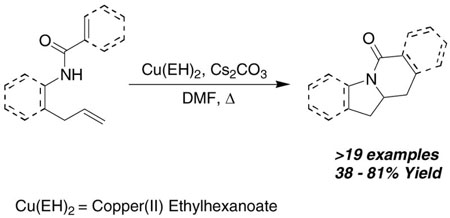
The copper(II) carboxylate promoted intramolecular carboamination reactions of variously substituted γ-alkenyl amides have been investigated. These oxidative cyclization reactions efficiently provide polycyclic lactams, useful intermediates in nitrogen heterocycle synthesis, in good to excellent yields. The efficiency of the carboamination process is dependent upon the structure of the amide backbone as well as the nitrogen substituent.
Nitrogen heterocycles are useful intermediates in complex molecule synthesis, as well as an abundant class of biologically active molecules. The rapid assembly of such molecules and the search for new molecular scaffolds provides a constant challenge to the synthetic and medicinal chemist, thus, new methods of entry into these systems are especially useful.1 One particularly attractive method is the intramolecular carboamination reaction of alkenes.2
In 2004, our lab disclosed the first copper(II) carboxylate promoted intramolecular carboamination reaction of N-arylsulfonyl 2-allyl anilines (Eq 1).2c More recently we reported further substrate versatility and diastereoselectivity, and provided an experimental analysis of the reaction mechanism.2f
 |
(1) |
In both of these accounts, a variety of γ and δ alkenyl N-aryl sulfonamides were shown to undergo the oxidative cyclization in the presence of copper(II) carboxylates to produce the corresponding polycyclic sultams in modest to good yield.2c, 2f In an effort to expand the utility of the method, a variety of aryl-, vinyl-, and aliphatic γ-alkenyl amides were investigated in the copper(II) promoted carboamination reaction. The results of our findings are described in this report.
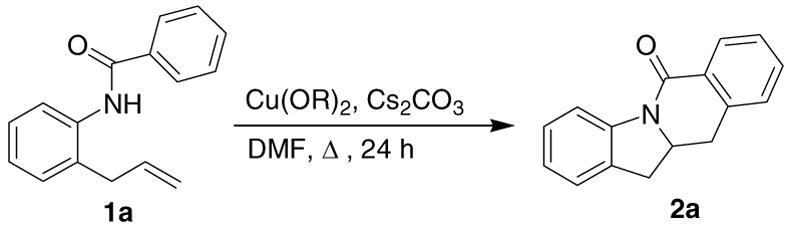
Under our original reaction conditions (e.g. Eq 1), 2-allyl aniline amides were poor substrates for the copper(II) carboxylate promoted intramolecular carboamination reaction, significantly less reactive in comparison to their aryl sulfonamide counterparts (Table 1, entries 1 and 2).2f Success in this matter was subsequently realized when N-benzoyl-2-allyl aniline was treated with more organic soluble copper(II) salts and slightly higher reaction temperatures, providing polycyclic lactam 2a in moderate yield (Table 1). The organic soluble copper salts, Cu(II) neodecanoate [Cu(ND)2] and Cu(II) ethylhexanoate [Cu(EH)2], were shown to be more reactive than Cu(OAc)2 (Table 1, entries 3 and 4).2f, 3 Cu(EH)2 was subsequently used throughout the substrate screening because of its lower cost and ease of use [Cu(EH)2 is purchased as a solid whereas Cu(ND)2 is purchased as a solution in toluene].
Table 1.
Reaction optimization.a
 | |||
|---|---|---|---|
| entry | copper salt | temperature | yieldb |
| 1c | Cu(OAc)2 | 170 °C | 16% |
| 2c | Cu(OAc)2 | 190 °C | 39% |
| 3 | Cu(ND)2 | 190 °C | 59% |
| 4 | Cu(EH)2 | 190 °C | 61% |
Conditions: Substrate in DMF (0.1 M) was treated with Cu(OR)2 (3 equiv) and Cs2CO3 (1 equiv). The mixture was heated to the indicated temperature for 24 h in a pressure tube.
Yields refer to product isolated by chromatography on SiO2.
DMSO (4 equiv) was used. The remainder of the material was either starting olefin or olefin-isomerized starting material. Cu(EH)2 = copper(II) ethylhexanoate, Cu(ND)2 = copper(II) neodecanoate.
