

Abstract
During the past several years, major advances have been made in understanding how reactive oxygen species (ROS) and nitrogen species (RNS) participate in signal transduction. Identification of the specific targets and the chemical reactions involved still remains to be resolved with many of the signaling pathways in which the involvement of reactive species has been determined. Our understanding is that ROS and RNS have second messenger roles. While cysteine residues in the thiolate (ionized) form found in several classes of signaling proteins can be specific targets for reaction with H2O2 and RNS, better understanding of the chemistry, particularly kinetics, suggests that for many signaling events in which ROS and RNS participate, enzymatic catalysis is more likely to be involved than non-enzymatic reaction. Due to increased interest in how oxidation products, particularly lipid peroxidation products, also are involved with signaling, a review of signaling by 4-hydroxy-2-nonenal (HNE) is included. This article focuses on the chemistry of signaling by ROS, RNS, and HNE and will describe reactions with selected target proteins as representatives of the mechanisms rather attempt to comprehensively review the many signaling pathways in which the reactive species are involved.
Keywords: signaling, glutathione, thioredoxin, oxidants, reactive oxygen species, thiols, peroxide, nitric oxide, peroxynitrite, 4-hydroxynonenal, cysteine, hydrogen peroxide, protein tyrosine phosphatase, reactive nitrogen species, eNOS, iNOS, nNOS, soluble guanylate cyclase, cGMP, tyrosine nitration, fatty acid nitration, NO-heme, NO-metal complexes, nitrite, protein kinase C, ERK, JNK, p38MAPK, tyrosine kinase receptors, calcium
2. OVERVIEW OF SIGNALING BY REACTIVE SPECIES
Understanding of the roles of reactive oxygen species (ROS), reactive nitrogen species (RNS) and the lipid peroxidation product, 4-hydroxy-2-nonenal (HNE) in signaling has evolved rapidly during the last decade. This has been markedly helped by identification of the specific targets in signaling pathways. In previous reviews, we defined how ROS, H2O2 in particular, act as second messengers (1–3); however, with increased understanding of the chemistry of signaling by reactive species, we will now append the description to include the likelihood that, despite the possibility of non-enzymatic reactions that can be demonstrated in vitro, most ROS and RNS modifications of signaling proteins must be enzyme catalyzed to account for what is observable in the biological context. Nonetheless, there may be some non-enzymatic reactions that occur rapidly enough to account for the biological reaction. In this review, we will focus on how selected signaling proteins are modified by reactive species. Thus, many other signaling proteins that are known to be regulated by reactive species will not be mentioned here as they could take up a whole book (for example (4)).
Generally, second messengers are produced in cells in response to receptor activation; however, as is well documented for nitric oxide (·NO) acting as endothelial derived relaxing factor, some molecules can move from the cell of origin to act as a second messenger in another. Recently, H2O2 has been suggested to act in such a paracrine manner (5). Regardless, second messengers are generally short-lived, and act specifically on effectors to transiently alter their activity. The transient alteration of signaling proteins may be seconds as for some phosphorylation of proteins or may be minutes to hours long as in the synthesis of cyclins and their binding to cell cycle kinases and then degradation. Superoxide (O2·−), H2O2 and ·NO are generated upon receptor activation and are short-lived, as are other second messengers.
Superoxide and H2O2 are generated by a variety of oxidoreductases (6) by a leak of electrons to O2 from the mitochondrial electron transport chain at Complex III (7,8), redox cycling of quinones (9) and numerous other autooxidation reactions. Although cells can respond to generation of H2O2 regardless of its source, the stimulated production of H2O2 that is associated with physiological signaling is more likely due to the oxidoreductases known as NADPH oxidases (NOXs). The NOX isoforms called DuOXs can generate H2O2 directly (10); however, most H2O2 production, results from the dismutation of O2·− produced by most NOX isoforms, five of which are found in mammals (10). Recent reviews of the NOX and DuOX enzymes provide a thorough description of the components, assembly and functions of these important sources of ROS for both signaling and pathology (11–15).
Nitric oxide (·NO) is synthesized in cells through a 5-electron oxidation of L-arginine by members of a family of heme containing enzymes called nitric oxide synthases (NOS). A brief review of how ·NO production is regulated is provided later in this article; however, for a thorough analysis of NOS enzymology the reader is referred to extensive reviews by others (16–19).
In contrast with ·NO and H2O2, HNE is generated as a secondary product in lipid peroxidation. HNE binds covalently to proteins and can form stable adducts even when acting as a signaling molecule. In those cases, the reversible aspect of HNE signaling likely involves degradation and resynthesis of proteins. Although the turnover of HNE-modified proteins has not been extensively investigated, recent studies suggest that HNE marks proteins for both ubiquitin-dependent and ubiquitin-independent degradation (20)
This article concerns the chemistry of signaling by reactive species. For this purpose, the kinetics of reaction needs to be considered. This is particularly important, as non-enzymatic reactions that can be demonstrated with pure enzymes in vitro may not have fast enough kinetics to occur in a cell where competing reactions would prevent the reaction. Therefore, to produce a redox dependent modification of the enzyme, enzymatic catalysis would be required to allow that modification under physiological conditions. Nonetheless, there is clear precedent for some non-enzymatic processes in signaling by reactive species such as the binding of ·NO to the heme of the regulatory domain of soluble guanylate cyclase, resulting in activation and production of cyclic GMP (21). Non-enzymatic reactions such as Michael addition of HNE to protein thiolates may also be fast enough to account for HNE-modification.
On the other hand, it is possible that a species may be so reactive that it lacks any specificity. For example, the hydroxyl radical (·OH) has no specificity, as it reacts at nearly the rate of diffusion with almost any molecule. Hydroxyl radical production is clearly involved in pathological processes but the lack of specificity makes it useless as a second messenger. Similarly, production of peroxynitrous acid while certainly capable of modifying proteins and thereby interfering in signaling when the target is involved in a signaling pathway is more likely to lead only indirectly to cell signaling. In fact, while evidence will be presented that HNE causes signaling by altering proteins, it also is more likely involved in adaptive responses to oxidative stress than in the physiological signaling that occurs under non-stress conditions. In contrast, the regulated production of H2O2 and ·NO in response to receptor-mediated stimulation are clearly part of normal physiology.
3. ROLE OF THIOLS IN SIGNALING BY ROS
The small peptide, glutathione (GSH (γ-L-glutamyl-L-cysteinyl-glycine)) and the small protein, thioredoxin (Trx), which has two critical cysteine residues, are intimately involved in signaling by reactive species. This is in part due to the enzymatic reduction of H2O2 by GSH, which is catalyzed by the glutathione peroxidases and peroxiredoxin (Prdx) 6 (see (22) for review of the glutathione peroxidase family and (23) for a review of Prdx 6).
Nonetheless, GSH and glutathione disulfide (GSSG) content in the cell is also dependent upon cellular export of GSH and GSSG, GSH synthesis, disulfide exchange with proteins that is generally catalyzed by a protein disulfide isomerase (PDI) or glutaredoxin. PDIs and glutaredoxin both contain two cysteines in their active sites, referred to as the thioredoxin fold (CXXC), which is essential to their function:
| <1> |
Glutathione S-transferase (GST) catalyzed conjugation reactions, which are important in metabolism and detoxification, can also modulate signaling by catalyzing Michael addition of GSH to HNE (reaction <2>):
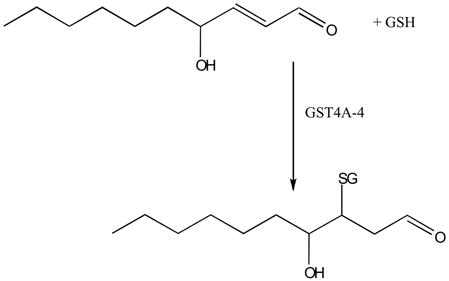 |
<2> |
Although glutathione could potentially react non-enzymatically in any of the reactions above, it would only do so in its thiolate form, GS−, which at pH 7.0 inside cells is 5 μM when GSH is 1 mM. With rate constants that are very slow for the non-enzymatic reactions, an enzyme that can catalyze each of these reactions is essential for any of them to proceed in a biological system.
The GSSG/2GSH ratio is a reflection of the total metabolism of the cell that involves GSH; however, as signal transduction reactions are localized rather than distributed throughout the cell, there is not a direct relationship between the redox state and any specific redox signaling reaction. Indeed, signaling by reactive species might be modulated by the reaction of the species with GSH through one of the enzymes described above but the first step is very unlikely to involve a direct interaction with GSH or GSSG. Rather, it is the modification of thiols in signaling proteins that is more likely directly involved. The modifications that have been best characterized involve protein cysteines in formation of protein-glutathione disulfide (PSSG) and protein nitrosothiol (PSNO).
The Trx system for removal of H2O2 is parallel to the GSH system. Five thioredoxin peroxidases, now called peroxiredoxin (Prdx) 1 through 5 catalyze the reduction of H2O2 to water using Trx:
| <3> |
where Trx-(SH)2 is the reduced Trx and Trx-(S)2 is Trx with an intramolecular disulfide between its two active site cysteines. Trx-(S)2 can be restored by the action of thioredoxin reductase:
| <4> |
As we will describe in detail below, GSH, Prdx and Trx play important roles in signaling beyond elimination of H2O2.
In the past few years, previously unrecognized biological functions for a range of redox-active sulfur species have been described. As intermediates and in some less well characterized systems, thiyl radicals (RS•), sulfenic (RSOH), sulfinic (RS(=O)OH) and sulfonic (RS(=O)2OH) acids, sulfenyl-amides, thiosulfinates, disulfide-S-monoxides, and polysulfides may play significant roles in signaling. These reactive sulfur species all possess exceptional chemical properties that allow them to participate in biochemical functions, such as redox sensing and response, catalysis, redox switching and intracellular redox signaling (24).
