

Abstract
Although nitrite (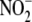 ) and nitrate
(
) and nitrate
(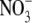 ) have been considered
traditionally inert byproducts of nitric oxide (NO) metabolism, recent studies
indicate that
) have been considered
traditionally inert byproducts of nitric oxide (NO) metabolism, recent studies
indicate that 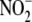 represents an
important source of NO for processes ranging from angiogenesis through hypoxic
vasodilation to ischemic organ protection. Despite intense investigation, the
mechanisms through which
represents an
important source of NO for processes ranging from angiogenesis through hypoxic
vasodilation to ischemic organ protection. Despite intense investigation, the
mechanisms through which 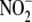 exerts its
physiological/pharmacological effects remain incompletely understood. We
sought to systematically investigate the fate of
exerts its
physiological/pharmacological effects remain incompletely understood. We
sought to systematically investigate the fate of
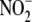 in hypoxia from cellular uptake
in vitro to tissue utilization in vivo using the Wistar rat
as a mammalian model. We find that most tissues (except erythrocytes) produce
free NO at rates that are maximal under hypoxia and that correlate robustly
with each tissue's capacity for mitochondrial oxygen consumption. By comparing
the kinetics of NO release before and after ferricyanide addition in tissue
homogenates to mathematical models of
in hypoxia from cellular uptake
in vitro to tissue utilization in vivo using the Wistar rat
as a mammalian model. We find that most tissues (except erythrocytes) produce
free NO at rates that are maximal under hypoxia and that correlate robustly
with each tissue's capacity for mitochondrial oxygen consumption. By comparing
the kinetics of NO release before and after ferricyanide addition in tissue
homogenates to mathematical models of
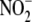 reduction/NO scavenging, we show
that the amount of nitrosylated products formed greatly exceeds what can be
accounted for by NO trapping. This difference suggests that such products are
formed directly from
reduction/NO scavenging, we show
that the amount of nitrosylated products formed greatly exceeds what can be
accounted for by NO trapping. This difference suggests that such products are
formed directly from 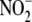 , without
passing through the intermediacy of free NO. Inhibitor and subcellular
fractionation studies indicate that
, without
passing through the intermediacy of free NO. Inhibitor and subcellular
fractionation studies indicate that 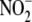 reductase activity involves multiple redundant enzymatic systems
(i.e. heme, iron-sulfur cluster, and molybdenum-based reductases)
distributed throughout different cellular compartments and acting in concert
to elicit NO signaling. These observations hint at conserved roles for the
reductase activity involves multiple redundant enzymatic systems
(i.e. heme, iron-sulfur cluster, and molybdenum-based reductases)
distributed throughout different cellular compartments and acting in concert
to elicit NO signaling. These observations hint at conserved roles for the
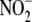 -NO pool in cellular processes such
as oxygen-sensing and oxygen-dependent modulation of intermediary
metabolism.
-NO pool in cellular processes such
as oxygen-sensing and oxygen-dependent modulation of intermediary
metabolism.
Nitric oxide (NO)3
is the archetypal effector of redox-regulated signal transduction throughout
phylogeny, from microorganisms to plants and animals
(1). The conserved influences
of NO extend from the regulation of basic cellular processes such as
intermediary metabolism (2),
cellular proliferation (3), and
apoptosis (4) to systemic
processes such as hypoxic vasoregulation
(5). Mammalian NO production
has been attributed to the enzymatic activity of NO synthases, nitrate
(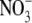 )/nitrite
(
)/nitrite
(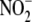 ) reductases and non-enzymatic
) reductases and non-enzymatic
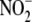 reduction
(6). The NO produced is
believed to act directly as a signaling molecule by binding to the heme of
soluble guanylyl cyclase or nitrosating peptide/protein cysteine residues
(7). More recently, it has
become apparent that
reduction
(6). The NO produced is
believed to act directly as a signaling molecule by binding to the heme of
soluble guanylyl cyclase or nitrosating peptide/protein cysteine residues
(7). More recently, it has
become apparent that 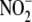 , previously
considered an inert byproduct of NO metabolism present in plasma (50–500
nm) and tissues (0.5–25 μm), is, under some
conditions, also a source of NO/nitrosothiol signaling
(6,
8). Although the importance of
, previously
considered an inert byproduct of NO metabolism present in plasma (50–500
nm) and tissues (0.5–25 μm), is, under some
conditions, also a source of NO/nitrosothiol signaling
(6,
8). Although the importance of
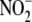 has received increasing
appreciation (9) as being
central to processes including exercise
(10), hypoxic vasodilation
(11), myocardial
preconditioning (12,
13), and angiogenesis
(14), controversy surrounds
the chemistry, kinetics, and tissue specificity of
has received increasing
appreciation (9) as being
central to processes including exercise
(10), hypoxic vasodilation
(11), myocardial
preconditioning (12,
13), and angiogenesis
(14), controversy surrounds
the chemistry, kinetics, and tissue specificity of
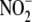 bioactivity
(15,
16). Perhaps the greatest
uncertainty pertains to the role of heme moieties in
bioactivity
(15,
16). Perhaps the greatest
uncertainty pertains to the role of heme moieties in
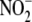 metabolism
(6,
10,
12,
13,
15–22).
We therefore sought to address systematically the path of
metabolism
(6,
10,
12,
13,
15–22).
We therefore sought to address systematically the path of
 biotransformation in hypoxic
tissues and its processing into NO and NO-related species across levels of
biological organization by employing an experimental paradigm that ranges from
cellular
biotransformation in hypoxic
tissues and its processing into NO and NO-related species across levels of
biological organization by employing an experimental paradigm that ranges from
cellular  uptake in vitro
to tissue
uptake in vitro
to tissue  utilization in
vivo. Our findings reveal that multiple heme, iron-sulfur cluster, and
molybdenum-based reductases, distributed among distinct subcellular
compartments, act in a cooperative manner to convert
utilization in
vivo. Our findings reveal that multiple heme, iron-sulfur cluster, and
molybdenum-based reductases, distributed among distinct subcellular
compartments, act in a cooperative manner to convert
 to NO and related signaling
products. The correlation between
to NO and related signaling
products. The correlation between 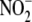 reductase activity and oxidative phosphorylation capacity across organs
suggests that
reductase activity and oxidative phosphorylation capacity across organs
suggests that 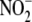 serves a cell
regulatory role (e.g. the modulation of intermediary metabolism)
beyond its capacity to elicit hypoxic vasodilatation.
serves a cell
regulatory role (e.g. the modulation of intermediary metabolism)
beyond its capacity to elicit hypoxic vasodilatation.
EXPERIMENTAL PROCEDURES
Animals and Reagents
Male Wistar rats (250–350 g) from Harlan (Indianapolis, IN) were allowed food (2018 rodent diet, Harlan) and water ad libitum and were maintained on a regular 12 h light/12 h dark cycle with at least 10 days of local vivarium acclimatization prior to experimental use. All protocols were approved by the Institutional Animal Care and Use Committee of the Boston University School of Medicine. All gasses and chemicals were of the highest available grade (see supplemental information for details).
Biological Sample Harvest and Preparation
Heparinized (0.07 units/g body weight, intraperitoneal) rats were
anesthetized with diethylether and euthanized by cervical dislocation. Whole
blood (3–5 ml) was collected from the inferior vena cava into
EDTA-containing tubes (1.8 mg/mL) and processed as detailed
(23,
24) to obtain RBC and plasma.
Isolated, packed RBC (1 blood vol-equivalent) were immediately lysed
hypotonically with 3 vol of water. After thoracotomy, a catheter was inserted
into the infrarenal portion of the abdominal aorta, and organs were flushed
free of blood by retrograde in situ perfusion at a rate of 10 ml/min
with air-equilibrated PBS before extirpation.
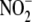 reductase activity was assessed by
measuring the NO generated immediately after addition of 100 μl of a 20
mm NaNO2 stock solution to tissue samples (200
μm final [
reductase activity was assessed by
measuring the NO generated immediately after addition of 100 μl of a 20
mm NaNO2 stock solution to tissue samples (200
μm final [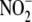 ]) using
gas-phase chemiluminescence.
]) using
gas-phase chemiluminescence.
Liver Homogenate Preparation
Hepatic tissue was acquired by standard techniques (see supplemental information). Liver homogenate was diluted 180-fold in LHM and preincubated at 37 °C for 4 min with either PBS (vehicle control) or a variety of inhibitors (supplemental Table S1) in a light-protected reaction vessel continuously purged with nitrogen. For analysis of inhibitor effects, the amount of NO generated within the first 4 min of incubation in the presence of test compound was compared with a parallel sample treated only with the respective inhibitor vehicle.
Nitrite Reductase Activity in Subcellular Fractions
Hepatic subcellular fractions were obtained by differential centrifugation
(25). Low-speed centrifugation
(1,000 × g, 10 min) of whole-liver homogenate (S0) was used to
remove undisrupted tissue, nuclei, and particulate debris into the resulting
pellet (P1), which was discarded. The supernatant (S1) was recovered and
re-centrifuged (10,000 × g, 10 min) to obtain a mitochondrial
fraction (P2). The corresponding supernatant (S2) was subjected to a final
centrifugation (105,000 × g, 60 min) to obtain microsomal (P3)
and cytosolic (S3) fractions. For analysis of
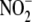 reductase activity across
fractions, each mitochondrial (P2) and microsomal (P3) pellet was re-suspended
in LHM to the same dilution as that of the supernatant fractions from which
they were derived and analyzed as described above. The contribution of each
subcellular fraction with respect to total hepatic
reductase activity across
fractions, each mitochondrial (P2) and microsomal (P3) pellet was re-suspended
in LHM to the same dilution as that of the supernatant fractions from which
they were derived and analyzed as described above. The contribution of each
subcellular fraction with respect to total hepatic
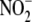 reductase activity was calculated
by accounting for the individual fractional product yield (6.2% for P2; 1.3%
for P3; 77.8% for S3, with 14.7% discarded as P1 debris). The influence of
NAD(P)H (100 μm) on the conversion of
reductase activity was calculated
by accounting for the individual fractional product yield (6.2% for P2; 1.3%
for P3; 77.8% for S3, with 14.7% discarded as P1 debris). The influence of
NAD(P)H (100 μm) on the conversion of
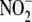 to NO by the subcellular fractions
was determined as the relative
to NO by the subcellular fractions
was determined as the relative 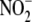 reductase activity in the absence or presence of pyridine nucleotide and
expressed as percent of control (with no exogenous pyridine nucleotide added).
reductase activity in the absence or presence of pyridine nucleotide and
expressed as percent of control (with no exogenous pyridine nucleotide added).
