

Abstract
Cell death/survival following CNS injury may be a result of alterations in the intracellular ratio of death and survival factors. Using immunohistochemistry, Western analysis and in situ hybridization, the expression of the anti-cell death protein, Bcl-2, and the pro-cell death protein, Bax, was evaluated following lateral fluid-percussion (FP) brain injury of moderate severity (2.3–2.6 atm) in adult male Sprague-Dawley rats. By 2 h post-injury, a marked reduction of cellular Bcl-2-immunoreactivity (IR) and a mild decrease in cellular Bax IR were observed in the temporal and occipital cortices, and in the hippocampal CA3 ipsilateral to the site of impact. These decreases in Bcl-2 and Bax IR appeared to precede the overt cell loss in these regions that was evident at 24 h. Immunoblot analysis supported the immunohistochemical data, with a modest but significant reduction in the intensities of both the Bcl-2 and Bax protein bands at 2 h (p < 0.05 compared to sham levels). However, the Bax:Bcl-2 ratio increased significantly at 2 h (2.28 ± 0.13) and remained elevated up to 7 days (2.05 ± 0.13) post-injury compared to sham-injured control tissue (1.62 ± 0.10, p < 0.05). Furthermore, cortical, but not hippocampal, levels of Bax protein increased by 25% (p < 0.05 compared to sham-injured controls) at 24 h post-injury, and returned to control levels by 7 days. In situ hybridization analysis of Bax mRNA revealed increased cellular grain density in the injured cortex (p < 0.05 compared to sham-injured brains), but not in the CA3 region of the injured hippocampus. No injury-induced changes in the expression of Bcl-2 mRNA were observed in any brain region. Taken together, these data suggest that the association between regional post-traumatic cell death and alterations in the cellular ratio of Bcl-2 and Bax may be, in part, due to alterations in mRNA and/or protein expression of the Bcl-2 family of proteins.
Keywords: apoptosis; βcl-2, Bax; cell death; head injury
INTRODUCTION
Neuronal cell death following traumatic brain injury (TBI) has been suggested, in part, to be responsible for post-injury mortality and morbidity. In humans, TBI results in neuronal loss in the cortex, hippocampus, cerebellum and thalamus (Adams et al., 1985; Kotapka et al., 1992; Ross et al., 1993). This pathology has been duplicated in various rodent models of TBI (Colicos et al., 1997; Dietrich et al., 1994; Hicks et al., 1996; Sutton et al., 1995). Changes in cellular morphology indicative of neurodegeneration are evident almost immediately following the impact in the injured cortex and hippocampus (Hicks et al, 1996; Colicos et al, 1996; Dietrich et al., 1994), and appear to continue for weeks to months following injury (Bramlett et al., 1997; Smith et al., 1997). Electron microscopic analysis revealed that some of these degenerating neurons appear necrotic, with swollen mitochondria, vacuolated cytoplasm and pyknotic nuclei (Dietrich et al., 1994), while others exhibit DNA fragmentation within shrunken cell bodies containing condensed chromatin and cellular structures characteristic of apoptosis (Colicos and Dash, 1996; Conti et al., 1998; Fox et al., 1998; Newcomb et al., 1999; Rink et al., 1995; Yakovlev et al., 1997). While the cellular mechanisms underlying regional cell death have yet to be elucidated, many neurochemical factors and proteins have been identified as potential canditates (McIntosh et al., 1998; Raghupathi et al., 1995; Yakovlev and Faden, 1996).
The Bcl-2 multigene superfamily include anti-apoptotic genes such as Bcl-2, Bcl-xL, and Bak (Boise et al., 1993; Chittenden et al., 1995) and pro-apoptotic genes such as Bax (Oltvai et al., 1993), Bad (Yang et al., 1995) and Bcl-xs (Gonzalez-Garcia et al., 1995). Although characterized as genes that are associated with developmental cell death, accumulating evidence suggests that these genes may participate in both pathologic apoptotic and necrotic cell death pathways (Bredesen, 1995). Bcl-2 is unique in that it has been shown to protect cells from a variety of insults such as treatment with calcium ionophores, glutamate, free radicals and withdrawal of trophic factors (Reed, 1998). Bax has been suggested to be necessary for developmental cell death (Deckwerth et al., 1996) and suppression of tumorigenesis (Yin et al., 1997). Recent studies have focused on the concomitant alterations in Bcl-2, Bcl-xL, and Bax in in vivo models of CNS injury. Decreased immunoreactivity for Bcl-2 and Bcl-xL and increased Bax immunoreactivity was observed in injured neurons following both focal (Gillardon et al., 1996b) and global ischemia (Chen et al., 1996; Krajewski et al., 1995). An increase in Bcl-2 immunoreactivity was observed in neurons, glia, and endothelial cells surrounding the infarcted cortex following focal ischemia (Chen et al., 1995), while Bcl-2 and Bcl-xL levels were relatively unchanged in neurons that survived the ischemic insult (Gillardon et al., 1996b; Honkaniemi et al., 1996). Nerve transection or administration of neurotoxins such as 1 methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or kainic acid resulted in increased Bax mRNA and protein in cells undergoing apoptosis (Gillardon et al., 1995, 1996a; Hassouna et al., 1996). Although these observations support the hypothesis that cell survival is dependent on the ratio of anti-apoptotic and proapoptotic Bcl-2 proteins (Korsmeyer, 1996), the specific mechanisms of action either Bcl-2 or Bax following CNS injury is unclear. In part, Bcl-2 and Bax may mediate cell death by regulating the activity of the cell death-inducing caspase family of proteases (Thornberry and Lazebnik, 1998).
The role of the Bcl-2 family proteins in TBI has been evaluated to a limited extent. In a model of combined TBI and hypoxemia, Clark et al. (1997) observed an up-regulation of Bcl-2 in cortical neurons that survived the traumatic injury, while increased Bax immunoreactivity was observed in apoptotic granule neurons in the dentate gyrus following controlled cortical impact injury (Kaya et al., 1999). Direct evidence for a potential role of Bcl-2 in the pathology of TBI was based on the attenuation of posttraumatic cortical neurodegeneration in transgenic mice overexpressing human Bcl-2 (Nakamura et al., 1999; Raghupathi et al., 1998). We postulated that the vulnerability of neurons to TBI could be related their endogenous levels of Bcl-2 and Bax and have used the well-characterized lateral fluid-percussion (FP) model of TBI in the rat to evaluate the regional post-injury expression of Bcl-2 protein and Bax protein and mRNA.
MATERIALS AND METHODS
Fluid-Percussion Brain Injury
Adult male Sprague-Dawley rats (350–400 g, n = 64) were anesthetized with sodium pentobarbital (60 mg/kg, i.p.), placed in a stereotactic frame and the scalp and temporal muscle were reflected. A hollow, female Luer-Lok fitting was rigidly fixed with dental cement to a 5.0-mm craniotomy centered between bregma and lambda sutures and over the left parietal cortex. The fluid-percussion injury device was connected to the animal via the LuerLok fitting and brain injury of moderate (2.3–2.6 atm, n = 44) severity was produced in animals as previously described (McIntosh et al., 1989). The device produces a pulse of 21–23 msec through the rapid injection of saline into the closed cranial cavity, resulting in the brief deformation of brain tissue. Sham-injured controls were surgically prepared but were not injured (n = 20).