Nonetheless, formation of glutathionylated cysteine residues in proteins (PSSG) has been paid the most attention. Mechanistically, PSSG formation by reactive oxygen species such as the superoxide radical anion and H2O2 raises a number of questions, highlighted in a detailed kinetic study (25). The reaction of H2O2 with low-molecular-weight thiol(ate)s, including glutathione (GSH), seems to be rather slow (approx. 20 M−1 s−1). This reaction produces glutathione sulfenic acid (GSOH). Protein-based thiols might be somewhat more reactive, but only in cases of an exceptionally low pKa value as in the active site of peroxiredoxins. The general reaction of protein thiolates with H2O2 is:
| <5> |
which can then go on to a reaction with GSH to yield a mixed disulfide:
| <6> |
The reaction of thiols with O2·− is faster (estimated as up to 1000 M−1 s−1), yet can rapidly regenerate O2·−. The underlying chain of reactions is complex. It starts with the formation of a sulfinyl radical, which reacts with a thiol to form a sulfenic acid and a thiyl radical. The latter reacts with a thiol to form the glutathione disulfide radical anion (GSSG•−), which loses an electron by reducing oxygen to O2·− (25).
The formation of PSSG is also called glutathionylation. It has been proposed that formation of PSSG during oxidative stress prevents the overoxidation of cysteine to sulfinic and sulfonic acids, which unlike sulfenic acid is not reducible by GSH. Apart from glutathionylation, another chemical modification to prevent cysteine overoxidation has recently been discovered in the form of sulfenyl-amide formation. It was shown that, the cysteine residue in protein tyrosine phosphatase 1B (PTP1B) can react after oxidation to a sulfenic acid by H2O2 in the absence of GSH (26,27). The sulfenic acid reacts further with an amide nitrogen of the backbone to form a cyclic sulfenyl-amide. While, as will be described below, the formation of the sulfenyl-amide in PTP1B is unlikely to occur in vivo, such intermediate formation may be found either during oxidative stress or in some proteins that actually do form a sulfenic acid intermediate in vivo. As with glutathionylation, formation of the cyclic sulfenyl-amide prevents overoxidation of cysteine to sulfinic and sulfonic acid. Sulfenyl-amide formation is also reversible, and GSH is able to rapidly reduce this species to the thiol.
Recent studies on the Prdx isoforms that have 2 cysteines in their active sites (2-cys-Prdx) have suggested that this cysteine-based redox system not only removes H2O2, but may play a role as a redox switch, which has been called the floodgate hypothesis (28). In this hypothesis, the inactivation of 2-cys-Prdx by overoxidation of an active site cysteine to a sulfinic acid would allow H2O2 to react with signaling proteins Although the reduction of the sulfinic acid form has been demonstrated to be catalyzed by sulfiredoxin (Srx) (29), in most systems, 2-cys-Prdx is not the rate-limiting enzyme for removal of H2O2 so that its inhibition would not provide a flood of H2O2. Also, the rate of formation of the sulfinic acid, which is second order in [H2O2], would need to out-compete reactions of H2O2 with target signaling proteins. It is more likely that 2-cys-Prdx contribute to signaling by oxidizing a Trx bound to a signaling protein than by the floodgate hypothesis. Indeed, some Prdx catalyzes the oxidation of Trx that, in its reduced form, binds to and inhibits the upstream kinase kinase kinase, ASK1 (30).
A documented example of where a Prdx rather than being an isolated redox-switch protein seems to be at the center of a more extensive Srx/Prdx redox network is the 2-Cys Prdx (Tpx1) in yeast, that interacts with signaling proteins such as Pap1 and the MAP kinase Sty1 (31–33). At low H2O2 concentrations, Tpx1 can oxidize Pap1 to its active disulfide form. This activated Pap1 translocates to the nucleus, where it binds to and increases the transcription of genes related to oxidative stress response proteins (31,33). At higher peroxide concentrations, however, overoxidation of Tpx1 increases and Pap1 activation is decreased (31). Instead, the Sty1 antioxidant defense pathway dominates (33), possibly regulated by Tpx1-Sty1 disulfide formation (32). The Tpx1-Pap1 pathway can be restored by Srx, which reduces overoxidized 2-Cys Prdx and thus increases the amount of catalytically active peroxidase. High H2O2 concentrations and the Sty1 response also result in the induction of Srx (33), which subsequently restores Prdx activity and hence Pap1 activation. Active Pap1 may then directly repress Sty1-dependent genes.
3a. Signaling proteins in which critical cysteines are modified
The mechanisms of protein thiol modification by RNS will be described in detail below. Those proteins are included here as the same critical cysteine(s) can be modified by either ROS or RNS and very likely by HNE as well when the cysteine is in the thiolate form. Among the signaling proteins (and their critical cysteines) that have been well characterized are the PTPs, PTP1B (cys215) (26,27,34–36), low molecular weight PTP (cys12,17) (37–39) and Src-homology 2 domain-containing PTP (SHP-2) (40), the small G protein, Ras (cys118) (41–44), the large G proteins, Gi (cys267) and Go (cys326) (45) and the lipid phosphatase, phosphatase and tensin homolog deleted on chromosome 10 (PTEN). In addition, as mentioned above, in its reduced form Trx binds and inhibits ASK1 and possibly other signaling proteins as well (46–53). When oxidized by formation of a disulfide between the two cysteines in its active site, Trx dissociates from ASK1, allowing it to become activated (46–53) (Figure 1). Also, GSTΠ inhibits c-Jun-N-terminal kinase (JNK) but dissociates and allows JNK activation when its critical active site cysteine is oxidized (54–56). The bacterial transcription factor, OxyR (57) and the eukaryotic transcription factors AP-1 (58) and NF-κB (59) also contain critical cysteine residues that are targets of signaling. Caspases that are involved in the signaling for apoptosis also have a critical cysteine in their active sites (60)).
FIGURE 1.
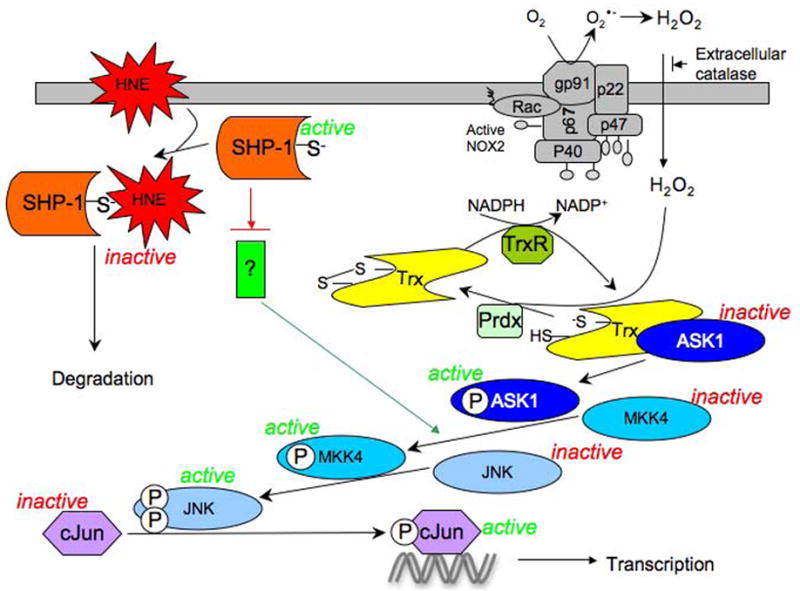
Model based on studies of the activation of JNK pathway upon generation of H2O2 in rat alveolar macrophages (30) and in human bronchial epithelial cells (HBE1) exposed to HNE (212). Several stimuli are able to induce the assembly of the membrane NADPH oxidase (NOX2) in macrphages resulting in the generation of extracellular superoxide that dismutes to H2O2. ASK1, in its inactive form, is bound to the reduced Trx. Oxidation of the bound Trx to the disulfide form by H2O2, which is catalyzed by a peroxiredoxin, causes dissociation of Trx from ASK1. ASK1 then can undergo dimerization and auto-phosphorylation that initiates the sequential phosphorylation of MKK4, JNK and cJun. Phospho- cJun, as part of the AP-1 transcription factor complex, can then bind to the TRE cis element resulting in the transcription of several genes.
HNE generated from the peroxidation of ω-6 polyunsaturated fatty acids under physiologic or pathologic conditions, can react with SHP-1 leading to its inactivation and degradation. As a consequence, the inhibitory effect of SHP-1 on an unknown protein upstream of MKK4 (currently under investigation), is released allowing the signal to be transmitted along the same pathway as above.
3b. An example of signaling by H2O2: mechanism of inhibition of the PTP family
The identification of the target proteins for H2O2 interaction has been a key to understanding how H2O2 mediates signaling processes. Despite this knowledge much work needs to be done to fully understand the mechanism by which H2O2 can modify the activity of key signaling proteins. Currently, the focus is largely on how cysteine residues on particular proteins work in the cells as redox sensors. The oxidative reaction of cysteine with H2O2 may change structure and function of protein mediating response to changes of redox state in the cell (61,62).
An important protein effector by which H2O2 signaling occurs is the family of PTPs (34,36–38,63,64). PTPs constitute a large family of important regulatory enzymes that play a key role in several physiological functions. Pairs of protein tyrosine kinase (PTKs) and PTPs regulate the degree of protein tyrosine phosphorylation of each target protein. Protein tyrosine phosphorylation is one of the mechanisms that enable the cell to respond to several extracellular stimuli, having a role in the regulation of most cellular functions such as growth, proliferation, differentiation and metabolism (65,66). While PTKs have been extensively studied, the past few years have seen an increase in interest in PTPs because of their recognized role as regulators of signal transduction under normal and pathophysiological conditions (66,67)
As thiol groups are crucial for the catalytic and structural functions of these proteins, their oxidation in normal cells is likely related to the physiological consequences of H2O2 production. The proteins belonging to the PTPs family share the same structure of catalytic site characterized by an 11-residue signature sequence (I/V)HCXAGXXR(S/T/G), which includes the catalytic cysteine (Cys215) (68,69). The catalytic mechanism involves a nucleophilic attack by an active site cysteine on a phosphotyrosine substrate that is finally hydrolyzed by a water molecule (70). The cysteine exists in reduced the thiolate anion form as it has a low pKa due to the presence of surrounding basic amino acids. The thiolate anion is the required form for the function of the PTPs but it is also more susceptible to oxidation by H2O2.