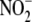 interaction with microsomal
cytochrome P450 was assessed by difference spectrophotometry and
chemiluminescence (see supplemental information).
interaction with microsomal
cytochrome P450 was assessed by difference spectrophotometry and
chemiluminescence (see supplemental information).
In Vitro Studies
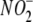 Uptake by RBCs and
Tissues—Rats were anesthetized with diethylether, and 5 ml of
arterial blood was collected via cardiac puncture. Blood was immediately
centrifuged at 1,400 × g (8 min, 4 °C). The supernatant was
discarded, and the RBC pellet was placed on ice. Blood-free heart and liver
tissue was obtained as described above and also kept on ice until use. Tissue
(0.5 g) was minced into 2-mm pieces and placed into 2 ml of air-saturated or
“hypoxic” (15 min bubbling with air or argon, respectively) PBS
and incubated at 37 °C. Following addition of 10 μm
NaNO2, aliquots of 50 μl were removed after 1, 3, 5, and 10 min
and centrifuged briefly (60 s at 16,100 × g) to clarify samples
prior to
Uptake by RBCs and
Tissues—Rats were anesthetized with diethylether, and 5 ml of
arterial blood was collected via cardiac puncture. Blood was immediately
centrifuged at 1,400 × g (8 min, 4 °C). The supernatant was
discarded, and the RBC pellet was placed on ice. Blood-free heart and liver
tissue was obtained as described above and also kept on ice until use. Tissue
(0.5 g) was minced into 2-mm pieces and placed into 2 ml of air-saturated or
“hypoxic” (15 min bubbling with air or argon, respectively) PBS
and incubated at 37 °C. Following addition of 10 μm
NaNO2, aliquots of 50 μl were removed after 1, 3, 5, and 10 min
and centrifuged briefly (60 s at 16,100 × g) to clarify samples
prior to
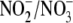 analysis. For uptake measurements by RBC, 4 ml of PBS were added to 1 ml of
packed RBC pellet and processed as above. No changes in the concentrations of
analysis. For uptake measurements by RBC, 4 ml of PBS were added to 1 ml of
packed RBC pellet and processed as above. No changes in the concentrations of
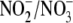 were observed in the absence of cells/tissue.
were observed in the absence of cells/tissue.
Tissue Homogenization and Incubation—Blood-free tissue
samples were homogenized in chilled PBS (1:5 w/v) using a Polytron
(PT10–35) homogenizer. Just prior to use, samples were brought with PBS
to a 6-fold final dilution (v/v) for tissues and a 4-fold final dilution (v/v,
equivalent to 2.5% hematocrit) for lysed RBC. For incubation, an equivalent
tissue/RBC sample volume was placed into the light-protected reaction vessel
of an ozone-chemiluminescence NO analyzer (CLD 77sp, EcoPhysics). The vessel
was maintained at 37 °C, and samples were purged sequentially with either
medical-grade air (normoxia) or nitrogen (hypoxia/near-anoxia). NO generation
was continuously monitored for 5 min following addition of 200
μm NaNO2 (final conc.). This
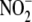 concentration has been established
as sufficient to support vasorelaxation in vitro, regional
vasodilation in vivo, and nitrosylation and S-nitrosation of
Hb in vitro and in vivo
(10). Aliquots of samples were
collected for protein determination, and all values were normalized to total
protein.
concentration has been established
as sufficient to support vasorelaxation in vitro, regional
vasodilation in vivo, and nitrosylation and S-nitrosation of
Hb in vitro and in vivo
(10). Aliquots of samples were
collected for protein determination, and all values were normalized to total
protein.
In Vivo Studies
Rats were administered 1.0 mg/kg NaNO2, intraperitoneal This
dosing regimen ensures that 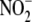 equilibrates rapidly across all major organ systems, RBC, and plasma to reach
a global steady-state prior to tissue sampling
(8). Three min after
equilibrates rapidly across all major organ systems, RBC, and plasma to reach
a global steady-state prior to tissue sampling
(8). Three min after
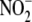 injection acute systemic hypoxia
was induced by cervical dislocation and maintained for 2 min. Normoxic
controls were examined in parallel without subjecting animals to global
hypoxia. In both cases, animals were sacrificed 5 min after
injection acute systemic hypoxia
was induced by cervical dislocation and maintained for 2 min. Normoxic
controls were examined in parallel without subjecting animals to global
hypoxia. In both cases, animals were sacrificed 5 min after
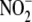 administration. The same in
situ retrograde perfusion technique was performed as for the in
vitro studies above except that the perfusate was air-equilibrated PBS
supplemented with NEM/EDTA (10 mm/2.5 mm) to eliminate
interference by exogenous
administration. The same in
situ retrograde perfusion technique was performed as for the in
vitro studies above except that the perfusate was air-equilibrated PBS
supplemented with NEM/EDTA (10 mm/2.5 mm) to eliminate
interference by exogenous 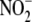 .
NO-related metabolites following
.
NO-related metabolites following 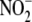 administration were profiled using previously validated methods
(23,
24). NO metabolites include
S-nitrosothiols (RSNO), N-nitrosamines (RNNO), and
iron-nitrosyls (NO-heme). Quantification of these species employed
group-specific derivatization, denitrosation, and gas-phase chemiluminescence.
administration were profiled using previously validated methods
(23,
24). NO metabolites include
S-nitrosothiols (RSNO), N-nitrosamines (RNNO), and
iron-nitrosyls (NO-heme). Quantification of these species employed
group-specific derivatization, denitrosation, and gas-phase chemiluminescence.
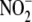 and
and
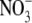 were quantified by ion
chromatography (ENO-20, Eicom).
were quantified by ion
chromatography (ENO-20, Eicom).
Data Analysis, Presentation, and Modeling
Unless otherwise noted, data are averages ± range from n = 3 individual experiments or means ± S.E. from n ≥ 3, as specified. Where appropriate, statistical analysis was performed by one-way analysis of variance with the Bonferroni post-hoc test. Least-squares regression analysis was used to characterize the slope and goodness-of-fit of model linear associations between data sets. Spearman rank correlation was applied to evaluate data co-variation. Statistical significance was set at p < 0.05. Origin 7.0 and Graph Pad Prism 4.0 were used for the statistical analyses. Modeling data were obtained through numeric integration using Mathematica, with a working precision of 20 digits.
RESULTS
Tissues Readily Take Up 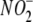 in an
Oxygen-independent
Manner—
in an
Oxygen-independent
Manner—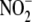 uptake by RBCs,
heart, and liver tissue was assessed under normoxic and hypoxic conditions by
measuring its disappearance from an external medium containing 10
μm
uptake by RBCs,
heart, and liver tissue was assessed under normoxic and hypoxic conditions by
measuring its disappearance from an external medium containing 10
μm 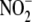 . Because
. Because
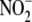 concentrations in these tissues are
≪10 μm (8,
23,
24), the initial disappearance
of
concentrations in these tissues are
≪10 μm (8,
23,
24), the initial disappearance
of 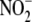 from PBS should reflect the flux
of this anion into the cells/tissues. Indeed,
from PBS should reflect the flux
of this anion into the cells/tissues. Indeed,
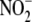 is taken up at similar rates by RBC
and heart tissue (0.31 μm/min and 0.24 μm/min,
respectively), and rates are roughly comparable under hypoxic conditions
(heart: 0.16 μm/min; RBC: 0.28 μm/min; see
supplemental Fig. S1). Similar data were obtained with liver (not shown). No
is taken up at similar rates by RBC
and heart tissue (0.31 μm/min and 0.24 μm/min,
respectively), and rates are roughly comparable under hypoxic conditions
(heart: 0.16 μm/min; RBC: 0.28 μm/min; see
supplemental Fig. S1). Similar data were obtained with liver (not shown). No
 accumulation was seen in the
extracellular medium.
accumulation was seen in the
extracellular medium.
Hypoxia Markedly Potentiates Tissue NO Production from
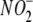 in Vitro—Under aerobic
conditions, NO production by RBC and blood-free tissues was minimal even in
the presence of 200 μM
in Vitro—Under aerobic
conditions, NO production by RBC and blood-free tissues was minimal even in
the presence of 200 μM 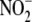 (Fig. 1A).
Hypoxia/near-anoxia (achieved by N2 purging) dramatically enhanced
NO formation from
(Fig. 1A).
Hypoxia/near-anoxia (achieved by N2 purging) dramatically enhanced
NO formation from 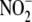 , particularly by
heart, liver, and vascular tissue. NO production by RBC lysate peaked after
∼3 min, whereas NO production by liver homogenate increased to reach
maximal levels after 50–60 min of hypoxia following a brief lag.
Near-anoxic,
, particularly by
heart, liver, and vascular tissue. NO production by RBC lysate peaked after
∼3 min, whereas NO production by liver homogenate increased to reach
maximal levels after 50–60 min of hypoxia following a brief lag.