Tissue Preparation
For histological and immunohistochemical analyses, one group of animals was reanesthetized and killed by transcardial perfusion with 4% paraformaldehyde at 2 h, 6 h, 24 h, and 7 days (n = 4 injured and 2 sham-injured per time point). Brains were removed from the skull, fixed for 4–5 h, cryoprotected with sucrose for 3–5 days, and frozen in isopentane at −30°C. Serial 40-μm sections were taken over the rostral-caudal extent of the brain. One series was mounted on gelatin-coated slides and stained with Toulidine Blue to determine cytoarchitectural boundaries. Parallel sections were subjected to immunohistochemical analysis for Bcl-2 and Bax. For Western blot analyses of tissue levels of Bax and Bcl-2 proteins, a second group of animals was anesthetized and killed by decapitation at 2 h, 6 h, 24 h, and 7 days (n = 4 injured and 2 sham-injured per time point). The brains were rapidly removed and the injured cortex and hippocampus were dissected and snap-frozen in liquid nitrogen. Corresponding areas from the hemisphere contralateral to the impact were also removed and frozen. For in situ hybridization experiments to evaluate postinjury expression of Bax and Bcl-2 mRNAs, a third group of animals was anesthetized and killed by decapitation at 24 h and 3 days (n = 6 injured and 2 sham-injured per time point). Brains were removed rapidly from the skull and frozen in isopentane at −30°C. Twelve micron sections were cut through the rostral-caudal extent of the brain on a cryostat.
Immunohistochemical Analysis
Sections were washed with 10 mM Tris (pH 7.4) containing 150 mM NaCl (TBS), incubated with 1% H2O2, and rinsed with TBS containing 0.01% Triton-X-100 (TBST). After blocking with 3% normal goat serum in TBST, sections were incubated overnight at 4°C with anti-Bcl-2 antibody (polyclonal, 1:500 dilution) or anti-Bax antibody (polyclonal, 1:500 dilution); both antibodies were generated against rat proteins and described in Krajewski et al., 1995. After washing with TBST, sections were incubated with biotinylated secondary antibody. The secondary abtibody was subsequently detected using the ABC technique (Vector Labs, Burlingame, CA), using the peroxidase-DAB system. Following the reaction with DAB, sections were washed in water, mounted on gelatin-coated slides and coverslipped. No immunoreactivity was observed when the primary antibody was omitted from the protocol. Specificity of the antibody was further elucidated since no immunostaining was observed when the primary antibody was preadsorbed with the immunizing antigen and subsequently incubated with the brain sections.
Western Blot Analysis
Frozen tissue representing the injured cortex and hippocampus, and corresponding regions from the hemisphere contralateral to the impact site were homogenized in RIPA bufffer (50 mm Tris, pH 7.0, 150 mM NaCl, 1% Triton-X-100) containing protease inhibitors (Boehringer Mannheim, Indianapolis, IN). Homogenates were nutated at 4°C for 30 min and centrifuged at 12,000 × g for 20 min. The supernatant fraction (50 μg total protein) was boiled in 1 × Laemmli buffer and electrophoresed on a 15% polyacrylamide gel containing SDS (SDS-PAGE), electroblotted onto Immobilon-P membranes (BioRad Labs, Richmond, CA) for 2 h at 60 V constant voltage. Membranes were washed in 0.1 M Tris (pH 7.4) blocked in 5% normal goat serum for 60 min and incubated with primary antibody (anti-Bcl-2, 1:1000; anti-Bax, 1:1000) overnight at 4°C. Blots were washed in Tris buffer and incubated with horseradish peroxidase–conjugated goat anti-rabbit IgG (Jackson Labs), and bands were visualized by enhanced chemiluminescence (ECL, Amersham, Arlington, IL). Following incubation with one antibody (either Bcl-2 or Bax), blots were stripped (0.5N NaOH for 30 min at 50°C) and reprobed using the second antibody. Equal loading of samples was evaluated by stripping the blots and reprobing them with an internal standard—an antibody to G-actin (clone AC40; Sigma Chemical Co., St. Louis, MO) or β-tubulin (Zymed Laboratories, San Francisco, CA). Autoradiographic films were analyzed using a densitometer (Molecular Dynamics, Sunnyvale, CA), and band intensities were evaluated using an image analysis system (MCID/M4, Imaging Research, St. Catherine's, Ontario, Canada), and presented as optical densities. The optical density values for Bcl-2 and Bax for each lane were normalized to that for either G-actin or β-tubulin from the same lane. Each sample was subjected to immunoblotting three times, and the final optical density value (relative to that for the internal standard) represents the average of these three separate analyses.
In Situ Hybridization
The bax riboprobe (370 bp) consisted of T7 transcripts off pGEM-5Zf+ (Promega) containing a bax PCR fragment from rat cDNA (Clontech) generated using primers at 190–212 (forward) and 559–537 (reverse) (Han et al., 1996; accession no. U49729). The bcl-2 riboprobe (639 bp) consisted of T7 transcripts off pGEM-5Zf+ containing a bcl-2 PCR fragment from rat cDNA generated using primers at 247-259 (forward) and 885–863 (reverse) (Tilly et al., 1995; accession no. L14680). The PCR fragments were sequenced and sequences were confirmed using a BLAST search in the GENBANK database.
Radiolabeled probes, synthesized in the presence [35S]-UTP (1250 mCi/mmol), were purified by extraction with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with ethanol, and utilized for in situ hybridization histochemistry using standard techniques (Chesselet et al., 1995). Tissue sections were brought to room temperature from −70°C under a stream of cool air, postfixed in 3% paraformaldehyde (containing 0.02% diethyl pyrocarbonate), acetylated, and dehydrated. Sections were incubated with radiolabeled probe (6–8 ng/section, 4 × 105 dpm/ng) in 4 × SSC containing 1 mg/mL salmon sperm DNA, 40% formamide, 8% dextran sulfate, 1 mg/mL tRNA, 10 mM DTT, 1 × Denhardt's solution. Hybridization was performed in humid chambers at 50°C for 3.5 h, which were empirically determined to be the optimal conditions for the Bax probe. Following hybridization, slides were washed in 50% formamide/2 × SSC (0.3 M NaCl/0.03 M sodium citrate) at 52°C, incubated with 100 μg/mL RNase A (30 min at 37°C) and rinsed in 2 × SSC/0.01% Triton X-100. Sections were dehydrated in a graded series of ethanols, defatted in xylene and desiccated. As control, adjacent sections were incubated with 100-fold excess of unlabeled cRNA for 60 min at 50°C, followed by incubation with the radio-labeled probe.
The sections were coated with Kodak NTB2 autoradiographic emulsion, developed, and counterstained with hematoxylin and eosin. In preliminary experiments, sections were exposed to emulsion for 3, 4, 5, 6, or 8 weeks, and it was determined that a 5-week exposure provided the optimal signal-to-noise ratio. Quantification of cellular grain density was therefore performed on sections that had been exposed to emulsion for 5 weeks. Cellular grain density was evaluated in the injured cortex, and area CA3 from two adjacent sections in each brain between 4.8 and 5.3 mm posterior to bregma, using an image analysis system (MCID, Ontario, Canada). Results were presented as mean number of grains per cell, using 50 cells per brain region. Background grain density was determined to be approximately 5 grains per cell in sections that were subjected to the hybridization protocol but were not incubated with radiolabeled cRNA, or in sections that were incubated with unlabeled cRNA prior to the hybridization with labeled cRNA.
Statistical Analysis
Intensities of bands from densitometric analysis of immunoblots and neuronal grain density in in situ hybridization experiments were analyzed using a one-way analysis of variance followed by a post-hoc Dunnett's test for comparison between sham-injured and brain-injured samples at the different time points. A p value of <0.05 was considered to be significant.
RESULTS
Pattern of Cortical and Hippocampal Cell Loss in the Injured Brain
Surgical preparation of rats did not result in overt damage to the cortex or hippocampus (Fig. 1A). No overt loss of Toluidine blue staining in any brain region was visible at 2 h following lateral fluid-percussion brain injury of moderate severity (Fig. 1B). By 6 h, decreased Nissl staining was observed in the cortex (data not shown), which had evolved into an overt lesion by 24 h post-injury (Fig. 1C, arrowheads), and by 7 days, intense Nissl stain in the injured cortex suggested that extensive gliosis had occurred (Fig. 1D, arrowhead). Hippocampal cell loss was restricted to the area CA3, and was observed at both 24 h (Fig. 1C, arrow) and 7 days (Fig. 1D, arrow) post-injury. No cell loss was observed in any region of the contralateral hemisphere (data not shown).