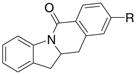
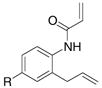
A variety of 2-allyl aniline amides were oxidatively cyclized in an efficient manner using the optimized reaction conditions (Table 2). The mildly electron deficient halogenated substrates 1b and 1c reacted efficiently (Table 2, entries 2 – 4 and 7). Worth noting, the flourine was displaced with dimethylamine when the reaction was run in DMF (Table 2, entry 3) (dimethylamine presumably arises from thermally decomposed DMF). When tert-butyl benzene was used as solvent, the carboamination adduct was obtained in good yield with the fluorine intact (Table 2, entry 4). 4-Cyano and 4-methoxy arylamides displayed comparatively lower reactivity (Table 2, entries 5 and 6). Meta-substituted aryl amides demonstrated a preference (ca. 1.8:1) for the ortho addition product over the para (Table 2, entries 7 and 8). This ortho preference is consistent with that of meta-substituted aryl sulfonamides2c, 2f and provides evidence for C-C bond formation via the addition of a carbon radical to an aromatic ring.2f, 4
Table 2.
Carboamination of o-allyl aryl amidesa
| entry | substrate | product(s) | yieldb(selectivity) | |
|---|---|---|---|---|
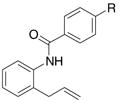
|
|
|||
| 1 | 1a, R = H | 2a, R = H | 61% | |
| 2 | 1b, R = Cl | 2b, R = Cl | 63% | |
| 3 | 1c, R = F | 2c, R = NMe2 | 73% | |
| 4c | 1c, R = F | 2d, R = F | 71% | |
| 5 | 1d, R = OMe | 2e, R = OMe | 56% | |
| 6 | 1e, R = CN | 2f, R = CN | 42% | |
| 7 |
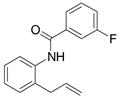
3 |
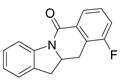
4 |
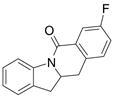
5 |
58%4:5 = 1.7:1 |
| 8 |
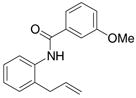
6 |
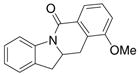
7 |
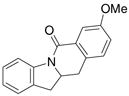
8 |
44%7:8 = 1.9:1 |
Conditions. Substrate in DMF (0.1 M) was treated with Cu(EH)2 (3 equiv) and Cs2CO3 (1 equiv). The mixture was heated to 190 °C for 24 h in a pressure tube.
Yields refer to the sum of products isolated by chromatography on SiO2. The remainder of the material was either starting olefin or olefin-isomerized starting material.
tButyl benzene was used as solvent. The structures of the products (e.g., regioisomer) were assigned by analysis of the aromatic region of the 1H NMR spectra.1
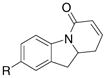
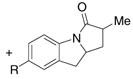
2-Allyl aniline derived vinyl amides are reactive carboamination substrates as well (Table 3). Interestingly, the unsubstituted vinyl amides 9a – c cyclized in 6-endo fashion at 140 °C to provide the polycyclic α,β-unsaturated lactones 10a – c in good yield (Table 3, entries 1–3). These observations are in contrast to similar palladium catalyzed processes where the 5-exo cyclization product (similar to 11) was the only observed regioisomer.2d,5 Increasing the reaction temperature to 190 °C, however, resulted in a 1.2:1 mixture of the 6-endo to 5-exo carboamination adducts (Table 3, entry 4). The 5-exo product 11a terminated in hydrogen atom capture rather than olefin formation. This observation presents an intriguing example of temperature control of regiochemistry.
Table 3.
Carboamination of o-allyl vinyl amidesa
| entry | substrate | product(s) | yieldb(selectivity) | |
|---|---|---|---|---|
|
|
|
||
| 1 | 9a, R = H | 10a, R = H | 11a, R = H | 72%(10a:11a = >20:1) |
| 2 | 9b, R = OMe | 10b, R = OMe | 11b, R = OMe | 74%(10b:11b = >20:1) |
| 3 | 9c, R = Cl | 10c, R = Cl | 11c, R = Cl | 71%(10c:11c = >20:1) |
| 4c | 9a, R = H | 10a, R = H | 11a, R = H | 56%(10a:11a = 1.2:1) |
| 5c |
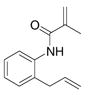
12 |
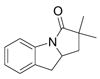
13 |
38% | |
| 6c |
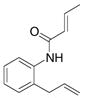
14 |
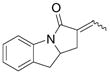
15 |
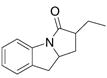
16 |
49%(15:16 = 2.5:1) |
Conditions. Substrate in DMF (0.1 M) was treated with Cu(EH)2 (3 equiv) and Cs2CO3 (1 equiv). The mixture was heated to 140 °C for 24 h in a pressure tube.