Near-anoxic, 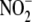 -dependent NO
production (standardized as mg protein/min) in all tissues surpassed that of
RBC (Fig. 1B). The
2-fold increase in RBC
-dependent NO
production (standardized as mg protein/min) in all tissues surpassed that of
RBC (Fig. 1B). The
2-fold increase in RBC 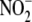 reduction
from normoxic to near-anoxic conditions is consistent with data previously
reported (10). No NO formation
was observed when 200 μm
reduction
from normoxic to near-anoxic conditions is consistent with data previously
reported (10). No NO formation
was observed when 200 μm
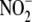 was substituted for
was substituted for
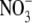 (not shown).
(not shown).
FIGURE 1.

Oxygen dependence of 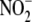 reduction to NO by different tissues in vitro. A,
original tracing from blood and tissues under normoxic (Air; 21%
O2) and near-anoxic (N2 containing ∼0.01%
O2) conditions. B, quantification of NO production (over 4
min) by RBC and different tissues from
reduction to NO by different tissues in vitro. A,
original tracing from blood and tissues under normoxic (Air; 21%
O2) and near-anoxic (N2 containing ∼0.01%
O2) conditions. B, quantification of NO production (over 4
min) by RBC and different tissues from
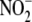 (200μm) under
normoxic and near-anoxic conditions. C, oxygen-dependence of
(200μm) under
normoxic and near-anoxic conditions. C, oxygen-dependence of
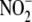 reduction to NO (n =
3–4 per cell/tissue compartment).
reduction to NO (n =
3–4 per cell/tissue compartment).
NO Generation from 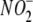 Is Profoundly
Oxygen-dependent—The oxygen-dependence of tissue
Is Profoundly
Oxygen-dependent—The oxygen-dependence of tissue
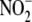 reductase activity was investigated
by adding 200 μm NaNO2 to liver and heart homogenates
and RBC lysate incubated at various oxygen concentrations (21, 10, 5, 1, 0.5,
and 0%) and monitoring of the resulting NO formation
(Fig. 1C). NO
production from
reductase activity was investigated
by adding 200 μm NaNO2 to liver and heart homogenates
and RBC lysate incubated at various oxygen concentrations (21, 10, 5, 1, 0.5,
and 0%) and monitoring of the resulting NO formation
(Fig. 1C). NO
production from 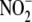 was maximal under
anoxia. Oxygen proved to be a potent inhibitor of
was maximal under
anoxia. Oxygen proved to be a potent inhibitor of
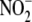 reduction in liver and heart with
>80% inhibition at 0.5% oxygen. Less pronounced changes in oxygen
dependence were observed with RBC lysate, with maximal rates of NO formation
occurring at ∼1% oxygen.
reduction in liver and heart with
>80% inhibition at 0.5% oxygen. Less pronounced changes in oxygen
dependence were observed with RBC lysate, with maximal rates of NO formation
occurring at ∼1% oxygen.
Acute Hypoxia Potentiates 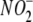 -dependent
NO Metabolite Formation in Vivo—To extend the above observations to
a more physiological context and to expand on existing observations that
hypoxia reduces endogenous brain
-dependent
NO Metabolite Formation in Vivo—To extend the above observations to
a more physiological context and to expand on existing observations that
hypoxia reduces endogenous brain 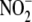 stores coincident with RSNO formation
(23), the in vivo
impact of
stores coincident with RSNO formation
(23), the in vivo
impact of 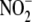 on NO-related metabolite
formation was assessed. To ensure initial
on NO-related metabolite
formation was assessed. To ensure initial
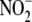 equilibration across all
compartments studied, we followed our previous protocol
(8) and administered to rats a
single intraperitoneal bolus of 1.0 mg/kg NaNO2 3 min prior to
inducing 2 min of global hypoxia. Acute global hypoxia attenuated
equilibration across all
compartments studied, we followed our previous protocol
(8) and administered to rats a
single intraperitoneal bolus of 1.0 mg/kg NaNO2 3 min prior to
inducing 2 min of global hypoxia. Acute global hypoxia attenuated
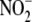 concentrations in heart, liver,
kidney, lung, and aorta (Fig.
2A) and enhanced NO metabolite formation in a tissue- and
product-selective manner (Fig. 2,
B–D). Tissue nitroso/nitrosyl products originated
from
concentrations in heart, liver,
kidney, lung, and aorta (Fig.
2A) and enhanced NO metabolite formation in a tissue- and
product-selective manner (Fig. 2,
B–D). Tissue nitroso/nitrosyl products originated
from 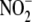 , since their levels under
identical hypoxic conditions were far less (<0.1%) without the supplied
, since their levels under
identical hypoxic conditions were far less (<0.1%) without the supplied
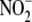 (23). Although each tissue
examined was capable of hypoxia-induced,
(23). Although each tissue
examined was capable of hypoxia-induced,
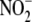 -dependent NO metabolite formation,
it was most pronounced in liver, heart, and the RBC. The tissue-specificity of
these responses to systemic hypoxia accords with previous demonstrations that
endogenous substrates and the turnover of NO-related oxidative and nitrosative
metabolites vary greatly among tissues
(23,
26). Although there is limited
NO production from
-dependent NO metabolite formation,
it was most pronounced in liver, heart, and the RBC. The tissue-specificity of
these responses to systemic hypoxia accords with previous demonstrations that
endogenous substrates and the turnover of NO-related oxidative and nitrosative
metabolites vary greatly among tissues
(23,
26). Although there is limited
NO production from 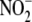 by hypoxic RBC
in vitro (Fig. 1), RBC
exhibited the greatest relative RSNO formation during acute hypoxia in
vivo. This finding is consistent with the S-nitrosation and
nitrosylation of hemoglobin (Hb) by
by hypoxic RBC
in vitro (Fig. 1), RBC
exhibited the greatest relative RSNO formation during acute hypoxia in
vivo. This finding is consistent with the S-nitrosation and
nitrosylation of hemoglobin (Hb) by 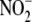 to form both, SNO-Hb and Hb-NO in vitro and in vivo
(10); indeed, SNO-Hb is
probably generated from circulating
to form both, SNO-Hb and Hb-NO in vitro and in vivo
(10); indeed, SNO-Hb is
probably generated from circulating 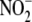 without the intermediate liberation of NO within the RBC
(27).
without the intermediate liberation of NO within the RBC
(27).
FIGURE 2.

Increased formation of tissue NO metabolites from
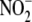 during acute global hypoxia in
vivo. Blood and tissues utilize
during acute global hypoxia in
vivo. Blood and tissues utilize
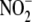 during global hypoxia in
vivo (A) to generate NO-related products including
S-nitrosothiols (RSNO) (B), N-nitrosamines (RNNO)
(C), and iron nitrosyls (NO-heme) (D). (n = 3); *,
p < 0.05 versus normoxia.
during global hypoxia in
vivo (A) to generate NO-related products including
S-nitrosothiols (RSNO) (B), N-nitrosamines (RNNO)
(C), and iron nitrosyls (NO-heme) (D). (n = 3); *,
p < 0.05 versus normoxia.
Kinetics and Concentration-dependence of
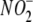 Reduction to NO—Liver effectively
generates NO from
Reduction to NO—Liver effectively
generates NO from 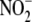 (Fig. 1), has a multiplicity of
potential reductases that can be pharmacologically investigated, and is
readily available and easily perfused free of blood. Accordingly, liver was
used to characterize more comprehensively the kinetics and chemistry of NO
production from
(Fig. 1), has a multiplicity of
potential reductases that can be pharmacologically investigated, and is
readily available and easily perfused free of blood. Accordingly, liver was
used to characterize more comprehensively the kinetics and chemistry of NO
production from 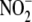 in tissue. The
in tissue. The
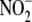 reductase activity of anoxic whole
liver homogenate was linearly dependent upon the concentration of supplied
reductase activity of anoxic whole
liver homogenate was linearly dependent upon the concentration of supplied
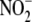 (25–1000 μm)
and, to a lesser extent, the amount of tissue protein employed
(Fig. 3A). Because the
generation of NO was measured over 4 min in each case and therefore represents
the initial rate of NO formation, this observation suggests that whole liver
homogenate grossly conforms to first order kinetics with respect to
(25–1000 μm)
and, to a lesser extent, the amount of tissue protein employed
(Fig. 3A). Because the
generation of NO was measured over 4 min in each case and therefore represents
the initial rate of NO formation, this observation suggests that whole liver
homogenate grossly conforms to first order kinetics with respect to
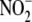 and, to a lesser extent, tissue
protein concentration. In contrast to the results obtained with whole liver
homogenate, no strict linearity was evident in the cytosolic fraction (S3)
between NO formation and
and, to a lesser extent, tissue
protein concentration. In contrast to the results obtained with whole liver
homogenate, no strict linearity was evident in the cytosolic fraction (S3)
between NO formation and 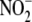 concentration, although the dependence upon protein concentration was similar
to that in whole liver homogenate (Fig.
3B).
concentration, although the dependence upon protein concentration was similar
to that in whole liver homogenate (Fig.
3B).
FIGURE 3.

Correlation between NO formation from
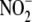 in different tissues and putative
in different tissues and putative
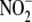 reductase activity. A,
apparent linear dependence of
reductase activity. A,
apparent linear dependence of 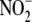 to NO
conversion by hypoxic whole liver homogenate (25–1000 μm
to NO
conversion by hypoxic whole liver homogenate (25–1000 μm
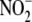 ). Inset, hypoxic NO
production from 200 μm
). Inset, hypoxic NO
production from 200 μm
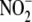 is not as linearly dependent upon
hepatic protein. B,
is not as linearly dependent upon
hepatic protein. B,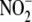 and
protein dependence deviate from linearity with the cytosolic fraction.
C, significant correlation exists between hypoxic
and
protein dependence deviate from linearity with the cytosolic fraction.