FIG. 1.

Histopathological alterations following lateral fluid-percussion (FP) brain injury. Brain sections (40 μm thick) were mounted on gelatin-coated glass slides and stained with Toulidine Blue. Representative sections from a sham-injured animal (A), at 2 h post-injury (B), 24h post-injury (C), and 7 days post-injury (D) are shown. Note the lack of overt cell loss in any brain region at 2 h post-injury. Arrowheads in C denote the cortical contusion that is typically observed at 24 h post-injury and in D denote the intense glial scar that is formed at 7 days post-injury. Arrows in C and D denote the selective loss of pyramidal cells in the CA3 region of the hippocampus. Boxes represent the areas of the cortex and hippocampus that are illustrated at higher magnification in Figures 2-4 and 6. Bar = 400 μm.
Alterations in Bcl-2 and Bax Immunoreactivities following Brain Injury
2 h post-injury
In the temporal cortex of sham animals, neurons appeared round and healthy with Toulidine Blue staining visible in the cytoplasm and nucleolus (Fig. 2A). By 2 h post-injury, neurons in the injured cortex appeared dark and shrunken (Fig. 2B), suggestive of damage, and at 6 h post-injury, an overt but mild loss of Toluidine Blue staining was observed (Fig. 2C). In sham animals, Bcl-2 and Bax immunoreactivities were observed as diffuse patterns of staining in the perinuclear cytoplasm including neuronal processes (Bcl-2, Fig. 2D; Bax, Fig. 2G). No immunoreactive material was observed when sections were either preincubated with immunizing antigen peptides, or when primary antibody was omitted from the protocol (data not shown). At 2 and 6 h following moderate brain injury, neurons in the cortex were characterized by a dramatic loss in cellular Bcl-2 immunoreactivity (Fig. 2E,F). In contrast to the pronounced decrease in cellular Bcl-2 staining, Bax IR in the same region (observed in adjacent sections) decreased to a very mild extent at 2 h post-injury (Fig. 2H). By 6 h, however, most neurons in the injured cortex no longer stained for Bax protein (Fig. 2I). Changes in Bcl-2 and Bax IR in injured brain regions at 2 h following injury appeared to precede loss of Toluidine Blue stain in affected cells (compare Fig. 2B,C with Fig. 2E,F,I).
FIG. 2.

Bcl-2 and Bax immunoreactivities in the cortex following lateral fluid-percussion brain injury. Photomicrographs from representative serial sections from sham-injured animals (A,D,G) and brain-injured animals at 2 h (B,E,H) and 6 h (C,F,I) post-injury stained with Toluidine Blue (A–C), and, anti-Bcl-2 (D–F) and anti-Bax (G–I) antibodies. At 2 h post-injury, Toluidine Blue staining revealed only shrunken neurons (B), but a dramatic loss of Bcl-2(+) cells (E), and mild loss of Bax(+) cells (H). At 6 h post-injury, mild but overt loss of Toluidine Blue staining was observed (C), which was accompanied by dramatic losses of Bcl-2(+) (F) and Bax(+) (I) cells. Bar = 100 μm.
Pyramidal neurons in the hippocampal CA3 region in sham animals appeared round with Toluidine Blue staining primarily in the nucleolus and cytoplasm (Fig. 3A). These neurons were immunoreactive for both Bcl-2 (Fig. 3F) and Bax (Fig. 3K). At 2 h (Fig. 3B) and 6 h (Fig. 3C) post-injury, Toluidine Blue staining revealed that the neurons in the CA3 region of the hippocampus ipsilateral to the site of impact appeared shrunken and elongated, suggestive of damage. In addition, an overt reduction in the number of Bcl-2 immunoreactive neurons was observed at both 2 and 6 h post-injury (Fig. 3G,H. At 2 h post-injury, the reduction in Bax immunoreactive cells was not as extensive as that observed for Bcl-2 (Fig. 3L), and areas of the pyramidal cell layer that did not contain Bax(+) cells were observed (Fig. 3L, arrow). However, in cells that did stain for Bax protein (Fig. 3L, arrowhead), Bax immunoreactive material was observed only in the cytoplasm (Fig. 3L, inset). By 6 h post-injury, an overt loss of Bax immunoreactive cells in the CA3 region was observed (Fig. 3M). The early loss of Bcl-2 immunoreactive cells (at 2 h post-injury) in the CA3 region occurred prior to that of Bax immunopositive cells (at 6 h post-injury), and prior to loss of Toluidine Blue staining, which was observed at 24 h post-injury (Fig. 4E).
FIG. 3.

Bcl-2 and Bax immunoreactivities in the hippocampus following lateral fluid-percussion brain injury. Photomicrographs from representative serial sections from the CA3 region of sham-injured animals (A,F,K) and brain-injured animals at 2 h (B,G,L) and 6 h (C,H,M) post-injury, stained with Toluidine Blue (A–C), and anti-Bcl-2 (F–H) and anti-Bax (K–M) antibodies. Photomicrographs from representative sections illustrating the hilus of the dentate gyrus in sham-injured animals (D,I,N) and at 2 h post-injury (E,J,O) stained with Toluidine Blue (D,E), and, anti-Bcl-2 (I,J) and anti-Bax (N,O) antibodies. At 2 h (B) and 6 h (C) after brain injury, neurons in the CA3 region take on a shrunken elongated shape suggestive of damage. While a significant number of cells in this region lose Bcl-2 immunoreactivity at 2 h (G) and 6 h (H), only a few cells lose Bax immunoreacivity at 2 h (L, arrow). Bax(+) cells in the CA3 region at 2 h post-injury (L, arrowhead) stain in a pattern similar to that in sham animals, with diffuse staining in the cytoplasm (L, inset). At 6 h post-injury, however, the number of cells that do not stain for Bax protein have increased (M). Bar = 100 μm (all panels) 170 μm (inset in L).
FIG. 4.

Bcl-2 and Bax immunoreactivities at 24 h and 7 days following lateral fluid-percussion brain injury. Adjacent brain sections (each 40 μm thick) from sham-injured rat brains were subjected to immunohistochemistry for Bcl-2 and Bax and Toluidine Blue staining. Representative photomicrographs illustrate sustained loss of Bcl-2 immnoreactivity in the cortex at 24 h post-injury (A), and a partial loss of Bax expression in the site of maximal injury (lower left hand corner in B). However, cells intensely immunoreactive for Bax were observed in the cortex surrounding the site of maximal injury (B, arrowheads). By 7 days, Toluidine Blue–stained sections reveal a glia-filled contusion (C); these glial cells do not appear to be immunoreactive for Bax (D). At 24 h following brain injury, a significant loss of CA3 neurons was observed (E), which was accompanied by a loss of cellular Bax immunoreactivity (F). Maximal loss of CA3 neurons was observed in Toluidine Blue–stained sections at 7 days post-injury (G). Scale bar = 100 μm (A–D), 70 μm (E–G).
Toluidine Blue–positive neurons were observed in the hilus of the dentate gyrus in both sham animals (Fig. 3D) and at 2 h post-injury (Fig. 3E). In sham animals, Bcl-2(+) and Bax(+) neurons were present in the hilar region (Bcl-2, Fig. 3I; Bax, Fig. 3N). In contrast to the injured cortex and hippocampal CA3 regions, where loss of Bcl-2 immunoreactivity in cells appeared to precede loss of Bax staining, neurons in the hilus of the dentate gyrus in the injured hemisphere appeared to lose Bcl-2 (Fig. 3J) and Bax (Fig. 3O) immunoreactivity.