Much of the work on the redox regulation of PTPs has been done with PTP1B. The cysteine thiolate of PTP1B can be readily oxidized to sulfenic acid, a reversible form of oxidation, but can be oxidized further to a sulfinic or sulfonic acid (27). As described above, Srx can reduce the sulfinic acid form of Prdx 1 (71) but thus far no evidence exists for the formation or reduction of the sulfinic acid form of PTP1B in vivo.
One of the characteristics of signal transduction is that the modification of effector proteins has to be transient. Nonetheless, “transient” may mean degradation and resynthesis; e.g., cyclins. For phosphorylation, transient is often seconds to minutes. In that case, the modifications of the protein kinases and phosphatases must be reversible. Reversible inactivation of several different protein tyrosine phosphatases has been shown in relevant cell types stimulated by PDGF, EGF, insulin, and B cell receptor ligands (40,72–74).
Two main mechanisms have been suggested as protection of irreversible inactivation of the protein effectors PTPs. One of the mechanisms involves the reaction of cysteine residue oxidized to sulfenic acid with an amide from the protein backbone. This reaction has been observed in purified proteins and in absence of glutathione in several PTPs (27,75). Thus, while feasible, this reaction has not yet been demonstrated to occur in vivo.
As described above, glutathionylation (formation of a mixed protein-glutathione disulfide) is one of the post-translational modifications that occurs in cells in response to oxidative stress that could prevent overoxidation (76); but, it also has been proposed in cell signaling (36,63,77). During this process, a critical cysteine sensitive to oxidation by H2O2 or other hydroperoxide may be oxidized and can then form a disulfide bond with glutathione (GSH), resulting in altered activity. Several signaling proteins have been shown to undergo glutathionylation, including PTPs and transcription factors (36,63,77–80). Although the mechanism by which glutathionylation occurs is not agreed upon, this process has been shown occur in vivo in relevant cell types (81–86). Although glutathionylation of PTP1B can be achieved in vitro with the purified enzyme through oxidation by H2O2 and addition of GSH (through the sulfenic acid and sulfenyl-amide as described above) (26) (27) or by addition of a very high concentration of GSSG (63), it is not known if glutathionylation occurs enzymatically or non-enzymatically in vivo and kinetic considerations clearly argue against the latter (87). While formation of glutathionylated PTP1B through disulfide exchange in the cytosol by reaction with GSSG enzymatically or non-enzymatically is unlikely due to the requirement for markedly increasing GSSG, a recent paper by Beer et al. demonstrated glutaredoxin 2 dependent catalysis of glutathionylation/deglutationylation of mitochondrial proteins (88). Interestingly, a paper by Seth and Rudolph of recent publication extended the diversity of mechanisms utilized by PTPs to prevent irreversible oxidation of the active site. They found that in MAP kinase phosphatase 3 (MKP3) the sulfenic acid formed by oxidation, did not form a sulfenyl-amide, or a disulfide with GSH but, reacted with one of several cysteines in N-terminal and C-terminal to form a disulfide (75). Further insight into cellular mechanisms of protein glutathionylation and deglutathionylation or sulfenyl-amide formation will enrich our understanding of redox signal transduction and potentially identify new therapeutic targets for diseases in which oxidative stress perturbs normal redox signaling.
4. OTHER SIGNALING MECHANISMS FOR ROS
While thiols appear to be the most likely direct target for action by H2O2, the participants in signaling downstream of the initial interaction of H2O2 include second messengers, such as calcium, diacylglycerol and ceramide. These second messengers in turn can signal for increased activity of a large variety of effectors. As the present article primarily concerns the chemistry of redox signaling rather than the downstream events, we will provide here several reviews that describe the involvement of those second messengers in hydroperoxide regulated signaling pathways (89–94). A caveat is that many of the studies cited in these reviews were done with exogenous H2O2 as a stimulant and thus, their physiological relevance remains to be established (87).
Another mechanism for signaling by ROS could modulation the binding of iron in the iron-sulfur clusters that are in the iron-response proteins that regulate iron metabolism through their binding to the iron-response element. Whether it is the iron oxidation state or oxidation of the sulfur in the clusters is not clearly understood but the effects are dramatic in terms of regulating iron metabolism. Regardless, this kind of ROS signaling appears to require higher levels of O2·− and/or H2O2 than is normally associated with physiological signaling. This important but incompletely understood aspect of ROS signaling has been the subject of an excellent review that implicates ROS-iron signaling in sepsis and acute respiratory distress syndrome (95,96).
5. SIGNALING BY RNS
Signal transduction via nitrogen oxide species is an important and burgeoning field that is entwined with the signaling associated with O2 and O2-derived species. Thus, a review of the cellular signaling of oxygen-derived species is not complete without including a discussion of nitrogen oxides since the chemical biology of these two classes of signaling molecules is intimately linked. The reason for this is due to the fact that oxygen (and derived species) are capable of reacting with •NO, leading to either activation or deactivation (depending on the cellular target) of its ability to elicit (patho)physiological responses. Also, due to similarities in their chemistries (metal ligation, electrophilicity, etc.), many nitrogen oxides and oxygen and/or oxygen-derived molecules act on similar (or the same) signaling targets. Thus, the intimate biochemical relationship between the nitrogen- and oxygen-derived species is unavoidable and profound and will be emphasized in the following discussions of nitrogen oxide signaling.
The first nitrogen oxide discovered as an endogenously generated species in mammalian systems was ·NO. This was an important discovery since it was the first report of the endogenous generation of a small, freely diffusible and otherwise toxic species for the purposes of signal transduction (for example, (97)).
6. REGULATION OF NITRIC OXIDE BIOSYNTHESIS – GENERAL CONSIDERATIONS
Nitric oxide is biosynthesized via the 5-electron oxidation of the amino acid L-arginine by a family of heme containing proteins referred to as the nitric oxide synthases (NOS). A comprehensive description of the enzymology of NO biosynthesis is beyond the scope of this article and has been reviewed extensively elsewhere (for example, (16–19)). However, of particular relevance to this article, the Km values of O2 for the three major isoforms differ significantly, indicating ·NO biosynthesis can be regulated by O2 levels, depending on the isoform (19). The apparent O2 Km values for nNOS, iNOS and eNOS are 350, 130 and 4 μM, respectively. This indicates that nNOS may be highly regulated by cellular O2 levels while eNOS will be maximally active at even extremely low O2 concentrations.
The regulation of NOS activity is an important aspect of nitrogen oxide signaling since ·NO is not easily stored (due to its high diffusability through membranes), has a short biological lifetime and is generally thought to be made only as needed (although there have been numerous reports of other stable nitrogen oxide species capable of serving as endogenous sources of ·NO, vide infra). Of special relevance to this review is that it appears that O2 (and O2-derived species) are important in NOS regulation/expression and examples of this will be given in order to illustrate and emphasize some of these relationships.
7. ACTIVATION OF SOLUBLE GUANYLATE CYCLASE AND cGMP SIGNALING
The most established biochemical target for the actions of NO is the enzyme soluble guanylate cyclase (sGC) (for reviews of sGC enzymology, see (98–100)). This enzyme catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). Thus, a large majority of the biological signaling associated with ·NO is due to the second messenger cGMP, an important messenger involved in a number of critical physiological processes in the cardiovascular system including regulation of vascular smooth muscle tone, cell proliferation, platelet aggregation and leukocyte recruitment (100,101). Thus, the regulation of cGMP levels has implications for the pathogenesis and treatment of numerous cardiovascular disorders including hypertension, sepsis, stroke and erectile dysfunction. In particular, since the development of atherosclerosis involves adhesion of monocytes to the vascular endothelial layer, platelet activation and smooth muscle proliferation (each inhibited by cGMP), manipulation of cGMP may also have important consequences in preventing the development of coronary artery disease (102,103).
8. INTERACTION OF NITROGEN OXIDES WITH THIOLS: S-NITROSYLATION AND OTHER PRODUCTS
The modification of cysteine residues on peptides and proteins via reaction with nitrogen oxides has been examined extensively and is considered to be an important aspect of nitrogen oxide signaling (104–107). The basic chemistry of thiol modification by nitrogen oxides has also been examined in detail and there are numerous mechanisms by which endogenously generated nitrogen oxides can react with and modify thiols. The reaction of ·NO with O2 generates several species that are capable of reacting with thiols (Reactions <7>–<11>).
| <7> |
| <8> |
| <9> |
| <10> |
| <11> |
The products of nitrogen oxide-mediated modification of thiols via the chemistry depicted above are S-nitrosothiols (RSNO, Reactions <10> and <11>) and the process of RSNO generation has been coined S-nitrosylation (a mechanistically ambiguous term). A fundamental problem with the biological relevance of the chemical processes leading to S-nitrosylation depicted in Reactions <7>–<11> is that generation of the ultimate thiol reactive species, ·NO2 or N2O3, requires high concentrations of ·NO since the generation of either is second order in ·NO (Reaction <7>). As the concentration of ·NO in biological systems (at least from endogenous generation) is likely to be very low or sub micromolar at the greatest, this 2nd order process seems very unlikely. However, both ·NO and O2 favorably partition into lipid/hydrophobic environments (i.e. cellular membranes) indicating that thiol modification via these reactions will be more prevalent in or near membranes (108).
The chemistry described above (Reactions <7>–<11>) is not the only conceivable sequence for the formation of S-nitrosothiols in biological systems as RS• may be formed by other reactions than reaction <9>. Indeed, it is becoming increasingly evident that cysteine thiyl radicals are present and crucial in some enzymes (for example, in ribonucleotide reductases (109), pyruvate formate lyase and benzylsuccinate synthase (110)) and are intermediates in thiol oxidation processes (i.e. thiol oxidation by one electron oxidants such as ·NO2. (111)). Thus, thiyl radicals can be generated in biological systems either as reactive species involved in enzyme-catalyzed reactions or as intermediates in thiol oxidation processes. Regardless, thiyl radicals would serve as precursors to S-nitrosothiols in the presence of ·NO.