C, significant correlation exists between hypoxic
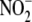 reductase activity and tissue XOR
activity (r = 0.892, p = 0.042). The correlation between
hypoxic
reductase activity and tissue XOR
activity (r = 0.892, p = 0.042). The correlation between
hypoxic 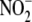 reductase activity and
mitochondrial inner surface area (D) is more striking, except for
kidney (r = 0.9998, p = 0.0001).
reductase activity and
mitochondrial inner surface area (D) is more striking, except for
kidney (r = 0.9998, p = 0.0001).
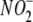 Reduction to NO Requires Enzymatic
Activity—To probe the involvement of enzymatic processes in hypoxic
NO formation, aliquots of liver homogenate were subjected to heat
pretreatment. Exposure of liver homogenate for 60 min to 56 or 80 °C
inhibited
Reduction to NO Requires Enzymatic
Activity—To probe the involvement of enzymatic processes in hypoxic
NO formation, aliquots of liver homogenate were subjected to heat
pretreatment. Exposure of liver homogenate for 60 min to 56 or 80 °C
inhibited 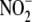 reductase activity by 72
and >90%, respectively, relative to control, unheated tissue samples. These
results are consistent with the conclusion that thermolabile enzymes mediate
the majority of hypoxic
reductase activity by 72
and >90%, respectively, relative to control, unheated tissue samples. These
results are consistent with the conclusion that thermolabile enzymes mediate
the majority of hypoxic 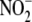 reduction
to NO, either directly or indirectly (e.g. by modulating reducing
equivalent supply).
reduction
to NO, either directly or indirectly (e.g. by modulating reducing
equivalent supply).
Putative Cellular 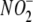 Reductases—To gain insight into potential mechanisms of tissue
Reductases—To gain insight into potential mechanisms of tissue
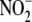 reduction,
reduction,
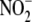 -dependent NO production was
quantified as a function of reported literature values for tissue XOR activity
(28)
(Fig. 3C) and/or
maximal oxidative phosphorylative capacity, indexed by Hulbert and Else
(29) as inner mitochondrial
membrane surface area (Fig.
3D). All tissues except kidney evidenced a strong linear
relationship between tissue
-dependent NO production was
quantified as a function of reported literature values for tissue XOR activity
(28)
(Fig. 3C) and/or
maximal oxidative phosphorylative capacity, indexed by Hulbert and Else
(29) as inner mitochondrial
membrane surface area (Fig.
3D). All tissues except kidney evidenced a strong linear
relationship between tissue 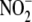 reductase activity as a function of mitochondrial membrane surface area
(Fig. 3D). Because
cytochrome oxidase activity directly correlates with inner mitochondrial
membrane surface area (29), a
linear relationship also exists between hypoxic tissue
reductase activity as a function of mitochondrial membrane surface area
(Fig. 3D). Because
cytochrome oxidase activity directly correlates with inner mitochondrial
membrane surface area (29), a
linear relationship also exists between hypoxic tissue
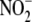 reduction to NO and cytochrome
oxidase activity. Rat kidney (especially, renal medulla) deviates
substantially from other organs in its greater dependence upon glycolysis
versus oxidative phosphorylation for ATP production
(30). Kidney
reduction to NO and cytochrome
oxidase activity. Rat kidney (especially, renal medulla) deviates
substantially from other organs in its greater dependence upon glycolysis
versus oxidative phosphorylation for ATP production
(30). Kidney
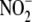 reductive capacity may be limited
to prevent NO-mediated mitochondrial inhibition due to the relatively high
local
reductive capacity may be limited
to prevent NO-mediated mitochondrial inhibition due to the relatively high
local 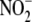 concentrations that arise
during renal anion filtration/secretion. The relatively weak relationship in
the kidney between
concentrations that arise
during renal anion filtration/secretion. The relatively weak relationship in
the kidney between 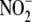 reductase
activity and maximal oxidative metabolic capacity
(Fig. 3D) supports the
overall concept of a robust interrelationship between oxidative intermediary
metabolism and
reductase
activity and maximal oxidative metabolic capacity
(Fig. 3D) supports the
overall concept of a robust interrelationship between oxidative intermediary
metabolism and 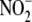 -dependent NO
formation. Mitochondrial inner surface area, cytochrome oxidase, and/or oxygen
utilization capacity reflect quantifiable, correlative parameters of this
interrelationship.
-dependent NO
formation. Mitochondrial inner surface area, cytochrome oxidase, and/or oxygen
utilization capacity reflect quantifiable, correlative parameters of this
interrelationship.
Chemical Sensitivity of 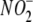 -dependent NO
Formation in Vitro—To evaluate further the enzyme systems that may
contribute to the formation of NO from
-dependent NO
Formation in Vitro—To evaluate further the enzyme systems that may
contribute to the formation of NO from
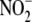 in hypoxic liver, tissue samples
were probed with an extensive array of inhibitors to target discrete enzymatic
activities (supplemental Table S1). Cyanide and oxygen were among the most
effective inhibitors, eliciting a concentration-dependent reduction of maximal
NO formation from
in hypoxic liver, tissue samples
were probed with an extensive array of inhibitors to target discrete enzymatic
activities (supplemental Table S1). Cyanide and oxygen were among the most
effective inhibitors, eliciting a concentration-dependent reduction of maximal
NO formation from 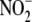 that reached
>95% (Fig. 4A),
suggesting the crucial involvement of metalloproteins and an oxygen-sensitive
component. The heme-oxidant oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) is also a
potent inhibitor (70% at 10 mm)
(Fig. 4A), suggesting
the involvement of reduced hemes. Similar inhibition by the thiol alkylator
N-ethylmaleimide (NEM) (∼60% at 10 mm)
(Fig. 4B) implies a role for
free thiols in liver
that reached
>95% (Fig. 4A),
suggesting the crucial involvement of metalloproteins and an oxygen-sensitive
component. The heme-oxidant oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) is also a
potent inhibitor (70% at 10 mm)
(Fig. 4A), suggesting
the involvement of reduced hemes. Similar inhibition by the thiol alkylator
N-ethylmaleimide (NEM) (∼60% at 10 mm)
(Fig. 4B) implies a role for
free thiols in liver 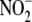 reductase
activity under hypoxia. The xanthine oxidoreductase (XOR) inhibitor,
oxypurinol (Oxy), partially (by 25%) inhibited hypoxic
reductase
activity under hypoxia. The xanthine oxidoreductase (XOR) inhibitor,
oxypurinol (Oxy), partially (by 25%) inhibited hypoxic
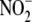 reduction, whereas preincubation of
liver homogenate with the flavin inhibitor, diphenyleneiodonium (DPI),
inhibited by as much as 62% (Fig.
4C), in further support of a role for XOR and/or
mitochondrial aldehyde dehydrogenase (ALDH2). The inhibitory effects of
cyanide and DPI are also consistent with an involvement of mitochondrial
respiratory complexes in
reduction, whereas preincubation of
liver homogenate with the flavin inhibitor, diphenyleneiodonium (DPI),
inhibited by as much as 62% (Fig.
4C), in further support of a role for XOR and/or
mitochondrial aldehyde dehydrogenase (ALDH2). The inhibitory effects of
cyanide and DPI are also consistent with an involvement of mitochondrial
respiratory complexes in 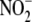 reduction.
Other inhibitors of mitochondrial respiration (i.e. rotenone (Rot),
antimycin A (Anti A), and myxothiazole (Myx)) also attenuated hypoxic NO
formation from
reduction.
Other inhibitors of mitochondrial respiration (i.e. rotenone (Rot),
antimycin A (Anti A), and myxothiazole (Myx)) also attenuated hypoxic NO
formation from 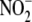 (Fig. 4D). Suppression
of NO formation by ethoxyresorufin (EthOx)
(Fig. 4E) implicates
yet another mitochondrial and/or microsomal route of hypoxic
(Fig. 4D). Suppression
of NO formation by ethoxyresorufin (EthOx)
(Fig. 4E) implicates
yet another mitochondrial and/or microsomal route of hypoxic
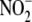 reduction: EthOx selectively
inhibits the cytochrome P450 CYP1A1, which is found in mitochondria
and endoplasmic reticulum. The nonselective cytochrome P450
monooxygenase inhibitor proadifen was without effect. The aggregate inhibitor
data allow conclusion that
reduction: EthOx selectively
inhibits the cytochrome P450 CYP1A1, which is found in mitochondria
and endoplasmic reticulum. The nonselective cytochrome P450
monooxygenase inhibitor proadifen was without effect. The aggregate inhibitor
data allow conclusion that 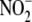 conversion to NO by hypoxic liver tissue is a multifactorial, metalloprotein-
and thiol-dependent process, which is highly susceptible to inhibition by
oxygen and involves XOR, several mitochondrial respiratory chain complexes,
and cytochrome P450. In further support of the cooperative nature
of this process, the combination of the three inhibitors rotenone, EthOx, and
DPI (Combi) inhibited hypoxic
conversion to NO by hypoxic liver tissue is a multifactorial, metalloprotein-
and thiol-dependent process, which is highly susceptible to inhibition by
oxygen and involves XOR, several mitochondrial respiratory chain complexes,
and cytochrome P450. In further support of the cooperative nature
of this process, the combination of the three inhibitors rotenone, EthOx, and
DPI (Combi) inhibited hypoxic 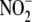 reduction to NO to an extent greater than the inhibition elicited by each
agent alone (Fig.
4F).
reduction to NO to an extent greater than the inhibition elicited by each
agent alone (Fig.
4F).
FIGURE 4.

Chemical sensitivity of hypoxic
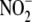 reduction to NO. Exposure of
whole liver homogenate to a variety of inhibitors targeting various cellular
components (see supplementary Table S1 for details) shows that hypoxic
reduction to NO. Exposure of
whole liver homogenate to a variety of inhibitors targeting various cellular
components (see supplementary Table S1 for details) shows that hypoxic
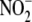 reduction to NO is carried out by
multiple enzymatic routes. Inhibitors for XOR, mitochondrial respiration,
hemes, and thiols all attenuate hypoxic
reduction to NO is carried out by
multiple enzymatic routes. Inhibitors for XOR, mitochondrial respiration,
hemes, and thiols all attenuate hypoxic
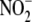 to NO conversion to varying
degrees, with virtually complete inhibition by KCN and oxygen implicating
metalloproteins as being most critical to the process (n = 3).