24 h post-injury
The area of the cortex exhibiting reduced cellular Bcl-2 IR at 2 h corresponded to that of maximal loss of Toluidine Blue staining observed at 24 h post-injury (Fig. 4A). In cells surrounding this lesion, Bax immunoreactivity appeared to be more intense compared to that in sham-injured animals (Fig. 4B, arrowheads). At 24 h post-injury, overt neuronal loss was also observed in area CA3 of the injured hippocampus (Fig. 4E), corresponding to the region which exhibited decreased cellular Bcl-2 IR at 2 h post-injury. Cellular Bax IR in the CA3 region of the hippocampus, which was relatively unaffected at 2 h, was reduced by 24 h post-injury (Fig. 4F).
7 days post-injury
By 7 days, the injured cortex contained primarily glia that stained with Toluidine Blue (Fig. 4C), but were not Bax-immunopositive (Fig. 4D). Hippocampal CA3 cell loss was almost complete at this time point post-injury (Fig. 4G).
No alterations in Bcl-2 or Bax IR were observed at any time post-injury in any region of the brain contralateral to the site of impact (data not shown).
Western Blot Analysis of Bcl-2 and Bax Protein Expression
The antibodies to Bcl-2 and Bax used in the immunohistochemical analyses generated single bands at 26 and 21 kDa, respectively, on immunoblots of Hela cell extracts (Fig. 5). In cortical and hippocampal homogenates, the most intensely stained bands comigrated with the 26-kDa (for Bcl-2) and 21-kDa (for Bax) bands observed with HeLa cell extracts. Minor, less intense bands were observed at higher molecular weight ranges, but did not appear to be different between sham and injured brain tissue (data not shown).
FIG. 5.
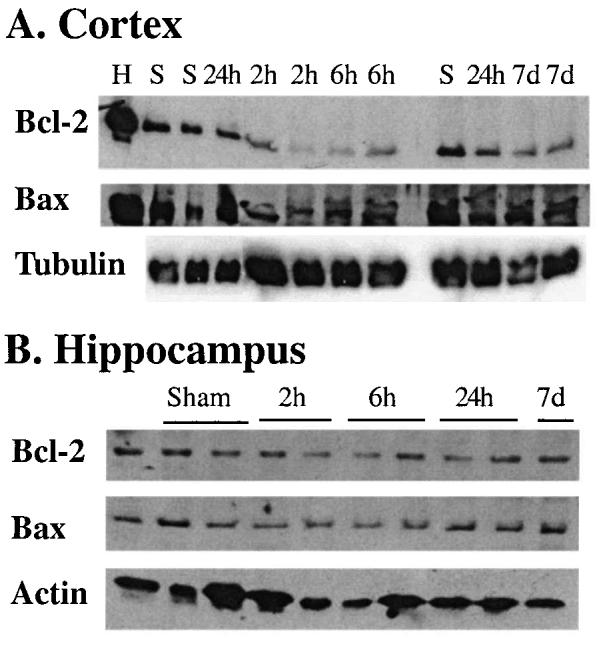
Immunoblot analysis of Bcl-2 and Bax levels in extracts from cortex (A) and hippocampus (B) following lateral fluid-percussion brain injury. Representative immunoblots of Bcl-2 and Bax expression in cortical and hippocampal extracts of sham-injured and of brain-injured animals at 2 h, 6 h, 24 h, and 7 days. In the immunoblot experiment shown here, blots were probed with anti-Bcl-2 antibody, stripped and reprobed with the anti-Bax antibody, and finally stripped and reprobed with either the anti-actin or the β-tubulin antibody. Only some samples from sham- and brain-injured rats are illustrated here in order to accommodate all samples on a single gel. H, HeLa cell extract.
Cortex
A modest decrease in the relative abundance of the Bcl-2 protein band was observed in cortical homogenates taken at 2 h and 6 h post-injury (Fig. 5A). This decrease was also evident on semi-quantitative densitometric analysis, where the average intensity of the bands (normalized for equal loading using the intensity of actin) decreased by almost 60% (p < 0.05 compared to sham-injured tissue; Table 1). The intensity of the Bcl-2 band in the injured cortex at 24 h and 7 days post-injury was statistically not different from that observed for sham animals (Table 1). The intensity of Bax staining in immunoblots of cortical samples was only mildly decreased at 2 h post-injury (to approximately 30% of sham controls), but appeared to recover to near control levels by 6 h and increased significantly by almost 30% at 24 h post-injury (p < 0.05 compared to sham controls; Fig. 5A and Table 1). By 7 days post-injury, tissue levels of Bax was not different from that in sham-injured animals (Fig. 5A and Table 1). A significant increase in Bax/Bcl-2 ratio was detected at all times post-injury (Table 1; p < 0.05 compared to the ratio in sham-injured rat brain). No differences in tissue levels for either Bcl-2 or Bax were observed in the contralateral hemisphere (data not shown).
Table 1.
Semi-Quantitative Analysis of Bcl-2 and Bax Protein Levels in Homogenates of Cortex and Hippocampus Taken from Hemispheres Ipsilateral and Contralateral to the Impact Site
|
Relative protein abundance |
|||
|---|---|---|---|
| Time | Bcl-2 | Bax | Ratio (Bax:Bcl-2) |
| Cortex | |||
| Sham | 0.35 ± 0.04 | 0.56 ± 0.11 | 1.62 ± 0.10 |
| 2 h | 0.13 ± 0.08* | 0.37 ± 0.15* | 2.28 ± 0.13* |
| 6 h | 0.16 ± 0.07* | 0.43 ± 0.08 | 2.32 ± 0.16* |
| 24 h | 0.28 ± 0.10 | 0.75 ± 0.05* | 2.13 ± 0.22* |
| 7 days | 0.29 ± 0.07 | 0.66 ± 0.07 | 2.05 ± 0.13* |
| Hippocampus | |||
| Sham | 0.42 ± 0.03 | 0.38 ± 0.03 | 0.95 ± 0.03 |
| 2 h | 0.38 ± 0.10 | 0.45 ± 0.08 | 0.91 ± 0.05 |
| 6 h | 0.40 ± 0.07 | 0.42 ± 0.05 | 0.90 ± 0.05 |
| 24 h | 0.45 ± 0.16 | 0.54 ± 0.12 | 0.98 ± 0.10 |
| 7 days | 0.42 ± 0.10 | 0.50 ± 0.16 | 0.91 ± 0.02 |
Intensities of protein bands corresponding to Bcl-2 (26 kDa) and Bax (21 kDa) were measured at the indicated times post-injury using computer-assisted densitometric analysis, normalized to the intensity of β-actin or tubulin, and compared to similar values from sham-injured tissue. Intensities of Bcl-2 and Bax were also compared to each other and presented as a ratio at each time point. Statistical analyses were performed using one-way ANOVA followed by a post-hoc Dunnett's test. *p < 0.05 compared to sham-injured values.
Hippocampus
The abundances of Bcl-2 and Bax proteins in hippocampal homogenates from injured animals taken at either 2 h, 6 h, 24 h, or 7 days post-injury was not significantly different from those in sham-injured animals (Fig. 5B).
In Situ Hybridization for Bax and Bcl-2 mRNAs
In situ hybridization histochemistry revealed that both Bax mRNA (Fig. 6A) and Bcl-2 mRNA (Fig. 6C) were constitutively expressed in the cortex of the sham-injured brain. Expression for both Bax and Bcl-2 mRNA was at a low level (8–12 grains per cell) albeit higher than background (typically 3–5 grains per cell). Qualitative analysis was suggestive of a mild increase in cellular Bax mRNA labeling in the cortex at 24 h post-injury (Fig. 6B), but not at 3 days post-injury (data not shown). In contrast, Bcl-2 mRNA expression in injured animals at 24 h post-injury did not appear to be different from that in sham-injured animals (compare labeled cells in Fig. 6C,D). Quantitative grain counting of Bax-labeled cells revealed a 20% increase in the number of grains per cell at the 24 h time-point (Table 2; p < 0.05 compared to sham-injured brains). Both Bax and Bcl-2 mRNAs were present in cells in the CA3 region of the hippocampus of sham-injured animals (data not shown), and no alterations in cellular grain density was observed at any time post-injury (Table 2).