Yields refer to the sum of products isolated by chromatography on SiO2. The remainder of the material was either starting olefin or olefin-isomerized starting material.
Heated to 190 °C for 24 h.
Disappointingly, the more substituted vinyl amides 12 and 14 were less reactive (Table 3, entries 5 and 6). These substrates required higher reaction temperature (190 °C) in order to consume most of the substrate, but in doing so, resulted in the concomitant formation of a mixture of 5-exo cyclization products and isomerized olefin starting material.
Our previous studies showed that the copper(II) carboxylate promoted intramolecular carboamination reaction is effective in cyclizing a variety of aliphatic N-aryl sulfonamides (e.g. Eq 2).2f
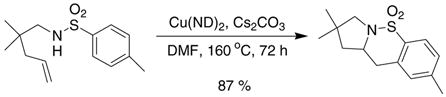 |
(2) |
This prompted us to investigate the cyclization reaction of a corresponding N-benzoyl aliphatic amide (Eq 3). Unfortunately, this substrate was shown to be unreactive upon heating to 190 °C for 24 h.
This result led us to examine a variety of structurally different imides and amides as shown Table 4. Imide 17 cyclized in high yield while the amide substrate in Eq. 3 did not react. This indicates that an sp2 hybridized
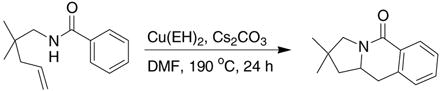 |
(3) |
carbon in the substrate’s backbone decreases the activation energy for cyclization. (This could be due to lower relative torsional strain in the five-membered ring of the transition state and product for substrate 17.) In addition, imide 17 cyclized at lower temperature and in higher yield and selectivity than its benzyl amide counterpart 19 (Table 4, entries 1 and 2). Thus, the imide is more reactive than the alkyl amide. Substrates with geminally disubstituted carbon backbones cyclized more efficiently due to the Thorpe-Ingold effect (compare entries 6 and 7, Table 4).6
Table 4.
Carboamination of aliphatic imides and amides.a
| entry | substrate | product(s) | yieldb(selectivity) | |
|---|---|---|---|---|
| 1 |
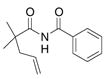
17 |
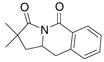
18 |
81% | |
| 2c,d |
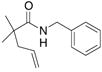
19 |
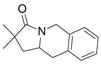
20 |
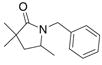
21 |
70%(20:21 = 1.6:2) |
| 3c,e |
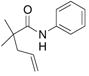
22 |
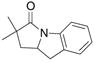
23 |
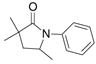
24 |
83%(23:24 = 3.3:1) |
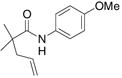
| 4c,e |
25 |
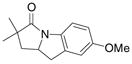
26 |
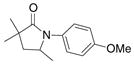
27 |
79%(26:27 = 3.4:1) |
| 5c |
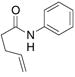
28 |
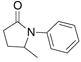
29 |
56% | |
| 6 |
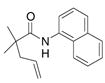
30 |
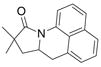
31 |
81% | |
| 7c |
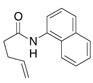
32 |
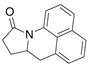
33 |
74% | |
Conditions. Substrate in DMF (0.1 M) was treated with Cu(EH)2 (3 equiv) and Cs2CO3 (1 equiv). The mixture was heated to 120 °C for 24 h in a pressure tube.
Yields refer to the sum of products isolated by chromatography on SiO2. The remainder of the material was either starting olefin or olefin-isomerized starting material.
Heated to 190 °C.
Reaction run for 72 h.
tbutyl benzene was used as solvent.