Two primary targets of nitrogen oxide chemistry in biological systems are metals and thiols. Both of these targets can be utilized for the generation of an S-nitrosothiol. As shown immediately below, ·NO can coordinate with a ferric ion (i.e. ferric porphyrin), to form a complex that can be depicted as either a ferric-nitrosyl or a ferrous-nitrosonium species (these can be viewed as resonance forms of the complex) (Reaction <12>). In the presence of a nucleophile (e.g. a thiol or water), nitrosation occurs, generating the corresponding ferrous metal and a nitrosated nucleophile (Nuc) (Reaction <13>). The ferrous porphyrin can then react directly with NO to form a ferrous nitrosyl (Reaction <14>). This process (Reactions <12>–<14>) is referred to as reductive nitrosylation (112).
| <12> |
| <13> |
| <14> |
In the event that the nucleophile in Reaction <13> is a thiol, this process generates an S-nitrosothiol. Significantly, this process is first order in ·NO and only requires the presence of an oxidized metal (i.e. a ferric porphyrin) and a nucleophilic thiol. From a kinetic perspective, metal-mediated formation of an S-nitrosothiol appears much more facile since 2nd order reactions in ·NO are not necessary.
S-Nitrosothiols are capable of further reactions leading to other possible products. For example, transnitrosation from one S-nitrosothiol to another thiol can occur (Reaction <15>). In a transnitrosation reaction, the equivalent of a nitrosonium ion (NO+) is transferred from one thiol to the other (although a ‘“free” nitrosonium ion is not likely formed under biological conditions). The reaction of a thiol and nitrosothiol can also have another outcome: generation of a disulfide and HNO (Reaction <16>).
| <15> |
| <16> |
The chemical factors that determine which pathway occurs (Reaction <15> or <16>) have not yet been elucidated.
Signaling associated with protein S-nitrosothiol formation (S-nitrosylation) has been hypothesized to be akin to other, more established signaling systems such as protein phosphorylation, ubiquitination, acetylation, etc. (for example, see (113)). However, at present there is an important distinction between, for example, phosphorylation and S-nitrosylation: Protein phosphorylation/dephosphorylation is a highly controlled signaling system with a variety of highly regulated protein kinase and phosphatase enzymes whose signaling specificity is derived from very discriminating protein-protein interactions. This degree of specificity and regulation has not yet been reported for S-nitrosylation and thus equating S-nitrosylation with phosphorylation, for example, seems unwarranted at this time. However, some recent work implicates specific S-nitrosylated enzymes (e.g. S-nitrosylated thioredoxin) in effecting the transnitrosation of, in this case, caspase 3, an event that would serve to regulate cellular apoptosis (114). This report indicates that there may indeed be specificity in S-nitrosylation signaling, although there is still not the body of evidence to warrant comparisons with many other, more established signaling pathways.
The reaction of ·NO with O2− is near diffusion controlled and generates a potent oxidant/pro-oxidant, peroxynitrite (ONOO−) (Reaction 17) that has the potential to oxidize thiols. Protonation of ONOO− (ONOOH pKa = 6.7) generates peroxynitrous acid (ONOOH), which can decompose to nitrate (NO3−) via a homolytic cleavage of the O-O bond (Reaction 18).
| <17> |
| <18> |
Peroxynitrite is also nucleophilic and capable of reacting with carbon dioxide (CO2) giving nitrosoperoxycarbonate (ONOOCO2−) (Reaction <19>). Nitrosoperoxycarbonate is fleeting as it, like peroxynitrous acid, also undergoes homolytic O-O bond cleavage to generate NO2 and the carbonate radical anion. Recombination of these two radical species leads to the generation of nitrocarbonate (Reaction <20>), which would eventually hydrolyze to give NO3− and bicarbonate (HCO3−).
| <19> |
| <20> |
Thus, the reaction of ·NO and O2− leads to the generation of a variety of one-electron oxidants (HO•, ·NO2, CO3•−), all of which are capable of oxidizing tyrosine to give a tyrosyl radical (allowing the possible generation of 3-nitrotyrosine).
Thiol oxidation by ONOO− can occur via either one- or two-electron processes (115). For example, the cysteine in albumin can be oxidized to the sulfenic acid by ONOO− (116) and thiyl radicals have been observed in the oxidation of glutathione and a hemoglobin cysteine when reacted with ONOO− (117). Thus, it is clear that ONOO−, derived from ·NO and O2·−, can lead to thiol oxidation. However, it should be noted that the general biological relevance of ONOO- generation has been questioned (118) and may be limited.
8a. Tyrosine nitration
It has been demonstrated that under certain (patho)physiological conditions, nitration of tyrosine occurs. The mechanism of tyrosine nitration is reported to occur predominantly via a free radical process through a tyrosyl radical intermediate (for example, (119,120)) (Reaction <21>).
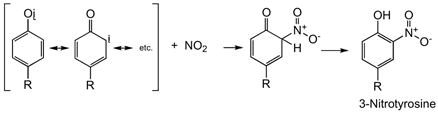 |
<21> |
As is the case with thiyl radicals, tyrosyl radicals are prevalent in a variety of enzyme catalyzed reactions, serving as one-electron oxidants (for example, see (121)). The generation of 3-nitrotyrosine via the mechanism depicted in Reaction 21 is dependent on the presence of a one-electron oxidant for the formation of a tyrosyl radical. There are several candidates and/or possible biologically relevant oxidants that would be capable of performing this oxidation. These include hydroxyl radical (HO•), carbonate radical anion (CO3•−), ·NO2 or peroxidase activity (e.g. myeloperoxidase/H2O2) (119). The generation of some of these oxidants can be formed via an initial reaction between ·NO and O2·− (vide supra).
Although peroxynitrite is capable of eliciting tyrosine nitration via the generation of the above-mentioned radical species, there are other mechanisms available that do not require ONOO−. For example, myeloperoxidase/H2O2 will oxidize NO2− to ·NO2, which can then nitrate tyrosine. Also, ·NO can trap the tyrosyl radical intermediate, leading to 3-nitrosotyrosine (122). Further oxidation of this intermediate by two-electrons gives 3-nitrotyrosine (Reaction <22>).
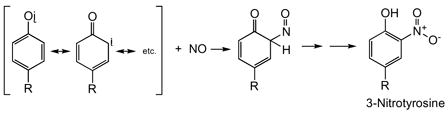 |
<22> |
The phosphorylation/dephosphorylation of protein tyrosines is an important and well-established mechanism of cell signaling. That is, the addition or elimination of phosphate at specific protein tyrosines results in changes in protein function that serve important signaling processes. Although tyrosine nitration may also alter protein function, there is currently an important distinction between tyrosine phosphorylation and nitration with regards to cell signaling. Unlike phosphorylation, nitration is currently viewed as an irreversible modification of tyrosine. Thus, if tyrosine nitration were part of a normal signaling process, it would represent a “trigger” rather than a reversible signal. Indeed, it is likely that tyrosine nitration is best viewed as a marker for ·NO-mediated oxidative stress. However, it is worth noting that “denitration” of protein nitrotyrosine has been reported (for example, (123–125)), lending credence to the idea that protein tyrosine nitration may not be merely a marker for nitrogen oxide/oxidative stress but could be a regulated and reversible signaling process.
8b. Fatty acid nitration
Many important cell signaling processes involve biochemical transformation of fatty acids for the generation of signaling molecules. For example, arachidonic acid serves as the precursor to myriad important signaling eicosanoids, including prostaglandins, thromboxanes and leukotrienes via the actions of specific enzymes. Unsaturated fatty acids are also subject to modification by nitrogen oxides and these products have been reported also to be possible signaling molecules. There appears to be little doubt that nitrated fatty acids (LNO2) are endogenously present at levels that can elicit biological responses (126). For example, nitrolinoleate is capable of inhibiting platelet and neutrophil function by a cAMP-dependent process (for example, (127)). Also, nitrolinoleate can serve as a storage form of ·NO since chemical decomposition releases ·NO and elicits vasorelaxation (128). Nitro derivatives of oleic acid have also been found to be capable of eliciting gene expression via activation of PPAR-gamma (126). Nitrolinoleic acid induces heme-oxygenase 1 by a predominantly ·NO-independent mechanism (129). Nitrated arachidonic acid derivatives have been shown to have anti-inflammatory properties via mechanisms that include the release of ·NO and an increase in cGMP (130).
The generation of nitrated fatty acids is chemically complex and likely involves fatty acid radical intermediates and ·NO2 (for example, (131)). As mentioned previously, the generation of ·NO2 from the reaction of ·NO and O2 is much more probable in membranes compared to the aqueous cytosol due to the partitioning of these species into hydrophobic environments (108). The release of ·NO from nitrated lipids has been examined and several mechanisms have been proposed involving homolytic cleavage of a precursor to generate ·NO and a relatively stable radical co-product (132,133). Regardless, the biological activity associated with nitrolipids is another example of the close relationship between ·NO and O2 signaling/biology since both species are required for the generation of nitrolipids, a process that is greatly enhanced by their abilities to favorably partition into membranes.