FIG. 6.
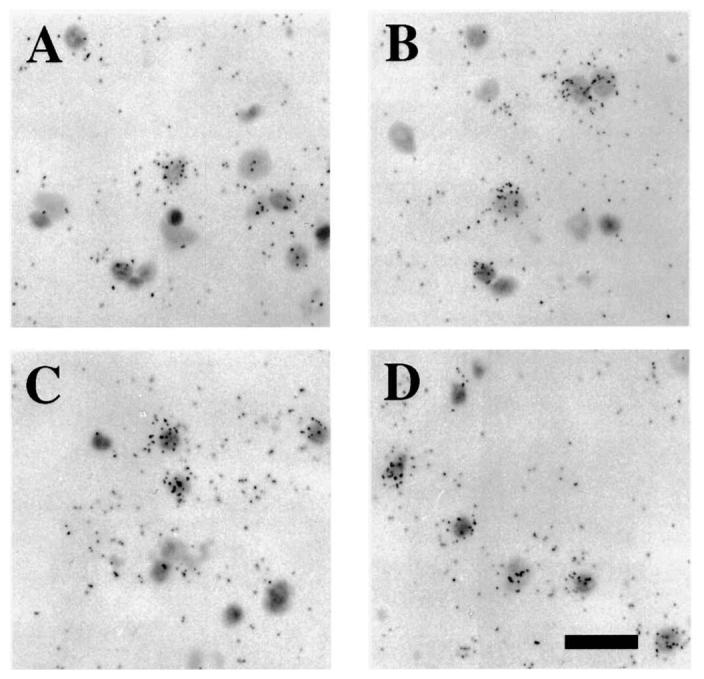
In situ hybridization histochemistry of Bax and Bcl-2 mRNA expression following lateral fluid-percussion brain injury. Representative bright field photomicrographs illustrating cellular localization of Bax mRNA (A,C) and Bcl-2 mRNA (B,D) in the temporal cortex of sham-injured rat brains (A,B), and at 24 h (C,D) following brain injury. Note accumulation of grains corresponding to Bax mRNA over cell bodies in the cortex at 24 h post-injury (C). Constitutive expression of Bcl-2 mRNA was observed in cortical cells in sham-injured rats (B), which did not appear to change at 24 h post-injury (D). Bar = 170 μm.
Table 2.
Quantitative Analyses of Bcl-2 and Bax mRNA Expression in Tissue Sections of Sham and Brain-Injured Rats
|
Cellular grain density (grains per cell) |
||
|---|---|---|
| Time | Bax | Bcl-2 |
| Cortex | ||
| Sham | 8 ± 2 | 12 ± 3 |
| 1 day | 14 ± 1* | 11 ± 4 |
| 3 days | 10 ± 3 | 13 ± 3 |
| Hippocampus | ||
| Sham | 14 ± 4 | 9 ± 1 |
| 1 day | 16 ± 3 | 8 ± 4 |
| 3 days | 17 ± 1 | 10 ± 3 |
Up to 50 cells containing a minimum of 5 grains (distributed over the cell body) were counted in the temporal cortex, and, in the CA3 region of hippocampus between 4.8 mm and 5.3 mm posterior to Bregma. For each probe, two adjacent brain sections (12 μm thick) were used, and the mean cellular grain density was obtained. Values presented represent the mean of two separate experiments. Statistical analyses were performed using one-way ANOVA followed by a post-hoc Dunnett's test. *p < 0.05 compared to sham-injured brains.
DISCUSSION
The data presented in this report suggest that the regional alterations in the cellular levels of Bcl-2 family of apoptosis-associated genes following lateral fluid-percussion (FP) brain injury may be associated with traumatic cell death. Lateral FP brain injury resulted in a marked loss of Bcl-2 immunoreactive cells in the injured cortex and hippocampus by 2 h following the initial trauma. Loss of Bcl-2 staining was observed in neurons of the parieto-temporal cortex, and area CA3 of the ipsilateral hippocampus in the absence of overt loss of staining for the pro-cell death protein, Bax. At 24 h post-injury, an increase in Bax mRNA and protein was observed in neurons in the peri-injured cortex. These observations suggest that neuronal survival may be compromised due to decreased expression of Bcl-2 and a concomitant increase in Bax levels, and support the “apostat” mechanism of cell death, which suggests that a shift in the cellular ratio of cell death activator proteins (such as Bax, Bad, Bcl-xS, c-Jun N-terminal kinase) and suppressor proteins (such as Bcl-2, Bcl-xL, extracellular signal regulated kinase) proteins regulates the fate of a cell (Oltvai et al., 1993; Xia et al., 1995).
The decrease in intensity of the Bcl-2 band at 2 and 6 h post-injury and the increase in intensity of the Bax band at 24 h post-injury in cortical homogenates, albeit mild to modest, appeared to confirm respectively, the decreased cellular Bcl-2 immnunoreactivity in the injured cortex (Fig. 2E) and the increased cellular Bax immunoreactivity (Fig. 4B). However, despite a robust loss of immunohistochemical staining for Bcl-2 and Bax in the core of the lesion at 24 h post-injury, we did not observe a similar pattern in the immunoblots. Similarly, despite decreases in the numbers of Bcl-2(+) and Bax(+) cells in the hippocampus, and the ensuing pattern of cell loss which were restricted to the CA3 and the hilus (Fig. 1C,D), no change in the abundance of Bcl-2 and Bax was observed in immunoblots of hippocampal homogenates. These apparent discrepancies underscore the differences in sensitivities between immunohistochemical and immunoblot analyses of protein expression, and the focal nature of the trauma-induced pathology.
In humans, TBI results in a variety of pathological changes, including cortical contusions, neuronal loss in the hippocampus and thalamus, and axonal injury in the acute (hours to days) period following injury (Adams et al., 1985; Gennarelli, 1997; Kotapka et al., 1992; Ross et al., 1993). In the acute period (hours) following experimental TBI, neurodegenerative processes are initiated, as indicated by morphological alterations such as pyknosis and dystrophy, and cellular changes such as increased silver impregnation (Colicos et al., 1996; Hicks et al., 1996; Sutton et al., 1993). Using TUNEL to detect DNA fragmentation and light microscopy to evaluate morphology, we and others have observed apoptotic and necrotic neurons in the traumatically injured cortex (Conti et al., 1998; Fox et al., 1998; Newcomb et al., 1999; Rink et al., 1995). The results in the present study suggest that loss of cortical Bcl-2 reactivity as early as 2 h post-injury appeared to precede overt cell loss. Furthermore, while Bax protein reactivity was only modestly decreased in the in the core of the lesion at 2 h post-injury with a more substantial decrease by 6 h post-injury, Bax mRNA and protein were upregulated in cortical neurons surrounding the lesion at 1 day post-injury. Although not tested in the present study, alterations in expression of Bax and Bcl-2 mRNA may occur at post-injury times prior to 24 h. However, the present findings appear to support the observations of an expanding cortical lesion reported by Smith et al. (1997) and lead to the speculation that neurodegeneration following TBI is an active process and that proteins associated with cell death may participate in this cascade.