(Oxygen: 21%, medical-grade air; Oxy: oxypurinol;
EthOxy: ethoxyresorufin; Rot: rotenone; Anti A:
antimycin A; Myx: myxathiozole; Combi: inhibitor mixture
containing DPI, ethoxyresorufin, and rotenone).
to NO conversion to varying
degrees, with virtually complete inhibition by KCN and oxygen implicating
metalloproteins as being most critical to the process (n = 3).
(Oxygen: 21%, medical-grade air; Oxy: oxypurinol;
EthOxy: ethoxyresorufin; Rot: rotenone; Anti A:
antimycin A; Myx: myxathiozole; Combi: inhibitor mixture
containing DPI, ethoxyresorufin, and rotenone).
To investigate whether tissue 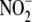 reductase activity is sensitive to tissue oxidation state, ferricyanide (5
mm) was introduced prior to or after
reductase activity is sensitive to tissue oxidation state, ferricyanide (5
mm) was introduced prior to or after
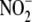 addition. Ferricyanide treatment
pre-oxidizes potential reductases (e.g. ferrous hemeproteins) to
diminish their reductase activity; ferricyanide exposure following
addition. Ferricyanide treatment
pre-oxidizes potential reductases (e.g. ferrous hemeproteins) to
diminish their reductase activity; ferricyanide exposure following
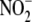 addition promotes the release of NO
equivalents bound to ferrous hemeproteins. As reported by Shiva et
al. (20) for heart
homogenate, ferricyanide treatment significantly alters hepatic
addition promotes the release of NO
equivalents bound to ferrous hemeproteins. As reported by Shiva et
al. (20) for heart
homogenate, ferricyanide treatment significantly alters hepatic
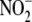 reductase activity (see
supplemental information), implicating the involvement of redox-sensitive heme
complexes. However, the enhanced reductase activity in the oxidized state
seems to contradict any simple conception of a ferrous heme-based
reductase activity (see
supplemental information), implicating the involvement of redox-sensitive heme
complexes. However, the enhanced reductase activity in the oxidized state
seems to contradict any simple conception of a ferrous heme-based
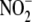 reductase. Addition of ferricyanide
after incubation with
reductase. Addition of ferricyanide
after incubation with 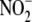 under hypoxic
(but not aerobic) conditions leads to large bursts of NO formation by liver
homogenates. The sudden rise and exponential decay of the chemiluminescence
signal is consistent with a reaction of ferricyanide with ferrous heme
nitrosyl complexes (31). A
comparison of the experimental data with mathematical models (see supplemental
information) simulating NO generation from
under hypoxic
(but not aerobic) conditions leads to large bursts of NO formation by liver
homogenates. The sudden rise and exponential decay of the chemiluminescence
signal is consistent with a reaction of ferricyanide with ferrous heme
nitrosyl complexes (31). A
comparison of the experimental data with mathematical models (see supplemental
information) simulating NO generation from
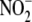 reduction and trapping by hemes
predicts that the majority of nitrosyl products formed from
reduction and trapping by hemes
predicts that the majority of nitrosyl products formed from
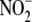 must be derived without the
intermediacy of free NO.
must be derived without the
intermediacy of free NO.
Multiple Intracellular Compartments Contribute to Hypoxic NO Production
from 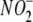 —To complement the inhibitor
studies performed in whole liver homogenate and gain insight into the
subcellular sites of hypoxic
—To complement the inhibitor
studies performed in whole liver homogenate and gain insight into the
subcellular sites of hypoxic 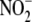 reductase activity in this organ, blood-free hepatic tissue was fractionated
into mitochondrial, cytosolic, and microsomal fractions by standard
differential centrifugation. The ability of each individual subfraction to
form NO when supplied with 200 μm NaNO2 was examined
during a 4-min incubation at 37 °C with nitrogen. Hepatic
reductase activity in this organ, blood-free hepatic tissue was fractionated
into mitochondrial, cytosolic, and microsomal fractions by standard
differential centrifugation. The ability of each individual subfraction to
form NO when supplied with 200 μm NaNO2 was examined
during a 4-min incubation at 37 °C with nitrogen. Hepatic
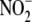 reductase activity under hypoxia
was distributed selectively among the three major liver subfractions studied,
with microsomes accounting for over half (∼63%) of the total activity
(Fig. 5A) when
adjusted for compartmental fractional yield. Cytosolic NO formation from
reductase activity under hypoxia
was distributed selectively among the three major liver subfractions studied,
with microsomes accounting for over half (∼63%) of the total activity
(Fig. 5A) when
adjusted for compartmental fractional yield. Cytosolic NO formation from
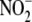 during hypoxia was sensitive to 100
μm oxypurinol (52% inhibition), suggesting the involvement of
XOR (and possibly other soluble proteins). Interestingly, subcellular
fractions produced more NO than whole liver homogenate
(Fig. 5B), implying
that, when liver subfractions are combined, some consume NO and/or inhibit NO
production by others. Consistent with the inhibitor studies, hypoxic
conversion of
during hypoxia was sensitive to 100
μm oxypurinol (52% inhibition), suggesting the involvement of
XOR (and possibly other soluble proteins). Interestingly, subcellular
fractions produced more NO than whole liver homogenate
(Fig. 5B), implying
that, when liver subfractions are combined, some consume NO and/or inhibit NO
production by others. Consistent with the inhibitor studies, hypoxic
conversion of 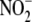 to NO by liver
mitochondria was stimulated (110 ± 31%) by 100 μm NADH
(and >70% by 100 μm NADPH)
(Fig. 5C), the
electron source for the mitochondrial respiratory chain, which can increase
the reduction potential of the system. The reason for the inhibition of
to NO by liver
mitochondria was stimulated (110 ± 31%) by 100 μm NADH
(and >70% by 100 μm NADPH)
(Fig. 5C), the
electron source for the mitochondrial respiratory chain, which can increase
the reduction potential of the system. The reason for the inhibition of
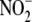 reduction in cytosol and microsomes
by NADPH is not obvious, but may reflect heightened consumption of NADPH by
non-mitochondrial anabolic pathways (e.g. cytosolic fatty acid
synthesis) stimulated by the added pyridine nucleotide. As a corollary to our
observation that hepatic microsomes significantly contribute to nitrite
reductase activity, spectral, and chemiluminescence studies confirm the
sequence of microsomal nitrite binding, formation of a Cyt P450
iron-nitrosyl complex and NO release (supplemental information and
Fig. 5D).
reduction in cytosol and microsomes
by NADPH is not obvious, but may reflect heightened consumption of NADPH by
non-mitochondrial anabolic pathways (e.g. cytosolic fatty acid
synthesis) stimulated by the added pyridine nucleotide. As a corollary to our
observation that hepatic microsomes significantly contribute to nitrite
reductase activity, spectral, and chemiluminescence studies confirm the
sequence of microsomal nitrite binding, formation of a Cyt P450
iron-nitrosyl complex and NO release (supplemental information and
Fig. 5D).
FIGURE 5.

Relative subcellular distribution and activity of hypoxic
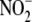 reduction to NO within liver
tissue. A, subcellular fractionation of hepatic tissue reveals
that
reduction to NO within liver
tissue. A, subcellular fractionation of hepatic tissue reveals
that 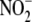 -dependent NO formation is
nonuniformly distributed among cell compartments. B, specific
activity of
-dependent NO formation is
nonuniformly distributed among cell compartments. B, specific
activity of 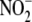 reduction to NO within
subcellular fractions reveals that both cytosolic (S3) and mitochondrial (P2)
compartments exhibit the highest specific
reduction to NO within
subcellular fractions reveals that both cytosolic (S3) and mitochondrial (P2)
compartments exhibit the highest specific
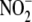 reductase activities. C,
pyridine nucleotide (NAD(P)H, 100 μm) enhances hypoxia-induced
reductase activities. C,
pyridine nucleotide (NAD(P)H, 100 μm) enhances hypoxia-induced
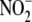 reduction to NO (70–110%
increase from control) within the mitochondrial fraction (n = 2).
D, upon supplying
reduction to NO (70–110%
increase from control) within the mitochondrial fraction (n = 2).
D, upon supplying 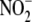 to
microsomal fractions (containing cyt P450), rapid formation of an
iron-nitrite complex precedes formation of an ironnitrosyl
(λmax = 448 nm) prior to release of NO. Inset,
microsomal NO generation from
to
microsomal fractions (containing cyt P450), rapid formation of an
iron-nitrite complex precedes formation of an ironnitrosyl
(λmax = 448 nm) prior to release of NO. Inset,
microsomal NO generation from 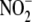 is
concentration-dependent. Increasing
is
concentration-dependent. Increasing 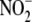 (0.1–1.0 mm) in hepatic microsomes potentiates NO formation
(n = 3).
(0.1–1.0 mm) in hepatic microsomes potentiates NO formation
(n = 3).