8c. ·NO-heme and ·NO-metal complexes
One of the most important chemical aspects of ·NO biology is its ability to coordinate metals such as the iron in iron-heme proteins. Indeed, the most established and certainly one of the most important biological targets for NO is the heme protein sGC (vide supra). Significantly, O2 also has a high affinity for many metals, including heme proteins (although it does not bind sGC). One important difference between ·NO and O2 with regards to reactions at iron-heme centers, however, is that ·NO can react with either ferrous or ferric hemes while O2 will only coordinate ferrous hemes. Regardless, it may be hypothesized that the chemical similarity between these two species allows them to share common targets or sites of biological reactivity. Although they may react or coordinate similar sites, the subsequent chemistry is likely to be quite different, indicating that ·NO may disrupt O2 biochemistry (and possibly vice-versa, although this is not the case with sGC). Numerous examples exist in the literature indicating that ·NO is capable of disrupting/regulating O2 biochemistry by competing with it at heme or metal binding sites. A prime example of this (and particularly relevant to this review) is the effect of ·NO on cytochrome c oxidase, the terminal electron acceptor of the mitochondrial electron transport chain and the site of O2 binding. Cytochrome c oxidase binds O2 via a ferrous heme. ·NO is also capable of binding the ferrous heme of cytochrome c oxidase in competition with O2 leading to an inhibition of respiration (·NO also inhibits non-competitively at a copper site as well) (for example, (134)). The competition between ·NO and O2 at the heme site has numerous important and profound physiological implications. Indeed, NO-mediated inhibition of O2 consumption via the mitochondrial electron transport chain has been reported to be an important regulator of H2O2 production and important in the control of O2 gradients (135). That is, ·NO binding to cytochrome c oxidase leads to an increased generation of O2·− that can result in either (or both) an increase in H2O2 (which may have normal signaling functions) or ONOO− production, with potential pathophysiological outcomes. Also, partial inhibition of cytochrome c oxidase by ·NO can extend O2 gradients, preventing hypoxia in tissue remote from an O2 source ((135) and references therein).
Recent studies indicate that ·NO can have numerous effects on the cellular response to hypoxia. The regulation of the expression of genes in response to hypoxia is due, in part, to the actions of hypoxia inducible factor 1 alpha (HIF1-α) (for example, (136)). Under normal oxygen conditions, HIF1-α protein levels are low due to the actions of O2-dependent, iron (non-heme) containing prolyl hydroxylases that oxidize it, leading to its degradation/elimination (for example (137)). However, under low O2 conditions (hypoxia), the activity of the prolyl hydroxylases is decreased (due to the lack of O2 as a co-substrate) leading to an increase in the levels of HIF1-α and, therefore, an increase in the expression of genes for the response to hypoxia (e.g. erythopoietin, vascular endothelial growth factor (VEGF), glycolytic proteins, etc.). The prolyl hydroxylases utilize iron to bind and activate O2 for the hydroxylation of HIF1-α. As ·NO is capable of binding to many sites that normally bind O2, ·NO can compete with O2 binding to numerous sites, altering the hypoxic response. Indeed, it is reported that ·NO can inhibit prolyl hydroxylase, leading to a possible hypoxic response under normoxic conditions (for example, (138)). On the other hand, ·NO binding to cytochrome c oxidase appears to result in a redistribution of O2 utilization resulting in an increase in prolyl hydroxylase activity and an inhibition of hypoxic responses (139). Thus, the effect of ·NO on the hypoxic response is complex and equivocal and appears to be dependent on numerous factors including the concentration of ·NO, the oxygen status of the cells and even the nature of the ·NO donor used in the studies.
9. NITRITE SIGNALING
Nitrite and nitrate comprise the oxidative fates of nitric oxide produced endogenously. NOS-derived ·NO is rapidly converted to nitrate by oxyhemoglobin or oxymyoglobin (Reaction <23>) (140).
| <23> |
In the absence of oxyhemoproteins, ·NO is oxidized to nitrite via Reactions <7> and <8> (above) and Reaction <24> (141) which, in addition to being a decomposition product of ·NO, may posses signaling properties through re-reduction to ·NO and participation in the formation of heme nitrosyls and nitrosothiols (vide infra).
| <24> |
The concentration of nitrite in the body is derived from three main sources; (1) oxidation of NOS derived ·NO (142), (2) reduction of nitrate in the mouth and gastrointestinal tract by bacteria (143), and (3) nutritional sources such as processed meats and green leafy vegetables (see (143)). Nitrite has a lifetime of hours in plasma; however, whole blood nitrite is rapidly oxidized by oxyhemoglobin and oxymyoglobin to nitrate with a half life of about 110 s while nitrate has a half life of 5–8 h (see (143)). This is in contrast to tissue metabolism where nitrite and nitrate have half-lives in the tens of minutes. For this reason, the concentration of nitrite in tissue is normally 1–2 orders of magnitude higher than blood (144–146). The reaction of nitrite with oxyhemoproteins has potential toxicological implications as it results in methemoprotein formation. It is a complex reaction characterized by a lag phase preceding an autocatalytic phase ((147) and references therein). The first step likely involves the oxidation of NO2− by the oxyhemoprotein and most likely accounts for the lag (Reaction <25>). This results in a ferric peroxo species that reacts with a second NO2− to form a high valence metal oxo species (Reaction <26>) that oxidizes another NO2− to ·NO2 and methemoprotein (Reaction <27>). Reaction of •NO2 with oxyhemoprotein to form a perferryl intermediate (Reaction <28>) carries the chain in the reaction allowing it to be autocatalytic.
| <25> |
| <26> |
| <27> |
| <28> |
The vasodilatory properties of nitrite have been known for some time. Furchgott and Bhadrakom in 1953 used supraphysiological concentrations of acidified nitrite to relax precontracted aortic strips (148). The idea that nitrite is an in vivo regulator of vascular tone was challenged years later when it was reported that physiological concentrations of nitrite are insufficient to regulate vascular tone (149). However, nitrite is now thought to serve as a source of ·NO under pathophysiological conditions such as hypoxia and ischemia where NOS derived ·NO is unavailable. Nitric oxide forms from nitrite nonenzymatically at low pH by a disproportionation reaction (Reactions <29>–<31>) but is limited to places where the pH may be close to the pKa of nitrous acid (3.3) such as ischemic tissue or the stomach.
| <29> |
| <30> |
| <31> |
·NO generated by this chemistry in the stomach is thought to play an important role in host defense and the regulation of gastric mucosal integrity (150). Nitrite reduction to nitric oxide is catalyzed by elements of the mitochondrial electron transport chain such as ubiquinol and cytochrome c oxidase (complex IV) under hypoxic conditions (151,152). The ·NO produced has a high affinity for cytochrome c oxidase and may serve as a reversible inhibitor of mitochondrial respiration (vide supra) preventing ischemic reperfusion injury. Xanthine oxidase, an enzyme normally responsible for reducing oxygen to O2·−, is also capable of reducing nitrite to ·NO (153–157). The maximal rate of this process is observed at low pH and low oxygen tension. The substrate oxygen is not only a competitive inhibitor but the product superoxide is also a diffusion-limited scavenger of ·NO. It was determined that the concentration of xanthine oxidase and nitrite were sufficient in no-flow ischemic cardiac tissue to generate ·NO levels comparable to NOS (158). Doyle and coworkers (140) were the first to describe nitrite reductase activity by deoxyhemoglobin (Reaction <32>).
| <32> |
More recently, it was found that deoxyhemoglobin converts nitrite to ·NO under hypoxic conditions resulting in vasodilation (159,160) with a maximum rate at 50% oxygenation providing a link between nitrite and hypoxic vasodilation (161). This link is controversial in light of the high affinity and stability constants associated with the reaction of ·NO produced and the increasing amount of deoxyhemoglobin (ka = 2 × 107 M−1 s−1, kd = 3 × 10−3 s−1, (162)). Therefore any ·NO produced from the nitrite reductase activity of deoxyhemoglobin could also be scavenged by deoxyhemoglobin or destroyed by oxyhemoglobin and not contribute to vasodilation. It has been proposed that the ferrous-nitrosyl-hemoglobin may be oxidized in vivo to the ferric-nitrosyl-hemoglobin to favor the release of ·NO (kd= 1 s−1, (162)). This is a very active area and more work is being done to address this phenomena.
10. LIPID PEROXIDATION PRODUCTS IN REDOX SIGNALING
Lipid peroxidation occurs as free radicals “steal” an electron from an unsaturated lipid molecule, which can result in a chain reaction as shown in the figure below:
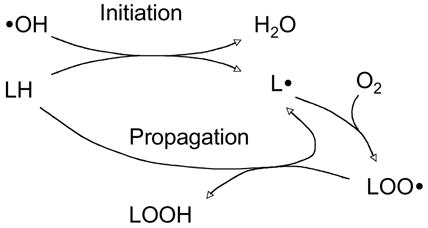 |
<33> |
As a result of the unavoidable production of free radicals and oxidants, lipid peroxidation occurs as a physiological phenomenon in aerobic organisms. Decomposition of the lipid peroxyl radicals or the primary free radical intermediate of lipid peroxidation generates reactive aldehydes. Accumulating evidence from the last decade has shown that some of aldehydes can function as messengers that activate or inhibit signaling pathways under physiologic or pathologic conditions. Here the focus is on the signaling pathways activated by HNE, the major unsaturated aldehyde derived from lipid peroxidation.
10a. HNE: production, reaction, and elimination
HNE is the major α, β-unsaturated aldehyde derived from peroxidation of ω-6 polyunsaturated fatty acids such as linoleic acid and arachidonic acid (163,164). The physiological concentration of HNE in the plasma is reportedly from 0.3–0.7 μM (164,165) but its concentration can reach as high 10 μM or more in plasma membrane under conditions of oxidative stress (166,167). While HNE is therefore considered as a biomarker of oxidative stress (167,168), it is also implicated in various oxidative stress-related diseases, including atherosclerosis (169), neurodegenerative diseases (170), and fibrosis (171).
Once formed, HNE is rapidly degraded by three major reactions: reduction to 1,4-dihydroxy-2-nonene by alcohol dehydrogenases (172), oxidation to 4-hydroxy-2-nonenoic acid by aldehyde dehydrogenase (173–175), or formation of the glutathione-conjugate (GS-HNE) catalyzed by glutathione-S-transferases as shown in equation <2>. The majority of HNE is metabolized through forming GS-HNE (172). Some GST isozymes; i.e., the human GSTA4-4 and GST5-8, have significantly higher catalytic efficiency in conjugation of HNE than other GSTs and are able to regulate the cellular effects of HNE (176–178). Cells also adapt to the need to eliminate HNE through induction of the rate-limiting enzyme for GSH synthesis, GSTs, and aldose reductase by HNE itself (179–181).