Our observations of decreased Bcl-2 immunoreactivity prior to decreases in Bax staining in hours following brain trauma support the suggestion that concomitant changes in intracellular expression or ratio of Bcl-2, Bcl-xL, and Bax may participate in the pathology of neuronal injury. Deprivation of nerve growth factor leading to programmed cell death in cultures of superior cervical ganglion cells resulted in a disappearance of neuronal Bcl-2, immedately prior to the appearance of DNA fragmentation (Greenlund et al., 1995). In models of axotomy or cerebral ischemia, decreased immunoreactivity for Bcl-2 and Bcl-xL and increased Bax immunoreactivity was observed in neurons in the injured regions that also labeled for DNA fragmentation (Gillardon et al., 1996a,b). Decreased Bcl-2 immunoreactivity and a concomitant upregulation of bax mRNA was also observed in the selectively vulnerable cells of the hippocampus (areas CA3 and CA4) 24–48 h after systemic administration of kainic acid (Gillardon et al., 1995). In contrast to these observations, an increase in both Bcl-2 mRNA expression and protein immunoreactivity was observed in neurons, glia and endothelial cells that survived an ischemic injury (Chen et al., 1995, 1997b). Increased expression of both Bcl-2 mRNA and protein has been observed in cortical neurons surrounding the contused cortex following cortical impact brain injury (Clark et al., 1997). However, this increase was observed only when brain trauma was accompanied by a subsequent hypoxemic injury, and in that study the effect of brain trauma alone on Bcl-2 expression was not discussed (Clark et al., 1997). Our observations that Bcl-2 mRNA or protein did not increase at any time post-injury underscore the complex nature of the cellular responses to mechanical injury and importantly highlight the differences in cellular responses to trauma and hypoxia-ischemia.
Upregulation of bax mRNA and protein has been observed in the selectively vulnerable neurons of the hippocampus, prior to cell death following global ischemia (Chen et al., 1996; Honkaniemi et al., 1996; Krajewski et al., 1995). Similarly, at 3–6 days following administration of the neurotoxin, MPTP, Bax expression (mRNA and protein) was increased in the neurons of the substantia nigra that were destined to die (Hassouna et al., 1996). Importantly, Kaya et al. (1999) have observed increased immunoreactivity for Bax in apoptotic granule cells of the denatate gyrus following controlled cortical impact injury. In contrast to the observations of Kaya et al. (1999), we observed an increase in Bax mRNA and protein only in the injured cortex and not in the hippocampus. At this time, the reason for this regional heterogenity in trauma-induced Bax alterations in not clear, but does underscore the regional differences in cellular responses to TBI. The tumor-suppressor gene, p53, has been implicated as a transcription factor controlling the expression of Bax (Miyashita and Reed, 1995). Recent evidence suggests that p53 expression may be upregulated in the injured cortex and hippocampus following experimental brain injury (Kaya et al., 1999; Napieralski et al., 1999). Although neuronal death following CNS injury has been associated with changes in expression of Bcl-2 and Bax genes, in vitro models of apoptotic cell death indicate that an alteration in the cellular localization of these proteins could result in cell death (Green and Reed, 1998). In response to a death signal, Bax may translocate to the mitochondria and may be associated with a release of Bcl-2 and cytochrome c from the inner mitochondrial membranes (Jurgensmeier et al., 1998). Recent preliminary data suggest that while cytochrome c is present in the soluble fraction of cortical cells as early as 2 h post-injury (Fulp et al., 2000), Bax levels in the mitochondrial fractions are not significantly altered at 3 h post-injury (Lifshitz et al., 2003). In our analyses of the nature of the immunohistochemical staining with antibodies to Bcl-2 and Bax, we did not observe the punctate intracellular labeling characteristic of mitochondria. In fact, both Bcl-2 and Bax immunoreactivities were characterized by a diffuse, perinuclear cytoplasmic pattern. These results do not preclude differential post-traumatic subcellular localization of Bcl-2 proteins, and may reflect the inability of the particular antibody used in our studies to detect these intracellular alterations.
The acute loss of Bcl-2 immunoreactivity in the injured brain regions following lateral FP brain injury was similar to that observed for cytoskeletal proteins such as MAP-2 and the neurofilament proteins (Hicks et al., 1994; Posmantur et al., 1996). Within 3 h of the initial trauma, immunoreactivity of MAP-2 and the 68-kDa neurofilament protein has been reported to significantly decline in the dendrites of neurons in the injured cortex and hippocampus. Moreover, the neurons in the denate hilar region of the hippocampus appear to be uniquely sensitive to a traumatic injury (Lowenstein et al., 1992) and lose immunoreactivity for MAP-2 and neurofilaments within 2 h post-injury (Hicks et al., 1994; Saatman et al., 1998). Our observations of a loss of both Bax and Bcl-2 immunoreactive material in hilar neurons appear to further support the sensitvity of these neurons to TBI. The proteolytic changes may be indicative of, or contribute to, neuronal damage. In this regard, both the calcium-activated protease, calpain, and the cell death–associated protease, caspase-3, have been observed to be activated following TBI in the rat (Clark et al., 2000; Newcomb et al., 1997; Pike et al., 1998; Posmantur et al., 1996; Saatman et al., 1996; Yakovlev et al., 1997). While it is not known if Bcl-2 can undergo calpain-mediated proteolysis, a recent report suggested that Bcl-2 may be a substrate for caspase-3, and that proteolysis of Bcl-2 resulted in its conversion to a Bax-like, pro-cell death protein (Cheng et al., 1997). Whatever the mechanism, the concomitant loss of cellular survival proteins such as Bcl-2, and the memebrane cytoskeleton proteins such as MAP-2 and neurofilament may irreparably damage neurons.
In addition to its role as an anti-apoptotic protein during development, Bcl-2 is rather unique in that it can prevent cell death caused by multiple pathways (Greenlund et al., 1995; Zhong et al., 1993). Overexpression of Bcl-2 in neuronal cultures has been reported to prevent necrotic and apoptotic cell death in vitro induced by calcium ionophores, glutamate, hydrogen peroxide, and hypoxia (Kane et al., 1993, 1995; Zhong et al., 1993). Furthermore, Bcl-2 has been reported to attenuate rises in intracellular calcium, presumably by increasing the capacity of mitochondria to take up calcium (Marin et al., 1996; Murphy et al., 1996). Because the acute neurochemical alterations following experimental brain injury include an increase in intracellular calcium, and generation of free radicals leading to oxidative damage (Chan et al., 1995; Fineman et al., 1993; Nilsson et al., 1996), it is tempting to speculate that the acute disappearance of Bcl-2 in injured neurons may exacerbate their vulnerability to trauma. One component of the chronic pathology reported in models of TBI includes secondary axotomy (Povlishock et al, 1992); overexpression of Bcl-2 leads to neuroprotection following axotomy in vivo (de Bilbao and Dubois-Dauphin, 1996). Loss of Bcl-2 IR may also explain, in part, the relative inability of the injured brain to exhibit signs of regeneration (Chen et al., 1997a).
Our data suggest that strategies to reverse the decrease in cellular Bcl-2 levels and/or block the increase in Bax mRNA (Gillardon et al., 1996) may prevent, in part, TBI-induced neurodegeneration. In this regard, we have recently reported that mice overexpressing human Bcl-2 in neurons have a markedly reduced cortical contusion following experimental brain injury (Raghupathi et al., 1998; Nakamura et al., 1999). Cell death following TBI can occur due to the activation of multiple pathways which include the differential expression of the Bcl-2 family of genes. Control of the expression of these genes in neurons may confer resistance to trauma-induced death, thereby attenuating behavioral morbidity.
ACKNOWLEDGMENTS
We would like to thank Joshua Leitner, Seamus Fernandez, and Suzanna de Vries for their expert technical assistance and Jeanne Marks for careful preparation of this manuscript. We would like to acknowledge Dr. Diane Merry for her thoughtful comments on the manuscript. These studies were supported, in part, by grants from the NINDS (P50-NS08803, R01-NS26818, and R01NS41561), the NIGMS (R01-GM34690), and a Merit Review from the VA.