The aryl amides 22 and 25 were able to undergo the cyclization in an efficient manner albeit with the formation of the hydroamination byproducts (Table 4, entries 3 and 4). In order to reduce hydroamination and maximize carboamination, a less hydrogen atom donating solvent, i.e. t-butyl benzene, was used. Although increased selectivity for carboamination was observed, formation of the hydroamination byproduct still occurred. Regardless, these are the first examples where two fused 5-membered rings are formed in the copper(II) carboxylate promoted intramolecular carboamination reaction. Substrate 28, which lacks the geminal methyl groups in the backbone, only produced hydroamination adduct 29 (Table 4, entry 5). Because of the apparent difficulty in forming a 5-membered ring with the aryl substituent7 (vide supra), naphthalene substrates 30 and 32 were investigated. Gratifyingly, both substrates underwent the oxidative cyclization in good yield without the simultaneous formation of the hydroamination byproduct (Table 4, entries 6 and 7).
During the synthesis of naphthalene substrate 30, an interesting observation was made. When the corresponding amide was treated under the coupling reaction conditions developed by Buchwald et al.8 (5 mol% CuI, DMDA, iodonaphthalene, K3PO4, in DMF at 120 °C), trace amounts of carboamination (ca. 2–3%) product (31) are observed in the 1H NMR. This occurrance is easily explained by the ability of Cu(I) to disproportionate into Cu(II) and Cu(0).9 Thus, in an effort to exploit this reactivity, a one-pot arylation/carboamination reaction was performed. Upon completion of the coupling reaction, 3 equivalents of Cu(EH)2 was added to the reaction mixture. After 24 h the carboamination adduct was obtained in 71% yield (Eq 4).
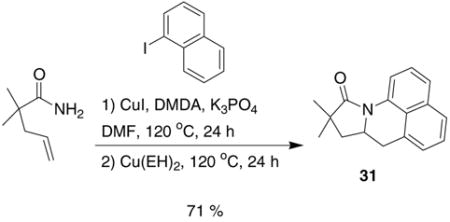 |
(4) |
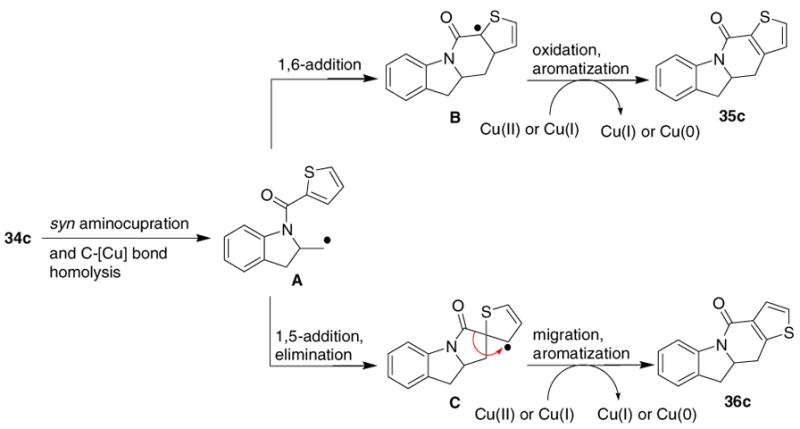
Another interesting observation was made when performing the carboamination reaction on heteroaromatic ring amides. Both the amido furan 34a and the amido pyrrole 34b derivatives of 2-allyl aniline were unreactive substrates. The amido thiophene 34c cyclized in 43% yielding two carboamination products 35c and 36c in a 1:1 ratio (Eq 5).
This observation can be rationalized based on the mechanism shown in Scheme 1. Thus, syn aminocupration followed by homolysis of the resulting
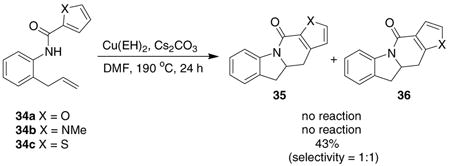 |
(5) |
unstable organocopper intermediate gives primary radical A.2f This radical can either add 1,6 to give the carboamination product 35c after oxidation of the aryl radical intermediate B, or 1,5 (ipso) giving rise to carboamination product 36c via intermediate C, radical migration and oxidative re-aromatization as shown in Scheme 1.10 Rearrangement of intermediate C via a carbocation is also possible.
Scheme 1.
Mechanistic rational for carboamination product 36c.
In conclusion, the intramolecular copper(II) carboxylate promoted carboamination reaction is an efficient method to form a variety of polycyclic lactams. The substrate scope has been expanded from sulfonamides, to include not only aryl amides, but vinyl amides and alkyl imides as well. Furthermore, a simple one-pot arylation/carboamination reaction sequence was achieved. The synthetic utility of this methodolgy and its application in natural product as well as biologically active molecule synthesis is currently ongoing in our lab and will be reported in due course.