The structure of HNE contains an electrophilic C=C double bond conjugated with the double bond of a carbonyl group, and a hydroxyl group that all contribute to making HNE highly readily react with nucleophiles such as the thiol of cysteine (which is a far better nucleophile when in the thiolate form), the imidazole group of histidine, and the primary amine of lysine in protein via Michael addition and/or Schiff base reaction (182–184). It is generally thought that the conjugation between HNE and protein is non-enzymatic and irreversible, and the resulting HNE-protein conjugates are subsequently degraded. Recent studies indicate that both ubiquitin-mediated and non-ubiquitin-mediated proteosomal degradation is responsible (20). Other studies have demonstrated that cross-linking between proteins may occur through bridging by HNE, which can form a Michael adduct and Schiff base sequentially, and that these cross-linked proteins may accumulate due to their resistance to proteosomal decomposition (185,186).
11. SIGNALING PATHWAYS ACTIVATED BY HNE
Numerous studies have shown that physiological concentrations of HNE can stimulate cellular proliferation (187–189), differentiation (190,191), and cytoprotective response (181,192–197), through affecting multiple signaling pathways. As low as 0.5 μM, HNE can induce transcription of glutamate cysteine ligase, the first enzyme of GSH synthesis, through activation of the Nrf2-EpRE signaling pathways (198). All these studies point to the potential signaling function of HNE in both physiological and pathological conditions. Although non-toxic concentrations of HNE are known to affect several signaling pathways, identification of the exact targets of HNE has a long way to go compared with H2O2 and ·NO.
11a. Activation of protein kinase C by HNE
PKCs are a family of 12 isozymes that are involved in many cellular processes such as proliferation, differentiation, stress response, and apoptosis. PKCs include three subfamilies; i.e., classical or conventional PKC (cPKC: α, β, γ), novel PKC (nPKC: δ, ε, θ, and η), and atypical PKC (aPKC: ζ and ι/λ). Many PKC isozymes are reportedly to be either activated or inhibited by HNE. These different effects appear to be isozyme- and HNE concentration-dependent.
Chiarpotto et al. first systematically investigated the dose-dependent effect of HNE on PKC isozyme activity. In rat hepatocyte, they demonstrated that PKC-βI and βII were activated by HNE at concentrations 0.1–1 μM, while they were inhibited when HNE concentration was higher than 1μM (1–10 μM) (199). This finding was supported by other reports, which, either by using PKC-βI and βII inhibitor GO6976, or by directly measuring kinase activity, demonstrated that these two PKC isozymes were activated by < 1 μM HNE (200). In contrast to PKC-β, the novel PKC-δ activity was inhibited by 0.1 μM HNE but increased by 1–10 μM HNE (199,200) . Numazawa also reported that HNE-mediated HO-1 induction was completely abrogated by dominant-negative expression of atypical PKC-ι (201), suggesting that HNE may also activate PKC-ι.
The underlying mechanism of how PKCs are modulated by HNE is largely unknown. PKCs contain several cysteine-rich regions in the regulatory domain and at the catalytic site, which are redox sensitive (202) and therefore potential targets for HNE conjugation. HNE could also potentially modulate PKC activity through forming adducts with lysine or histidine residuals that are essential for enzyme activity. Obviously further research is required to clarify how PKC activity is affected by HNE including whether PKCs are directly modified or indirectly from upstream signaling events.
11b. MAPK activation by HNE
Mitogen activated protein kinases (MAPK) are a group of serine-threonine kinases that include extracellular signal regulated kinase (ERK), p38MAPK, and c-Jun N-terminal kinase (JNK). The redox regulation of MAPK pathways has been well known (203) and numerous studies have clearly shown that pathophysiological concentration of HNE (1–25 μM) could increase the phosphorylation/activity of the major three MAPK pathways (204–210).
Using antibodies specific to HNE-histidine adduct, Parola et al. found JNK-HNE adducts in nucleus with treatment of 1 μM HNE. Based on the evidence that no upstream kinase in JNK cascade was activated and that the nuclear JNK was not in a phosphorylated form, the authors concluded that histidine-modification in JNK is sufficient for JNK nuclear translocation and activation (211). In contrast, Song et al. reported that HNE more likely activated JNK through targeting upstream kinases. They found that HNE-mediated JNK activation could be abrogated with the dominant-negative inhibition of stress-activated protein kinase kinase 1 (SAPKK1 or SEK1), the upstream kinase of JNK that was also activated by HNE (205). It should be noted however, that modification of the upstream protein kinases by HNE directly has not been demonstrated. Direct modification of a PTP that modulates upstream signaling for the JNK pathway seems more likely than is direct modification of a protein kinase because the former has a thiolate in its active site that is inactivated by modification. This inactivation of the phosphatase would result in increased phosphorylation of its target protein. Recently, HNE-induced inactivation and degradation of SHP-1, which is upstream of JNK has been demonstrated (212) (Figure 1); however, it is only presumed but not yet demonstrated that the inactivation of SHP-1 that precedes its degradation resulted from active site cys conjugation with HNE. Similarly, if a kinase had a cys residue in its catalytic site that reacted with HNE, then the kinase activity would likely be inactivated and less phosphorylation of its target protein would occur. The modulation of a regulatory his in JNK that results in its activation described above (211) has not been shown with other kinases.
The mechanism of activation of ERK and p38MAPK by HNE is even less studied compared to JNK. HNE activation of ERK appears through targeting upstream kinases. In most studies of ERK activation by HNE, an ERK pathway inhibitor PD98059 is usually used. PD98059 inhibits ERK through inactivating MEK1/2, the upstream kinase of ERK phosphorylation. In most cases, PD98059 blocked HNE-mediated ERK activation, suggesting that HNE activates ERK through targeting upper kinases in ERK cascade. Regarding p38MAPK, Kumagai et al. reported that both p38MAPK and its upstream kinase MKK3/6, were phosphorylated (activated) upon exposure to 25 μM HNE (210). Other researchers also noticed that HNE could markedly increase p38MAPK phosphorylation (196,213). These suggest that HNE activate p38MAPK through targeting upstream signaling molecules, which again have not yet been identified.
11c. Tyrosine kinase receptors
Tyrosine kinase receptors (RTKs), such as epidermal growth factor (EGFR) and platelet-derived growth factor receptor (PDGFR), are a group of transmembrane proteins that display tyrosine kinase activity upon ligands binding. Studies have shown that some RTKs could be activated via redox modification in ligand-independent mechanism (214–217). Liu et al. first reported that conjugation of HNE with EGFR caused the clustering and autophosphorylation of EGFR, and triggered the EGFR-related RTK signaling cascade, including phosphorylation and activation of subsequent Src, ERK, and JNK1/2 pathways (218).
Several laboratories have confirmed the HNE activation of RTKs. In one study, HNE exposure triggered activation of PDGFR and EGFR, through an antioxidant-insensitive and ROS independent mechanism, suggesting that HNE may activate RTKs through direct conjugation that was confirmed by the presence of HNE-PDGFR adducts in atherosclerotic areas (219). The derivatization of RTKs by HNE was also confirmed by an in vitro assay that found HNE-conjugated with PDGFR-β (220).
11d. HNE and calcium signaling
The calcium ion is a well-known second messenger that is involved in many cellular functions such as muscle contraction, cellular motility, and regulation of enzyme activity. At rest state, the calcium level is maintained at 10–100 nM in the cytosol with the accumulation of calcium in endoplasmic reticulum and mitochondria. Calcium signaling occurs when the Ca2+ concentration in the cytosol increases suddenly to 500–1000 nM with the entry of Ca2+ from intracellular storage sites or extracellular compartment via Ca2+ channels. Accumulating evidence suggests that redox modification of Ca2+channels could affect the channel activity and interfere with Ca2+ signaling (221). Studies have shown that HNE may also modulate cell signaling network by affecting Ca2+ signaling. Micromolar HNE could cause Ca2+ accumulation in the cytoplasm (222,223), and this may be result from the inhibitory effect of HNE on (Ca2+/Mg2+)-ATPase (224) and Ca2+-ATPase (225). Lu et al. recently reported that 2 h after exposure to 1–10 μM HNE Ca2+ current was increased by 40% in hippocampal neurons, and the cytosolic Ca2+ level was increased to about 400 nM. Further study showed that HNE activated voltage-dependent Ca2+ channel (VDCC) through tyrosine kinase-mediated phosphorylation instead of direct conjugation to VDCC (226). The HNE activation of VDCC was further confirmed by Akaishi et al., who found that 10 μM HNE stimulated VDCC in dentate granule cells (227).
11e. Future of HNE signaling studies
In addition to aforementioned signaling pathways, HNE has also been reportedly involved in the functional modulation of a diversity of other signaling proteins such as phospholipase D (228), PIP2-phospholipase C (229), PI3K (230), NF-κB signaling (231), and other transcription factors, with more to come. As HNE readily forms adduct with proteins via Michael addition, many redox sensitive signaling molecules are also good targets of HNE, although the hydrophobic feature of HNE may favor modification of proteins associated with membranes and/or those proteins that have hydrophobic residues surrounding the target amino acid. As noted above, HNE modulation of signaling protein activity does not necessarily means direct conjugation, as an upstream target needs to be considered. It is essential that future studies identify the actual target of HNE modification. Future studies should also clarify how HNE signaling is integrated with signaling caused by other reactive oxygen/nitrogen species. The toxicity of H2O2 can be partly explained by increased lipid peroxidation resulting in HNE production but this is less likely to occur under conditions of signaling by H2O2. Although it has been demonstrated that low HNE can cause an increase in O2·− production by neutrophils while higher concentrations of HNE inhibit (232), other studies have shown that HNE can activate uncoupling proteins and thereby decrease mitochondrial ROS production (233,234). Thus, the relationship between HNE and H2O2 in terms of signaling versus oxidative injury requires further examination.