REFERENCES
- ADAMS JH, DOYLE D, GRAHAM DI, et al. The contusion index: a reappraisal in human and experimental non-missile injury. Neuropathol. Appl. Neurobiol. 1985;11:299–308. doi: 10.1111/j.1365-2990.1985.tb00027.x. [DOI] [PubMed] [Google Scholar]
- BOISE LH, GONZALEZ-GARCIA M, POSTEMA CE, et al. bcl–x, a bcl-2–related gene that functions as a dominant regulator of apoptic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- BRAMLETT HM, DIETRICH WD, GREEN EJ, et al. Chronic histopathological consequences of fluid-percussion brain injury in rats: effects of posttraumatic hypothermia. Acta Neuropathol. 1997;93:190–199. doi: 10.1007/s004010050602. [DOI] [PubMed] [Google Scholar]
- BREDESEN DE. Neural apoptosis. Ann. Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- CHAN PH, EPSTEIN CJ, LI Y, et al. Transgenic mice and knockout mutants in the study of oxidative stress in brain injury. J. Neurotrauma. 1995;12:815–824. doi: 10.1089/neu.1995.12.815. [DOI] [PubMed] [Google Scholar]
- CHEN J, GRAHAM SH, CHAN PH, et al. Bcl-2 is expressed in neurons that survive focal ischemia in the rat. NeuroReport. 1995;6:394–398. doi: 10.1097/00001756-199501000-00040. [DOI] [PubMed] [Google Scholar]
- CHEN J, ZHU RL, NAKAYAMA M, et al. Expression of the apoptosis-effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J. Neurochem. 1996;67:1–8. doi: 10.1046/j.1471-4159.1996.67010064.x. [DOI] [PubMed] [Google Scholar]
- CHEN DF, SCHNEIDER GE, MARTINOU J-C, et al. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature. 1997a;385:434–439. doi: 10.1038/385434a0. [DOI] [PubMed] [Google Scholar]
- CHEN J, GRAHAM SH, NAKAYAMA M, et al. Apoptosis repressor genes Bcl-2 and Bcl-x-long are expressed in the rat brain following global ischemia. J. Cereb. Blood Flow Metab. 1997b;17:2–10. doi: 10.1097/00004647-199701000-00002. [DOI] [PubMed] [Google Scholar]
- CHENG EHY, KIRSCH DG, CLEM RJ, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1969. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- CHESSELET M-F, MACKENZIE L, EBERLE-WANG K. In situ hybridization with 35S- and digoxigenin-labeled RNA probes. In: Henderson Z, editor. In Situ Hybridization Techniques for the Brain. Wiley; New York: 1996. pp. 79–110. [Google Scholar]
- CHITTENDEN T, HARRINGTON EA, O'CONNOR R, et al. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 1995;374:733–736. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- CLARK RSB, CHEN J, WATKINS SC, et al. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J. Neurosci. 1997;17:9172–9182. doi: 10.1523/JNEUROSCI.17-23-09172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK RSB, KOCHANEK PM, WATKINS SC, et al. Caspase-3–mediated neuronal cell death after traumatic brain injury in rats. J. Neurochem. 2000;74:740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- COLICOS MA, DASH PK. Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res. 1996;739:120–131. doi: 10.1016/s0006-8993(96)00824-4. [DOI] [PubMed] [Google Scholar]
- COLICOS MA, DIXON CE, DASH PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111–119. doi: 10.1016/s0006-8993(96)00819-0. [DOI] [PubMed] [Google Scholar]
- CONTI AC, RAGHUPATHI R, TROJANOWSKI JQ, et al. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J. Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE BILBAO F, DUBOIS-DAUPHIN M. Time course of axotomy-induced apoptotic cell death in facial motoneurons of neonatal wild type and bcl-2 transgenic mice. Neuroscience. 1996;71:1111–1119. doi: 10.1016/0306-4522(95)00505-6. [DOI] [PubMed] [Google Scholar]
- DECKWERTH TL, ELLIOTT JL, KNUDSON CM, et al. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- DIETRICH WD, ALONSO O, HALLEY M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J. Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- FINEMAN I, HOVDA DA, SMITH M, et al. Concussive brain injury is associated with a prolonged accumulation of calcium: a 45Ca autoradiographic study. Brain Res. 1993;624:94–102. doi: 10.1016/0006-8993(93)90064-t. [DOI] [PubMed] [Google Scholar]
- FOX GB, FAN L, LEVASSEUR RA, et al. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J. Neurotrauma. 1998;15:599–614. doi: 10.1089/neu.1998.15.599. [DOI] [PubMed] [Google Scholar]
- FULP CT, RAGHUPATHI R, SIMAN R, et al. Traumatic brain injury induces upregulation of cytosol-localized procaspase-3 and mitochondrial-localized activated caspase-3. Soc. Neurosci. Abst. 2000;30 Abstr. 863.2. [Google Scholar]
- GENNARELLI TA. The pathobiology of traumatic brain injury. Neuroscientist. 1997;3:73–81. [Google Scholar]
- GILLARDON F, WICKERT H, ZIMMERMANN M. Up-regulation of bax and down-regulation of bcl-2 is associated with kainate-induced apoptosis in mouse brain. Neurosci. Lett. 1995;192:85–88. doi: 10.1016/0304-3940(95)11619-8. [DOI] [PubMed] [Google Scholar]
- GILLARDON F, KLIMASCHEWSKI L, WICKERT H, et al. Expression pattern of candidate cell death effector proteins Bax, Bcl-2, Bcl-x and c-Jun in sensory and motor neurons following sciatic nerve transection in the rat. Brain Res. 1996a;739:244–250. doi: 10.1016/s0006-8993(96)00829-3. [DOI] [PubMed] [Google Scholar]
- GILLARDON F, LENZ C, WASCHKE KF, et al. Altered expression of Bcl-2, Bcl-X, Bax, and c-Fos colocalizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Mol. Brain Res. 1996b;40:254–260. doi: 10.1016/0169-328x(96)00059-9. [DOI] [PubMed] [Google Scholar]
- GILLARDON F, ZIMMERMANN M, UHLMANN E, et al. Antisense oligonucleotides to bax mRNA promote survival of rat sympathetic neurons in culture. J. Neurosci. Res. 1996c;43:726–734. doi: 10.1002/(SICI)1097-4547(19960315)43:6<726::AID-JNR9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- GONZALEZ-GARCIA M, GARCIA I, DING L, et al. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc. Natl. Acad. Sci. USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREEN DR, REED JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- GREENLUND LJS, KORSMEYER SJ, JOHNSON EM., Jr. Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- HAN J, SABBATINI P, PEREZ D, et al. The E1b 19k protein blocks apoptosis by interacting with and inhibiting the p53–inducible and death-promoting Bax protein. Genes Dev. 1996;10:461–477. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- HASSOUNA I, WICKERT H, ZIMMERMAN M, et al. Increase in bax expression in substantia nigra following 1–methyl-4–phenyl-1,2,3,6–tetrahydropyridine (MPTP) treatment of mice. Neurosci. Lett. 1996;204:85–88. doi: 10.1016/0304-3940(96)12323-5. [DOI] [PubMed] [Google Scholar]
- HICKS RR, SMITH DH, McINTOSH TK. Temporal response and effects of excitatory amino acid antagonism on microtubule-associated protein 2 immunoreactivity following experimental brain injury. Brain Res. 1995;678:151–160. doi: 10.1016/0006-8993(95)00179-t. [DOI] [PubMed] [Google Scholar]
- HICKS RR, SOARES HD, SMITH DH, et al. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- HONKANIEMI J, MASSA SM, BRECKINRIDGE M, et al. Global ischemia induces apoptosis-associated genes in hippocampus. Mol. Brain Res. 1996;42:79–88. doi: 10.1016/s0169-328x(96)00121-0. [DOI] [PubMed] [Google Scholar]