DISCUSSION
The present study provides intriguing new insights into the
oxygen-dependent processing of nitrite by mammalian tissues. The major
findings are as follows: (a)
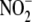 is transported into cells rapidly
irrespective of ambient oxygen tension. (b) While
is transported into cells rapidly
irrespective of ambient oxygen tension. (b) While
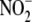 bioconversion to NO in
vitro is limited under aerobic conditions, all tissues (especially the
vasculature) readily convert
bioconversion to NO in
vitro is limited under aerobic conditions, all tissues (especially the
vasculature) readily convert 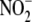 to NO
during hypoxia, and this is accompanied by nitrosation and nitrosylation of
cell constituents. (c) This pattern of
to NO
during hypoxia, and this is accompanied by nitrosation and nitrosylation of
cell constituents. (c) This pattern of
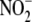 bioconversion to NO-related
signaling products is recapitulated in vivo.(d) Although
tissue
bioconversion to NO-related
signaling products is recapitulated in vivo.(d) Although
tissue 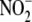 reduction may be facilitated
by non-enzymatic (e.g. disproportionation) and enzymatic mechanisms,
tissue
reduction may be facilitated
by non-enzymatic (e.g. disproportionation) and enzymatic mechanisms,
tissue 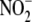 reductase activity is
largely (>80%) heat labile, suggesting that enzymatic mechanisms
predominate. (e) The kinetics of NO generation from
reductase activity is
largely (>80%) heat labile, suggesting that enzymatic mechanisms
predominate. (e) The kinetics of NO generation from
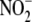 appears to be first-order with
respect to
appears to be first-order with
respect to 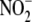 concentration, but do
not obey simple first-order kinetics with respect to protein concentration.
Hepatic and cardiac
concentration, but do
not obey simple first-order kinetics with respect to protein concentration.
Hepatic and cardiac 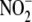 -dependent NO
production under hypoxia commences after a delay and is sustained for a
prolonged period of time. (f) Tissue
-dependent NO
production under hypoxia commences after a delay and is sustained for a
prolonged period of time. (f) Tissue
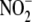 reductase activity is associated
with mitochondrial indices of oxidative phosphorylative capacity. (g)
Inhibitor and subfractionation studies suggest that tissue
reductase activity is associated
with mitochondrial indices of oxidative phosphorylative capacity. (g)
Inhibitor and subfractionation studies suggest that tissue
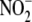 conversion to NO is multifactorial,
and
conversion to NO is multifactorial,
and 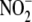 reductase activity is
distributed throughout different cell compartments. In liver, microsomal Cyt
P450 moieties appear to be the dominant reductases, with
significant contributions from mitochondria and cytosol (i.e. XOR).
(h) Ferricyanide and modeling studies demonstrate that
reductase activity is
distributed throughout different cell compartments. In liver, microsomal Cyt
P450 moieties appear to be the dominant reductases, with
significant contributions from mitochondria and cytosol (i.e. XOR).
(h) Ferricyanide and modeling studies demonstrate that
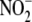 bioconversion under hypoxic
conditions leads to the formation of nitrosyl products largely without the
intermediacy of free NO.
bioconversion under hypoxic
conditions leads to the formation of nitrosyl products largely without the
intermediacy of free NO.
The proposal that hypoxic vasodilation is a result of RBC-dependent
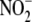 conversion to NO
(6,
10,
18,
19,
21,
22) proved to be a stimulus
for investigating
conversion to NO
(6,
10,
18,
19,
21,
22) proved to be a stimulus
for investigating 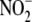 bioconversion. We
(23) and others
(15) have offered some initial
characterization of
bioconversion. We
(23) and others
(15) have offered some initial
characterization of 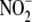 metabolism in
tissues. Increasing recognition of the biological significance of
metabolism in
tissues. Increasing recognition of the biological significance of
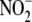 and its ubiquitous presence in
mammalian systems (8) mandates
further detailing of the elusive mechanisms through which
and its ubiquitous presence in
mammalian systems (8) mandates
further detailing of the elusive mechanisms through which
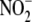 is converted to NO and, perhaps,
bioactive NO metabolites. Our present results and those of others
(15) suggest that, at
physiological
is converted to NO and, perhaps,
bioactive NO metabolites. Our present results and those of others
(15) suggest that, at
physiological 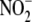 levels, hypoxic RBC
do not liberate significant amounts of NO. Instead, vascular tissue appears to
have the greatest capacity to generate NO from
levels, hypoxic RBC
do not liberate significant amounts of NO. Instead, vascular tissue appears to
have the greatest capacity to generate NO from
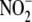 . We now show that hypoxic
. We now show that hypoxic
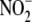 reductase activity in tissues is
accompanied by the nitrosation and nitrosylation of cellular targets,
suggesting that some of the resulting NO metabolites may represent
reductase activity in tissues is
accompanied by the nitrosation and nitrosylation of cellular targets,
suggesting that some of the resulting NO metabolites may represent
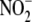 -related tissue effectors of (or
markers for) hypoxic signaling.
-related tissue effectors of (or
markers for) hypoxic signaling.
Formation of NO from 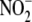 is an
inefficient process on the order of 0.05 nmol/h/g wet tissue/μm
is an
inefficient process on the order of 0.05 nmol/h/g wet tissue/μm
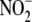 in liver and heart, with most of
the reductase activity being thermolabile (i.e. enzymatic)
(15). Accordingly, in tissues
such as liver and heart with
in liver and heart, with most of
the reductase activity being thermolabile (i.e. enzymatic)
(15). Accordingly, in tissues
such as liver and heart with 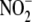 at a
steady-state concentration of ∼500 nm
(8,
23,
24), the expected NO
production rate would be 0.025 μmol/kg/h. The average rate of whole body NO
production in rats and humans is significantly greater, ∼1 μmol/kg/h
(32). Tissues with higher
at a
steady-state concentration of ∼500 nm
(8,
23,
24), the expected NO
production rate would be 0.025 μmol/kg/h. The average rate of whole body NO
production in rats and humans is significantly greater, ∼1 μmol/kg/h
(32). Tissues with higher
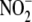 concentrations such as aorta
(∼20 μm
concentrations such as aorta
(∼20 μm 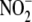 ) and with
rates of
) and with
rates of 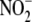 conversion to NO at least
twice that of the heart or liver, the NO production under hypoxic conditions
could reach ∼2 μmol/kg/h. It is thus conceivable that intrinsic NO
generation from
conversion to NO at least
twice that of the heart or liver, the NO production under hypoxic conditions
could reach ∼2 μmol/kg/h. It is thus conceivable that intrinsic NO
generation from 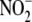 may have
significant physiological importance in vascular tissue as an autonomous
mediator of hypoxic vasodilation
(11,
16). Conversely, one might
question how the modest rates of
may have
significant physiological importance in vascular tissue as an autonomous
mediator of hypoxic vasodilation
(11,
16). Conversely, one might
question how the modest rates of
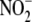 -dependent NO production in heart
and liver could account for the protective effects of
-dependent NO production in heart
and liver could account for the protective effects of
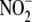 against ischemia-reperfusion injury
in these organs. Relevant insight may be obtained from our ferricyanide
experiment, where the amount of free NO trapped by hemes accounts for only a
small percentage of
against ischemia-reperfusion injury
in these organs. Relevant insight may be obtained from our ferricyanide
experiment, where the amount of free NO trapped by hemes accounts for only a
small percentage of 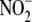 -derived NO.
This result suggests that the production of free NO from
-derived NO.
This result suggests that the production of free NO from
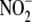 under hypoxia may represent a
relatively minor component of a potent chemical pathway that generates
bioactive (i.e. tissue-protective) NO metabolites directly from
under hypoxia may represent a
relatively minor component of a potent chemical pathway that generates
bioactive (i.e. tissue-protective) NO metabolites directly from
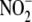 .
.
Given the well-recognized interplay among oxygen, NO, and the mitochondrial
electron transport chain (ETC) at the interface between tissue oxygen
consumption and oxygen-dependent energy conservation, the mitochondrion is
well-positioned to act as a “metabolic coordinator”
(2,
33). Our observation that
rates of 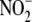 conversion to NO correlate
robustly with maximal mitochondrial respiratory capacity accords with the
hypothesis that tissue oxygen demand and ambient oxygen concentration are
operationally related through
conversion to NO correlate
robustly with maximal mitochondrial respiratory capacity accords with the
hypothesis that tissue oxygen demand and ambient oxygen concentration are
operationally related through
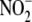 -dependent NO formation by, or more
provocatively for, the ETC
(12,
34,
35). This link between
-dependent NO formation by, or more
provocatively for, the ETC
(12,
34,
35). This link between
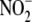 -dependent NO formation and
oxidative intermediary metabolism is further underlined by the notable
exception of renal tissue, the renal medulla relying largely on anaerobic
metabolism (30). While these
observations underscore the significance of mitochondria in
-dependent NO formation and
oxidative intermediary metabolism is further underlined by the notable
exception of renal tissue, the renal medulla relying largely on anaerobic
metabolism (30). While these
observations underscore the significance of mitochondria in
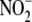 bioconversion, the profile of
bioconversion, the profile of
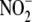 reductases differs among tissues,
and the mitochondrial compartment does not account for the majority of
reductases differs among tissues,
and the mitochondrial compartment does not account for the majority of
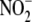 bioconversion. Far greater
subcellular complexity is observed. At least in the liver, our combined
inhibitor and cell-fractionation studies suggest that hepatic
bioconversion. Far greater
subcellular complexity is observed. At least in the liver, our combined
inhibitor and cell-fractionation studies suggest that hepatic
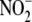 reductase activity occurs in the
cytosol, the mitochondrion (along the ETC), and, predominantly, the microsomes
(Cyt P450), with thiols and metalloproteins playing a crucial
cooperative role. Microsomal heme-containing cytochromes are effective
reductase activity occurs in the
cytosol, the mitochondrion (along the ETC), and, predominantly, the microsomes
(Cyt P450), with thiols and metalloproteins playing a crucial
cooperative role. Microsomal heme-containing cytochromes are effective
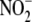 reductases
(36,
37), as confirmed by the
spectral studies herein.
reductases
(36,
37), as confirmed by the
spectral studies herein.