12. SUMMARY AND PERSPECTIVE
Signaling by reactive species depends to a great extent on the chemical properties of these molecules with target proteins and lipids. Nonetheless, kinetic considerations suggest that non-enzymatic reactions may not account for the observed modifications of proteins in many cases. Indeed, the necessity of enzymatic processes allows regulation that is a hallmark of physiological signaling. The identification of such signaling enzymes is one of the major challenges to the field.
Of equal importance is the identification of the site of action of reactive species both in terms of the target protein in a pathway identified as affected by the reactive species and the actual site of modification in that protein. Advances in mass spectroscopy and techniques that can be used in other aspects of signaling, such as siRNA and antibodies that recognize these modifications will bring needed rigor to this area of signaling as they are doing in phosphorylation and other better recognized post-translational modifications that are key to signal transduction.
The field of signaling by reactive species has advanced considerably. But, what remains as an obstacle is still a lack of appreciation by many scientists that reactive species are not merely instruments of cellular torture but of normal cellular physiology.
Acknowledgments
This work was supported in part by ES05511 from the National Institutes of Health and 14RT-0059 from the California Tobacco Related Diseases Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Forman HJ, Torres M, Fukuto J. Mol Cell Biochem. 2002;234–235(1–2):49–62. [PubMed] [Google Scholar]
- 2.Forman HJ, Fukuto J, Torres M. Am J Physiol Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 3.Forman HJ, Torres M. Am J Respir Crit Care Med. 2002;166(12 Pt 2):S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 4.Forman HJ, Fukuto J, Torres M. Signal transduction by reactive oxygen and nitrogen species: pathways and chemical principles. Kluwer Academic Publishers, Dordrecht; Boston: 2003. [Google Scholar]
- 5.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ. Faseb J. 2005;19(7):854–856. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 6.Massey V, Strickland S, Mayhew SG, Howell LG, Engel PC, Matthews RG, Schulman M, Sullivan PA. Biochemical and Biophysical Research Communications. 1969;36:891. doi: 10.1016/0006-291x(69)90287-3. [DOI] [PubMed] [Google Scholar]
- 7.Loschen G, Azzi A, Richter C, Flohe L. FEBS Letters. 1974;42:68. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 8.Forman HJ, Kennedy JA. Biochemical and Biophysical Research Communications. 1974;60:1044–1050. doi: 10.1016/0006-291x(74)90418-5. [DOI] [PubMed] [Google Scholar]
- 9.McCord JM, Fridovich I. J Biol Chem. 1970;245(6):1374–1377. [PubMed] [Google Scholar]
- 10.Lambeth JD. Curr Opin Hematol. 2002;9(1):11–17. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Lambeth JD. Free Radic Biol Med. 2007 doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bedard K, Krause KH. Physiological reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 13.Lambeth JD, Kawahara T, Diebold B. Free Radic Biol Med. 2007 doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiszt M. Cardiovasc Res. 2006;71(2):289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Nauseef WM. Histochem Cell Biol. 2004 doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 16.Griffith OW, Stuehr DJ. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Poulos TL. J Inorg Biochem. 2005;99(1):293–305. doi: 10.1016/j.jinorgbio.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 18.Stuehr DJ. Annu Rev Pharmacol Toxicol. 1997;37:339–359. doi: 10.1146/annurev.pharmtox.37.1.339. [DOI] [PubMed] [Google Scholar]
- 19.Stuehr DJ. J Nutr. 2004;134(10 Suppl):2748S–2751S. doi: 10.1093/jn/134.10.2748S. discussion 2765S–2767S. [DOI] [PubMed] [Google Scholar]
- 20.Botzen D, Grune T. Redox Rep. 2007;12(1):63–67. doi: 10.1179/135100007X162130. [DOI] [PubMed] [Google Scholar]
- 21.Ignarro LJ. Semin Hematol. 1989;26(1):63–76. [PubMed] [Google Scholar]
- 22.Flohé L. Sulfur and selenium catalysis as paradigms for redox regulations. In: Forman HJ, Fukuto J, Torres M, editors. Signal transduction by reactive oxygen and nitrogen species: pathways and chemical principles. Kluwer Academic Publishers, Dordrecht; Boston: 2003. [Google Scholar]
- 23.Manevich Y, Fisher AB. Free Radic Biol Med. 2005;38(11):1422–1432. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Jacob C, Knight I, Winyard PG. Biol Chem. 2006;387(10–11):1385–1397. doi: 10.1515/BC.2006.174. [DOI] [PubMed] [Google Scholar]
- 25.Winterbourn CC, Metodiewa D. Free Radic Biol Med. 1999;27(3–4):322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 26.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Nature. 2003;423(6941):769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 27.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Nature. 2003;423(6941):773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 28.Wood ZA, Poole LB, Karplus PA. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 29.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Curr Opin Cell Biol. 2005;17(2):183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Zhang H, Iles KE, Rinna A, Merrill G, Yodoi J, Torres M, Forman HJ. Free Radic Res. 2006;40:865–874. doi: 10.1080/10715760600758514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bozonet SM, Findlay VJ, Day AM, Cameron J, Veal EA, Morgan BA. J Biol Chem. 2005;280(24):23319–23327. doi: 10.1074/jbc.M502757200. [DOI] [PubMed] [Google Scholar]
- 32.Veal EA, Findlay VJ, Day AM, Bozonet SM, Evans JM, Quinn J, Morgan BA. Mol Cell. 2004;15(1):129–139. doi: 10.1016/j.molcel.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 33.Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. Proc Natl Acad Sci U S A. 2005;102(25):8875–8880. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li S, Whorton AR. Arch Biochem Biophys. 2003;410(2):269–279. doi: 10.1016/s0003-9861(02)00696-3. [DOI] [PubMed] [Google Scholar]
- 35.Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. J Biol Chem. 1999;274(49):34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 36.Lee SR, Kwon KS, Kim SR, Rhee SG. J Biol Chem. 1998;273(25):15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 37.Raugei G, Ramponi G, Chiarugi P. Cell and Molecular Life Sciences. 2002;59(6):941–949. doi: 10.1007/s00018-002-8481-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiarugi P, Fiaschi T, Taddei ML, Talini D, Giannoni E, Raugei G, Ramponi G. J Biol Chem. 2001;276(36):33478–33487. doi: 10.1074/jbc.M102302200. [DOI] [PubMed] [Google Scholar]
- 39.Cirri P, Chiarugi P, Camici G, Manao G, Raugei G, Cappugi G, Ramponi G. Eur J Biochem. 1993;214(3):647–657. doi: 10.1111/j.1432-1033.1993.tb17965.x. [DOI] [PubMed] [Google Scholar]
- 40.Meng TC, Fukada T, Tonks NK. Mol Cell. 2002;9(2):387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 41.Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Journal of Biological Chemistry. 1995;270(13):7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 42.Deora AA, Hajjar DP, Lander HM. Biochemistry. 2000;39(32):9901–9908. doi: 10.1021/bi992954b. [DOI] [PubMed] [Google Scholar]
- 43.Lander HM, Ogiste JS, Teng KK, Novofrodsky A. Journal of Biological Chemistry. 1995;270(36):21195–21198. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 44.Williams JG, Pappu K, Campbell SL. Proceedings National Academy of Sciences, USA. 2003;100(11):6376–6381. doi: 10.1073/pnas.1037299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishida M, Schey KL, Takagahara S, Kontani K, Katada T, Urano Y, Nagano T, Nagao T, Kurose H. Journal of Biological Chemistry. 2002;277(11):9036–9042. doi: 10.1074/jbc.M107392200. [DOI] [PubMed] [Google Scholar]
- 46.Song JJ, Lee YJ. Biochemical Journal. 2003;373(Pt 3):845–853. doi: 10.1042/BJ20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. EMBO Rep. 2001;2(3):222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gotoh Y, Cooper JA. J Biol Chem. 1998;273(28):17477–17482. doi: 10.1074/jbc.273.28.17477. [DOI] [PubMed] [Google Scholar]
- 49.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Embo J. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Journal of Biological Chemistry. 2003;278(35):33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- 51.Sun QA, Gladyshev VN. Methods in Enzymology. 2002;347:451–461. doi: 10.1016/s0076-6879(02)47045-0. [DOI] [PubMed] [Google Scholar]
- 52.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Nat Cell Biol. 2002;4(10):743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 53.Sumbayev VV. Arch Biochem Biophys. 2003;415(1):133–136. doi: 10.1016/s0003-9861(03)00199-1. [DOI] [PubMed] [Google Scholar]
- 54.Yin Z, Ivanov VN, Habelhah H, Tew K, Ronai Z. Cancer Res. 2000;60(15):4053–4057. [PubMed] [Google Scholar]
- 55.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Embo J. 1999;18(5):1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elsby R, Kitteringham NR, Goldring CE, Lovatt CA, Chamberlain M, Henderson CJ, Wolf CR, Park BK. Journal of Biological Chemistry. 2003;278(25):22243–22249. doi: 10.1074/jbc.M301211200. [DOI] [PubMed] [Google Scholar]
- 57.Aslund F, Zheng M, Beckwith J, Storz G. Proc Natl Acad Sci U S A. 1999;96(11):6161–6165. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Faseb J. 1999;13(12):1481–1490. doi: 10.1096/fasebj.13.12.1481. [DOI] [PubMed] [Google Scholar]