- JURGENSMEIER JM, XIE Z, DEVERAUX Q, et al. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANE DJ, SARAFIAN TA, ANTON R, et al. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- KANE DJ, ORD T, ANTON R, et al. Expression of bcl-2 inhibits necrotic neural cell death. J. Neurosci. Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- KAYA SS, MAHMOOD A, LI Y, et al. Apoptosis and expression of p53 response proteins and cyclin D1 after cortical impact in rat brain. Brain Res. 1999;818:23–33. doi: 10.1016/s0006-8993(98)01204-9. [DOI] [PubMed] [Google Scholar]
- KORSMEYER SJ. Regulators of cell death. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- KOTAPKA MJ, GRAHAM DI, ADAMS JH, et al. Hippocampal pathology in fatal non-missile human head injury. Acta Neuropathol. 1992;83:530–534. doi: 10.1007/BF00310031. [DOI] [PubMed] [Google Scholar]
- KRAJEWSKI S, MAI JK, KRAJEWSKA M, et al. Upregulation of Bax protein levels in neurons following cerebral ischemia. J. Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIFSHITZ J, FRIBERG H, NEUMAR RW, et al. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: evidence for differentially sensitive populations in the cortex and hippocampus. J. Cereb. Blood Flow Metab. 2003;23:219–231. doi: 10.1097/01.WCB.0000040581.43808.03. [DOI] [PubMed] [Google Scholar]
- LOWENSTEIN DH, THOMAS MJ, SMITH DH, et al. Selective vulnerabilty of dentate hilar neurons following TBI: a potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 1992;12:4846–4853. doi: 10.1523/JNEUROSCI.12-12-04846.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARIN MC, FERNANDEZ A, BICK RJ, et al. Apoptosis suppression by bcl-2 is correlated with the regulation of nuclear and cytosolic Ca2+ Oncogene. 1996;12:2259–2266. [PubMed] [Google Scholar]
- McINTOSH TK, VINK R, NOBLE L, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- McINTOSH TK, JUHLER M, WIELOCH T. Novel pharmacological strategies in the treatment of experimental brain injury. J. Neurotrauma. 1998;15:731–769. doi: 10.1089/neu.1998.15.731. [DOI] [PubMed] [Google Scholar]
- MIYASHITA M, REED JC. Tumor suppressor, p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- MURPHY AN, BREDESEN DE, CORTOPASSI G, et al. Bcl-2 potentiates the maximum calcium uptake capacity of neural cell mitochondria. Proc. Natl. Acad. Sci. USA. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKAMURA M, RAGHUPATHI R, MERRY DE, et al. Overexpression of Bcl-2 is neuroprotective after experimental brain injury in transgenic mice. J. Comp. Neurol. 1999;412:681–692. doi: 10.1002/(sici)1096-9861(19991004)412:4<681::aid-cne9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- NAPIERALSKI JA, RAGHUPATHI R, McINTOSH TK. The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Mol. Brain Res. 1999;71:78–86. doi: 10.1016/s0169-328x(99)00155-2. [DOI] [PubMed] [Google Scholar]
- NEWCOMB JK, KAMPFL A, POSMANTUR RM, et al. Immunohistochemical study of calpain-mediated breakdown products to a-spectrin following controlled cortical impact injury in the rat. J. Neurotrauma. 1997;14:369–383. doi: 10.1089/neu.1997.14.369. [DOI] [PubMed] [Google Scholar]
- NEWCOMB JK, ZHAO X, PIKE BR, et al. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in rats. Exp. Neurol. 1999;158:76–88. doi: 10.1006/exnr.1999.7071. [DOI] [PubMed] [Google Scholar]
- NILSSON P, LAURSEN H, HILLERED L, et al. Calcium movements in traumatic brain injury: the role of glutamate receptor-operated ion channels. J. Cereb. Blood Flow Metab. 1996;16:262–270. doi: 10.1097/00004647-199603000-00011. [DOI] [PubMed] [Google Scholar]
- OLTVAI ZN, MILIMAN CL, KORSMEYER SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- POSMANTUR RM, KAMPFL A, LIU SJ, et al. Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. J. Neuropathol. Exp. Neurol. 1996;55:68–80. doi: 10.1097/00005072-199601000-00007. [DOI] [PubMed] [Google Scholar]
- POVLISHOCK JT, ERB DE, ASTRUC J. Axonal response to TBI: reactive axonal change, deafferentation, and neuroplasticity. J. Neurotrauma. 1992;9:S189–S200. [PubMed] [Google Scholar]
- RAGHUPATHI R, McINTOSH TK, SMITH DH. Cellular responses to brain injury. Brain Pathol. 1995;5:437–442. doi: 10.1111/j.1750-3639.1995.tb00622.x. [DOI] [PubMed] [Google Scholar]
- REED JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- RINK AD, FUNG KM, TROJANOWSKI JQ, et al. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am. J. Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- ROSS DT, GRAHAM DI, ADAMS JH. Selective loss of neurons from the thalamic reticular nucleus following severe human head injury. J. Neurotrauma. 1993;10:151–165. doi: 10.1089/neu.1993.10.151. [DOI] [PubMed] [Google Scholar]
- SAATMAN KE, BOZYCZKO-COYNE D, MARCY VR, et al. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J. Neuropathol. Exp. Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- SAATMAN KE, GRAHAM DI, McINTOSH TK. The neuronal cytoskeleton is at risk following mild and moderate brain injury. J. Neurotrauma. 1998;15:1047–1058. doi: 10.1089/neu.1998.15.1047. [DOI] [PubMed] [Google Scholar]
- SMITH DH, CHEN X-H, PIERCE JES, et al. Progressive atrophy and neuronal cell death for one year following experimental brain trauma in the rat. J. Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- STRAUSS KI, RAGHUPATHI R, KRAJEWSKI S, et al. Apoptosis-related genes are modulated in a model of traumatic brain injury. J. Neurotrauma. 1996;13:620. [Google Scholar]
- SUTTON RL, LESCAUDRON L, STEIN DG. Unilateral cortical contusion injury in the rat: vascular disruption and temporal development of cortical necrosis. J. Neurotrauma. 1993;10:135–149. doi: 10.1089/neu.1993.10.135. [DOI] [PubMed] [Google Scholar]
- THORNBERRY NA, LAZEBNIK Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- TILLY JL, TILLY KI, KENTON ML, et al. Expression of members of the bcl-2 gene family in the immature rat ovary: equine chorionic gonadotropin-mediated inhibition of granulosa cell apoptosis is associated with decreased bax and constitutive bcl-2 and bcl-xlong messenger ribonucleic acid levels. Endocrinology. 1995;136:232–241. doi: 10.1210/endo.136.1.7828536. [DOI] [PubMed] [Google Scholar]
- XIA Z, DICKENS M, RAINGEAUD J, et al. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- YAKOVLEV AG, FADEN AI. Molecular strategies in CNS injury. J. Neurotrauma. 1995;12:767–777. doi: 10.1089/neu.1995.12.767. [DOI] [PubMed] [Google Scholar]
- YAKOVLEV AG, KNOBLACH SM, FAN L, et al. Activation of CPP32–like caspases contributes to neuronal apoptosis and neurlogical dysfunction after traumatic brain injury. J. Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG E, ZHA J, JOCKEL J, et al. Bad, a heterodimeric partner for Bcl-Xl and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- YIN C, KNUDSON CM, KORSMEYER SJ, et al. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature. 1997;385:637–640. doi: 10.1038/385637a0. [DOI] [PubMed] [Google Scholar]
- ZHONG L-T, SARAFIAN T, KANE DJ, et al. Bcl-2 inhibits death of central neural cells induced by multiple agents. Proc. Natl. Acad. Sci. USA. 1993;90:4533–4537. doi: 10.1073/pnas.90.10.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]