Except for oxygen and cyanide, no one agent tested completely inhibited
hypoxic 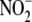 reduction. However, a
combination of three enzyme inhibitors (rotenone, EthOx, and DPI) virtually
blocked all
reduction. However, a
combination of three enzyme inhibitors (rotenone, EthOx, and DPI) virtually
blocked all 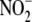 -dependent NO formation
by liver homogenates. These observations are not easily reconciled with those
of Li et al. (15),
who failed to demonstrate any effect of rotenone on
-dependent NO formation
by liver homogenates. These observations are not easily reconciled with those
of Li et al. (15),
who failed to demonstrate any effect of rotenone on
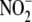 reductase activity and identified
XOR and ALDH2 as important components of the cardiac and hepatic
reductase activity and identified
XOR and ALDH2 as important components of the cardiac and hepatic
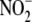 reductase system. Inhibitor
nonspecificity undoubtedly accounts for some of these differences: although
raloxifene has been used as an ALDH2 inhibitor
(37), it also has potent
effects on some microsomal Cyt P450 species (e.g. 3A4)
(38). Similarly, DPI is
promiscuous and inhibits a variety of enzymes exhibiting flavin-dependent
electron transfer including XOR, ALDH2, and other oxidoreductases. This
demonstrates the weakness of isolated inhibitor studies and in part explains
why our own inhibitor studies do not entirely accord with cell fractionation
studies. For example, although rotenone inhibited 60% of the
reductase system. Inhibitor
nonspecificity undoubtedly accounts for some of these differences: although
raloxifene has been used as an ALDH2 inhibitor
(37), it also has potent
effects on some microsomal Cyt P450 species (e.g. 3A4)
(38). Similarly, DPI is
promiscuous and inhibits a variety of enzymes exhibiting flavin-dependent
electron transfer including XOR, ALDH2, and other oxidoreductases. This
demonstrates the weakness of isolated inhibitor studies and in part explains
why our own inhibitor studies do not entirely accord with cell fractionation
studies. For example, although rotenone inhibited 60% of the
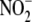 bioconversion in total liver
homogenate, cell fractionation studies suggest that mitochondria contribute
relatively modestly (14%) to overall hepatic
bioconversion in total liver
homogenate, cell fractionation studies suggest that mitochondria contribute
relatively modestly (14%) to overall hepatic
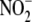 reductase activity. We reconcile
these observations by recognizing the limitations of inhibitor pharmacology,
the nature of cell fractionation as a methodology that artificially divides a
physiologically integrated, heuristic
reductase activity. We reconcile
these observations by recognizing the limitations of inhibitor pharmacology,
the nature of cell fractionation as a methodology that artificially divides a
physiologically integrated, heuristic
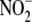 reductase, thereby preventing
cooperative effects, and the potential for the inhibitor itself to enter into
subfraction-selective reactions. Nonetheless, the inhibitor data do indicate
that hepatic “
reductase, thereby preventing
cooperative effects, and the potential for the inhibitor itself to enter into
subfraction-selective reactions. Nonetheless, the inhibitor data do indicate
that hepatic “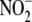 reductase” represents a biochemically and spatially complex activity
that involves heme, iron-sulfur cluster, and molybdenum enzymes distributed
among a number of organelles that cooperate to reduce
reductase” represents a biochemically and spatially complex activity
that involves heme, iron-sulfur cluster, and molybdenum enzymes distributed
among a number of organelles that cooperate to reduce
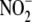 to NO.
to NO.
While there is compelling evidence herein and elsewhere that Hb is unlikely
to contribute significantly to
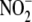 -dependent NO formation in hypoxia
due to the voracious capacity of deoxy/oxyHb to scavenge free NO, the
significance of heme proteins as intracellular
-dependent NO formation in hypoxia
due to the voracious capacity of deoxy/oxyHb to scavenge free NO, the
significance of heme proteins as intracellular
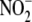 reductases remains unknown. To
address this issue, we combined the use of ferricyanide (which putatively
oxidizes iron from ferrous to ferric hemes) with modeling to dissect the role
of hemes in hepatic and cardiac
reductases remains unknown. To
address this issue, we combined the use of ferricyanide (which putatively
oxidizes iron from ferrous to ferric hemes) with modeling to dissect the role
of hemes in hepatic and cardiac 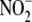 reductase activity. Our data suggest that: (a) Ferrous heme moieties
within cells contribute substantially to NO scavenging. (b) Much of
the NO liberated by ferricyanide is likely to have originated from nitrosyl
groups generated directly from
reductase activity. Our data suggest that: (a) Ferrous heme moieties
within cells contribute substantially to NO scavenging. (b) Much of
the NO liberated by ferricyanide is likely to have originated from nitrosyl
groups generated directly from
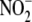 -dependent nitrosylation.
(c) The enhanced effect on NO formation by ferricyanide pretreatment
reflects either the limited participation of ferrous heme in
-dependent nitrosylation.
(c) The enhanced effect on NO formation by ferricyanide pretreatment
reflects either the limited participation of ferrous heme in
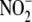 bioconversion or, more likely, a
mixed scavenging and liberating role for ferrous heme. Both the concentrations
and reactivities of the different heme and other reductase moieties
(microsomal, mitochondrial and cytosolic) will determine the net response to
exogenous ferricyanide. NO liberation from microsomal ferrous
cyt-P450 nitrosyls is well-known
(36) and may be
counterbalanced by scavenging from other ferrous heme nitrosyl complexes
slower in releasing NO and the influence of other reductases (e.g.
XOR/ALDH2) (15). Pretreatment
with ferricyanide may alter this balance (possibly across compartments) and
favor early NO liberation. In other tissues (e.g. vasculature) where
the heme profile and ratio of
bioconversion or, more likely, a
mixed scavenging and liberating role for ferrous heme. Both the concentrations
and reactivities of the different heme and other reductase moieties
(microsomal, mitochondrial and cytosolic) will determine the net response to
exogenous ferricyanide. NO liberation from microsomal ferrous
cyt-P450 nitrosyls is well-known
(36) and may be
counterbalanced by scavenging from other ferrous heme nitrosyl complexes
slower in releasing NO and the influence of other reductases (e.g.
XOR/ALDH2) (15). Pretreatment
with ferricyanide may alter this balance (possibly across compartments) and
favor early NO liberation. In other tissues (e.g. vasculature) where
the heme profile and ratio of 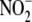 to
heme proteins (and the activity of ferri-heme reductases) differ, the impact
of scavenging may be attenuated, and NO release from heme moieties more
profound. Indeed, recent experimental evidence points to a role for heme
moieties in vascular
to
heme proteins (and the activity of ferri-heme reductases) differ, the impact
of scavenging may be attenuated, and NO release from heme moieties more
profound. Indeed, recent experimental evidence points to a role for heme
moieties in vascular 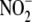 bioconversion
(16).
bioconversion
(16).
Hypoxic 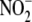 bioconversion to NO is
effected with tissue-selectivity by an involved interplay of heme, iron-sulfur
cluster, and molybdenum-containing enzymes, the nature and ratio of
heme-dependent proteins (and other enzymes),
bioconversion to NO is
effected with tissue-selectivity by an involved interplay of heme, iron-sulfur
cluster, and molybdenum-containing enzymes, the nature and ratio of
heme-dependent proteins (and other enzymes),
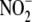 and oxygen concentration, and redox
state varying with tissue, time, and ambient conditions. The intricacy of
these interactions and the redundancy exhibited by different
and oxygen concentration, and redox
state varying with tissue, time, and ambient conditions. The intricacy of
these interactions and the redundancy exhibited by different
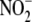 reducing enzymes in different cell
compartments within tissues raise important questions regarding the biological
role of reductive
reducing enzymes in different cell
compartments within tissues raise important questions regarding the biological
role of reductive 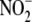 metabolism to NO.
Oxygen-dependent conversion of
metabolism to NO.
Oxygen-dependent conversion of 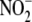 to
NO renders it more suitable for hypoxic vasodilation
(11) than
l-arginine-driven, NO synthase-mediated hypoxic vasodilation, since
the latter requires oxygen as a co-substrate to produce NO. However, the
identification of redundant
to
NO renders it more suitable for hypoxic vasodilation
(11) than
l-arginine-driven, NO synthase-mediated hypoxic vasodilation, since
the latter requires oxygen as a co-substrate to produce NO. However, the
identification of redundant 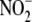 reductase activities in diverse tissues with varying functions and the
association of this
reductase activities in diverse tissues with varying functions and the
association of this 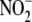 bioconversion
with tissue oxidative phosphorylation capacity suggest that the biology of
bioconversion
with tissue oxidative phosphorylation capacity suggest that the biology of
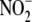 extends well beyond vasodilation
and tissue protection. Based on the evidence presented herein, it is tempting
to speculate that conversion of
extends well beyond vasodilation
and tissue protection. Based on the evidence presented herein, it is tempting
to speculate that conversion of 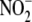 to
NO is part of a conserved regulatory mechanism that acutely matches oxygen
homeostasis to intermediary metabolism
(37). As well acting directly
on mitochondria (12,
35), the resulting nitrosation
of master transcription factors such as HIF-1α may have a profound
impact on the cellular metabolic milieu
(39). Different tissue
compartments might affect their oxygen-sensing through the common path of
to
NO is part of a conserved regulatory mechanism that acutely matches oxygen
homeostasis to intermediary metabolism
(37). As well acting directly
on mitochondria (12,
35), the resulting nitrosation
of master transcription factors such as HIF-1α may have a profound
impact on the cellular metabolic milieu
(39). Different tissue
compartments might affect their oxygen-sensing through the common path of
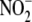 conversion to NO. The resulting NO
then acts to modify mitochondrial function and the cellular metabolic and
transcriptional milieu. This hypothetical paradigm represents a tuning
mechanism through which eukaryotic cells might optimize their use of oxygen
and carbon units for maximally efficient energy provision
(2,
33,
35). Whether
conversion to NO. The resulting NO
then acts to modify mitochondrial function and the cellular metabolic and
transcriptional milieu. This hypothetical paradigm represents a tuning
mechanism through which eukaryotic cells might optimize their use of oxygen
and carbon units for maximally efficient energy provision
(2,
33,
35). Whether
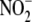 is indeed involved in such
processes and whether there are differences in bioactivity between endogenous
and exogenous
is indeed involved in such
processes and whether there are differences in bioactivity between endogenous
and exogenous 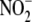 will require further
investigation.
will require further
investigation.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants HL 69029 (to M. F.) and the Kirschstein National Research Service Award Cardiovascular Training Grant (to B. O. F. and N. S. B.). This work was also supported by the Medical Research Council (MRC Strategic Appointment Scheme, to M. F.), and a Wellcome Trust CVRI Fellowship (to H. A.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental information, Table S1, and Figs. S1 and S2.