Supplementary Material
Acknowledgments
We thank Mr. Dov Shalman (Northwestern University), Ms. Fatima Sequiera (University at Buffalo, ACS administered PRF Summer Scholar 40968-G1), and Ms. Melantha Jackson (from Utica College, NSF REU Fellowship at SUNY, Buffalo, CHE-0453206) for preliminary studies. This work was
References
- 1.Zeni G, Larock RC. Chem Rev. 2004;104:2285. doi: 10.1021/cr020085h. [DOI] [PubMed] [Google Scholar]
- 2.(a) Larock RC, Yang H, Weinreb SM, Herr RJ. J Org Chem. 1994;59:4172. [Google Scholar]; (b) Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J Org Chem. 1997;62:2113. doi: 10.1021/jo961988b. [DOI] [PubMed] [Google Scholar]; (c) Sherman ES, Chemler SR, Tan TB, Gerlitis O. Org Lett. 2004;6:1573. doi: 10.1021/ol049702+. [DOI] [PubMed] [Google Scholar]; (d) Yip KT, Yang M, Law KL, Zhu NY, Yang D. J Am Chem Soc. 2006;8:3130. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]; (e) Scarborough CC, Stahl SS. Org Lett. 2006;8:3251. doi: 10.1021/ol061057e. [DOI] [PubMed] [Google Scholar]; (f) Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Peng J, Lin W, Yuan S, Chen Y. J Org Chem. 2007;72:3145. doi: 10.1021/jo0625958. [DOI] [PubMed] [Google Scholar]; (h) Nakhla JS, Wolfe JP. Org Lett. 2007;9:3279. doi: 10.1021/ol071241f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Bertrand MB, Leathen ML, Wolfe JP. Org Lett. 2007;9:457. doi: 10.1021/ol062808f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Wolfe JP. Eur J Org Chem. 2007:571. and references therein. [PMC free article] [PubMed] [Google Scholar]
- 3.Antilla JC, Buchwald SL. Org Lett. 2001;3:2077. doi: 10.1021/ol0160396.Baran PS, Richter JM. J Am Chem Soc. 2004;126:7450. doi: 10.1021/ja047874w.(c) For a review of copper-facilitated C-N and C-C bond formation, see Chemler SR, Fuller PH. Chem Soc Rev. 2007;36:1153. doi: 10.1039/b607819m. and references therein.
- 4.(a) Ito R, Migita T, Morikawa N, Simamura O. Tetrahedron. 1965;21:955. [Google Scholar]; (b) Pryor WA, Davis WH, Gleaton JH. J Org Chem. 1975;40:2099. [Google Scholar]
- 5.(a) Hegedus LS, McKearin JM. J Am Chem Soc. 1982;104:2444. [Google Scholar]; (b) Danishefsky S, Taniyama E. Tetrahedron Lett. 1983;24:15. [Google Scholar]
- 6.Beesley RM, Ingold CK, Thorpe JF. J Am Chem Soc. 1915;107:1080. [Google Scholar]
- 7.Stevens CV, Meenen EV, Masschelein KGR, Eeckhout Y, Hooghe W, D’hondte B, Nemkyin VN, Zhdankin VV. Tetrahedron Lett. 2007;48:7108. [Google Scholar]
- 8.Klapars A, Antilla JC, Huang X, Buchwald SL. J Am Chem Soc. 2001;123:7727. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- 9.Cotton FA, Wilkinson G, Murillo CA, Bochmann M. Advanced Inorganic Chemistry. John Wiley & Sons; New York: 1999. [Google Scholar]
- 10.(a) Studer A, Bossart M. Tetrahedron. 2001;57:9649. [Google Scholar]; (b) Guindeuil S, Zard SZ. Chem Comm. 2006:665. doi: 10.1039/b516020k. [DOI] [PubMed] [Google Scholar]; (c) Gagosz F, Moutrille C, Zard SZ. Org Lett. 2002;4:2707. doi: 10.1021/ol026221m. [DOI] [PubMed] [Google Scholar]; (d) Kyei AS, Tchabanenko K, Baldwin JE, Adlington RM. Tetrahedron Lett. 2004;45:8931. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.