Footnotes
The abbreviations used are: NO, nitric oxide; ALDH2 (mtALDH2),
mitochondrial aldehyde dehydrogenase 2; DPI, diphenyleneiodonium; EthOx,
ethoxyresorufin; Hb, hemoglobin; i.p., intraperitoneal; NaNO2,
sodium nitrite; 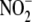 , nitrite;
, nitrite;
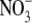 , nitrate; RBC, red blood cells;
RNNO, N-nitrosamines; RSNO, S-nitrosothiols; SNO-Hb,
S-nitrosohemoglobin; XOR, xanthine oxidoreductase; PBS,
phosphate-buffered saline.
, nitrate; RBC, red blood cells;
RNNO, N-nitrosamines; RSNO, S-nitrosothiols; SNO-Hb,
S-nitrosohemoglobin; XOR, xanthine oxidoreductase; PBS,
phosphate-buffered saline.
References
- 1.Schmidt, H. H., and Walter, U. (1994) Cell 78 919–925 [DOI] [PubMed] [Google Scholar]
- 2.Erusalimsky, J. D., and Moncada, S. (2007) Arterioscler. Thromb. Vasc. Biol. 27 2524–2531 [DOI] [PubMed] [Google Scholar]
- 3.Lancaster, J. R., Jr., and Xie, K. (2006) Cancer Res. 66 6459–6462 [DOI] [PubMed] [Google Scholar]
- 4.Li, C. Q., and Wogan, G. N. (2005) Cancer Lett. 226 1–15 [DOI] [PubMed] [Google Scholar]
- 5.Edmunds, N. J., Moncada, S., and Marshall, J. M. (2003) J. Physiol. 546 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lundberg, J. O., Weitzberg, E., and Gladwin, M. T. (2008) Nat. Rev. Drug Discov. 7 156–167 [DOI] [PubMed] [Google Scholar]
- 7.Stamler, J. S., Lamas, S., and Fang, F. C. (2001) Cell 106 675–683 [DOI] [PubMed] [Google Scholar]
- 8.Bryan, N. S., Fernandez, B. O., Bauer, S. M., Garcia-Saura, M. F., Milsom, A. B., Rassaf, T., Maloney, R. E., Bharti, A., Rodriguez, J., and Feelisch, M. (2005) Nat. Chem. Biol. 1 290–297 [DOI] [PubMed] [Google Scholar]
- 9.Mazzone, M., and Carmeliet, P. (2008) Nature 453 1194–1195 [DOI] [PubMed] [Google Scholar]
- 10.Cosby, K., Partovi, K. S., Crawford, J. H., Patel, R. P., Reiter, C. D., Martyr, S., Yang, B. K., Waclawiw, M. A., Zalos, G., Xu, X., Huang, K. T., Shields, H., Kim-Shapiro, D. B., Schechter, A. N., Cannon, R. O., III, and Gladwin, M. T. (2003) Nat. Med. 9 1498–1505 [DOI] [PubMed] [Google Scholar]
- 11.Maher, A. R., Milsom, A. B., Gunaruwan, P., Abozguia, K., Ahmed, I., Weaver, R. A., Thomas, P., Ashrafian, H., Born, G. V., James, P. E., and Frenneaux, M. P. (2008) Circulation 117 670–677 [DOI] [PubMed] [Google Scholar]
- 12.Shiva, S., Sack, M. N., Greer, J. J., Duranski, M., Ringwood, L. A., Burwell, L., Wang, X., MacArthur, P. H., Shoja, A., Raghavachari, N., Calvert, J. W., Brookes, P. S., Lefer, D. J., and Gladwin, M. T. (2007) J. Exp. Med. 204 2089–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez, F. M., Shiva, S., Vincent, P. S., Ringwood, L. A., Hsu, L. Y., Hon, Y. Y., Aletras, A. H., Cannon, R. O., III, Gladwin, M. T., and Arai, A. E. (2008) Circulation 117 2986–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar, D., Branch, B. G., Pattillo, C. B., Hood, J., Thoma, S., Simpson, S., Illum, S., Arora, N., Chidlow, J. H., Jr., Langston, W., Teng, X., Lefer, D. J., Patel, R. P., and Kevil, C. G. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 7540–7545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li, H., Cui, H., Kundu, T. K., Alzawahra, W., and Zweier, J. L. (2008) J. Biol. Chem. 283 17855–17863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alzawahra, W. F., Talukder, M. A., Liu, X., Samouilov, A., and Zweier, J. L. (2008) Am. J. Physiol. Heart Circ. Physiol., 295 H499-H508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu, S., Grubina, R., Huang, J., Conradie, J., Huang, Z., Jeffers, A., Jiang, A., He, X., Azarov, I., Seibert, R., Mehta, A., Patel, R., King, S. B., Hogg, N., Ghosh, A., Gladwin, M. T., and Kim-Shapiro, D. B. (2007) Nat. Chem. Biol. 3 785–794 [DOI] [PubMed] [Google Scholar]
- 18.Grubina, R., Huang, Z., Shiva, S., Joshi, M. S., Azarov, I., Basu, S., Ringwood, L. A., Jiang, A., Hogg, N., Kim-Shapiro, D. B., and Gladwin, M. T. (2007) J. Biol. Chem. 282 12916–12927 [DOI] [PubMed] [Google Scholar]
- 19.Isbell, T. S., Gladwin, M. T., and Patel, R. P. (2007) Am. J. Physiol. Heart Circ. Physiol. 293 H2565–H2572 [DOI] [PubMed] [Google Scholar]
- 20.Shiva, S., Huang, Z., Grubina, R., Sun, J., Ringwood, L. A., MacArthur, P. H., Xu, X., Murphy, E., Darrley-Usmar, V. M., and Gladwin, M. T. (2007) Circ. Res. 100 654–661 [DOI] [PubMed] [Google Scholar]
- 21.Crawford, J. H., Isbell, T. S., Huang, Z., Shiva, S., Chacko, B. K., Schechter, A. N., rley-Usmar, V. M., Kerby, J. D., Lang, J. D., Jr., Kraus, D., Ho, C., Gladwin, M. T., and Patel, R. P. (2006) Blood 107 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, K. T., Keszler, A., Patel, N., Patel, R. P., Gladwin, M. T., Kim-Shapiro, D. B., and Hogg, N. (2005) J. Biol. Chem. 280 31126–31131 [DOI] [PubMed] [Google Scholar]
- 23.Bryan, N. S., Rassaf, T., Maloney, R. E., Rodriguez, C. M., Saijo, F., Rodriguez, J. R., and Feelisch, M. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4308–4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feelisch, M., Rassaf, T., Mnaimneh, S., Singh, N., Bryan, N. S., Jourd'Heuil, D., and Kelm, M. (2002) FASEB J. 16 1775–1785 [DOI] [PubMed] [Google Scholar]
- 25.Rickwood, D. (1993) Preparative Centrifugation: A Practical Approach, Oxford University Press, New York
- 26.Rodriguez, J., Maloney, R. E., Rassaf, T., Bryan, N. S., and Feelisch, M. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 336–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angelo, M., Singel, D. J., and Stamler, J. S. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 8366–8371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parks, D. A., and Granger, D. N. (1986) Acta Physiol. Scand. Suppl 548 87–99 [PubMed] [Google Scholar]
- 29.Hulbert, A. J., and Else, P. L. (1989) Am. J. Physiol. 256 R63–R69 [DOI] [PubMed] [Google Scholar]
- 30.Cohen, J. J. (1979) Am. J. Physiol. 236 F423–F433 [DOI] [PubMed] [Google Scholar]
- 31.Gladwin, M. T., Wang, X., Reiter, C. D., Yang, B. K., Vivas, E. X., Bonaventura, C., and Schechter, A. N. (2002) J. Biol. Chem. 277 27818–27828 [DOI] [PubMed] [Google Scholar]
- 32.Castillo, L., Beaumier, L., Ajami, A. M., and Young, V. R. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 11460–11465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moncada, S., and Erusalimsky, J. D. (2002) Nat. Rev. Mol. Cell Biol. 3 214–220 [DOI] [PubMed] [Google Scholar]
- 34.Kozlov, A. V., Staniek, K., and Nohl, H. (1999) FEBS Lett. 454 127–130 [DOI] [PubMed] [Google Scholar]
- 35.Benamar, A., Rolletschek, H., Borisjuk, L., Velange-Macherel, M. H., Curien, G., Mostefai, H. A., Andriantsitohaina, R., and Macherel, D. (2008) Biochim. Biophys. Acta, 1777 1268–1275 [DOI] [PubMed] [Google Scholar]
- 36.Kozlov, A. V., Dietrich, B., and Nohl, H. (2003) Br. J. Pharmacol. 139 989–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, H., Liu, X., Cui, H., Chen, Y. R., Cardounel, A. J., and Zweier, J. L. (2006) J. Biol. Chem. 281 12546–12554 [DOI] [PubMed] [Google Scholar]
- 38.Zhou, S., Yung, C. S., Cher, G. B., Chan, E., Duan, W., Huang, M., and McLeod, H. L. (2005) Clin. Pharmacokinet. 44 279–304 [DOI] [PubMed] [Google Scholar]
- 39.Kaelin, W. G., Jr., and Ratcliffe, P. J. (2008) Mol. Cell 30 393–402 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.