- 59.Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, Aizawa S, Tanaka H, Kataoka K, Watanabe H, Handa H. J Biol Chem. 2002;277(46):44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 60.Brady KD, Giegel DA, Grinnell C, Lunney E, Talanian RV, Wong W, Walker N. Bioorganic & Medicinal Chemistry. 1999;7(4):621–631. doi: 10.1016/s0968-0896(99)00009-7. [DOI] [PubMed] [Google Scholar]
- 61.Barford D. Curr Opin Struct Biol. 2004;14(6):679–686. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 62.Rhee S, Bae YS, Lee SR, Kwon J. Sci STKE. 2000;53:PE1. doi: 10.1126/stke.2000.53.pe1. [DOI] [PubMed] [Google Scholar]
- 63.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Biochemistry. 1999;38(20):6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 64.Krejsa CM, Nadler SG, Esselstyn JM, Kavanagh TJ, Ledbetter JA, Schieven GL. J Biol Chem. 1997;272(17):11541–11549. doi: 10.1074/jbc.272.17.11541. [DOI] [PubMed] [Google Scholar]
- 65.Hunter T. Harvey Lect. 1999;94:81–119. [PubMed] [Google Scholar]
- 66.Tonks NK. FEBS Lett. 2003;546(1):140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- 67.Dube N, Cheng AMLT. Proc Natl Acad Sci U S A. 2004;101(7):1834–1839. doi: 10.1073/pnas.0304242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barford D. Curr Opin Struct Biol. 1995;5(6):728–734. doi: 10.1016/0959-440x(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 69.Neel B, Tonks NK. Curr Opin Cell Biol. 1997;2:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 70.Pannifer A, Flint AJ, Tonks NK, Barford D. J Biol Chem. 1998;273(17):10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 71.Biteau B, Labarre J, Toledano MB. Nature. 2003;425(6961):980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 72.Mahadev K, Wu X, Motoshima H, Goldstein BJ. Cell Signal. 2004;16(3):323–331. doi: 10.1016/j.cellsig.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 73.Singh DK, Kumar D, Siddiqui Z, Basu SK, Kumar V, Rao KV. Cell. 2005;121(2):281–293. doi: 10.1016/j.cell.2005.02.036. [DOI] [PubMed] [Google Scholar]
- 74.Tonks N. Cell. 2005;121(5):667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 75.Seth D, Rudolph J. Biochemistry. 2006;45(28):8476–8487. doi: 10.1021/bi060157p. [DOI] [PubMed] [Google Scholar]
- 76.Schuppe-Koistinen I, Gerdes R, Moldeus P, Cotgreave IA. Arch Biochem Biophys. 1994;315(2):226–234. doi: 10.1006/abbi.1994.1494. [DOI] [PubMed] [Google Scholar]
- 77.Kil IS, Park JW. J Biol Chem. 2005;280(11):10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- 78.Brar SS, Grigg C, Wilson KS, Holder WD, Jr, Dreau D, Austin C, Foster M, Ghio AJ, Whorton AR, Stowell GW, Whittall LB, Whittle RR, White DP, Kennedy TP. Mol Cancer Ther. 2004;3(9):1049–1060. [PubMed] [Google Scholar]
- 79.Fratelli M, Goodwin LO, Orom UA, Lombardi S, Tonelli R, Mengozzi M, Ghezzi P. Proc Natl Acad Sci U S A. 2005;102(39):13998–14003. doi: 10.1073/pnas.0504398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klatt P, Lamas S. Methods Enzymol. 2002;348:157–174. doi: 10.1016/s0076-6879(02)48635-1. [DOI] [PubMed] [Google Scholar]
- 81.Cruz C, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. J Biol Chem. 2007;282(5):2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Newman S, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, Klein JB, Turner DM, Butterfield DA. J Neurosci Res. 2007;85(7):1506–1514. doi: 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]
- 83.Rinna A, Torres M, Forman HJ. Free Radic Biol Med. 2006;41(1):86–91. doi: 10.1016/j.freeradbiomed.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rossi R, Giustarini D, Milzani A, Dalle-Donne I. Blood Cells Mol Dis. 2006;37(3):180–187. doi: 10.1016/j.bcmd.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 85.Sparaco M, Gaeta LM, Tozzi G, Bertini E, Pastore A, Simonati A, Santorelli FM, Piemonte F. J Neurosci Res. 2006;83(2):256–263. doi: 10.1002/jnr.20729. [DOI] [PubMed] [Google Scholar]
- 86.Velu C, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS. Biochemistry. 2007;46(26):7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Forman HJ. Free Radic Biol Med. 2007;42(7):926–932. doi: 10.1016/j.freeradbiomed.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beer S, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. J Biol Chem. 2004;279(46):47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 89.Suzuki YJ, Forman HJ, Sevanian A. Free Radic Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 90.Hoyal CR, Giron-Calle J, Forman HJ. J Toxicol Environ Health B Crit Rev. 1998;1(2):117–134. doi: 10.1080/10937409809524547. [DOI] [PubMed] [Google Scholar]
- 91.Marchesini N, Hannun YA. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2004;82(1):27–44. doi: 10.1139/o03-091. [DOI] [PubMed] [Google Scholar]
- 92.Carrasco MA, Jaimovich E, Kemmerling U, Hidalgo C. Biological research. 2004;37(4):701–712. doi: 10.4067/s0716-97602004000400028. [DOI] [PubMed] [Google Scholar]
- 93.Goldkorn T, Ravid T, Khan EM. Antioxid Redox Signal. 2005;7(1–2):119–128. doi: 10.1089/ars.2005.7.119. [DOI] [PubMed] [Google Scholar]
- 94.Franklin RA, Rodriguez-Mora OG, Lahair MM, McCubrey JA. Antioxid Redox Signal. 2006;8(9–10):1807–1817. doi: 10.1089/ars.2006.8.1807. [DOI] [PubMed] [Google Scholar]
- 95.Quinlan GJ, Chen Y, Evans TW, Gutteridge JM. Clin Sci (Lond) 2001;100(2):169–182. [PubMed] [Google Scholar]
- 96.Quinlan GJ, Evans TW, Gutteridge JM. Free Radic Biol Med. 2002;33(10):1306–1313. doi: 10.1016/s0891-5849(02)00903-6. [DOI] [PubMed] [Google Scholar]
- 97.Ignarro LJ. Nitric Oxide, Biology and Pathobiology. Academic Press; San Diego: 2000. [Google Scholar]
- 98.Andreopoulos S, Papapetropoulos A. Gen Pharmacol. 2000;34(3):147–157. doi: 10.1016/s0306-3623(00)00062-8. [DOI] [PubMed] [Google Scholar]
- 99.Denninger JW, Marletta MA. Biochim Biophys Acta. 1999;1411(2–3):334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 100.Hobbs AJ. Trends Pharmacol Sci. 1997;18(12):484–491. doi: 10.1016/s0165-6147(97)01137-1. [DOI] [PubMed] [Google Scholar]
- 101.Moncada S, Palmer RMJ, Higgs EA. Pharmacological reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- 102.Clancy RM, Abramson SB. Arthritis Rheum. 2000;43(10):2260–2264. doi: 10.1002/1529-0131(200010)43:10<2260::AID-ANR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 103.Lloyd-Jones DM, Bloch KD. Annual review of medicine. 1996;47:365–375. doi: 10.1146/annurev.med.47.1.365. [DOI] [PubMed] [Google Scholar]
- 104.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Nature reviews. 2005;6(2):150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 105.Martinez-Ruiz A, Lamas S. Cardiovasc Res. 2004;62(1):43–52. doi: 10.1016/j.cardiores.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 106.Martínez-Ruiz AaLS. Cardiovascular Research. 2007;75(2):9. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Y, Hogg N. Free Radic Biol Med. 2005;38(7):831–838. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 108.Liu X, Miller MJ, Joshi MS, Thomas DD, Lancaster JR., Jr Proc Natl Acad Sci U S A. 1998;95(5):2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kolberg M, Strand KR, Graff P, Andersson KK. Biochim Biophys Acta. 2004;1699(1–2):1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 110.Giles NM, Giles GI, Jacob C. Biochem Biophys Res Commun. 2003;300(1):1–4. doi: 10.1016/s0006-291x(02)02770-5. [DOI] [PubMed] [Google Scholar]
- 111.Ford E, Hughes MN, Wardman P. Free Radic Biol Med. 2002;32(12):1314–1323. doi: 10.1016/s0891-5849(02)00850-x. [DOI] [PubMed] [Google Scholar]
- 112.Lim MD, Lorkovic IM, Ford PC. J Inorg Biochem. 2005;99(1):151–165. doi: 10.1016/j.jinorgbio.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 113.Tannenbaum SR, White FM. ACS chemical biology. 2006;1(10):615–618. doi: 10.1021/cb600439h. [DOI] [PubMed] [Google Scholar]
- 114.Mitchell DA, Marletta MA. Nature chemical biology. 2005;1(3):154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 115.Pryor WA, Squadrito GL. Am J Physiol. 1995;268(5 Pt 1):L699–722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 116.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Biochemistry. 2003;42(33):9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 117.Augusto O, Lopes de Menezes S, Linares E, Romero N, Radi R, Denicola A. Biochemistry. 2002;41(48):14323–14328. doi: 10.1021/bi0262202. [DOI] [PubMed] [Google Scholar]
- 118.Fukuto JM, Ignarro LJ. Accounts of Chemical Research. 1997;30:149–152. [Google Scholar]
- 119.Bartesaghi S, Ferrer-Sueta G, Peluffo G, Valez V, Zhang H, Kalyanaraman B, Radi R. Amino acids. 2007;32(4):501–515. doi: 10.1007/s00726-006-0425-8. [DOI] [PubMed] [Google Scholar]
- 120.Radi R. Proc Natl Acad Sci U S A. 2004;101(12):4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stubbe J, van Der Donk WA. Chem Rev. 1998;98(2):705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 122.Gunther MR, Hsi LC, Curtis JF, Gierse JK, Marnett LJ, Eling TE, Mason RP. J Biol Chem. 1997;272(27):17086–17090. doi: 10.1074/jbc.272.27.17086. [DOI] [PubMed] [Google Scholar]
- 123.Koeck T, Fu X, Hazen SL, Crabb JW, Stuehr DJ, Aulak KS. J Biol Chem. 2004;279(26):27257–27262. doi: 10.1074/jbc.M401586200. [DOI] [PubMed] [Google Scholar]
- 124.Kuo WN, Kanadia RN, Shanbhag VP, Toro R. Mol Cell Biochem. 1999;201:11–16. doi: 10.1023/a:1007024126947. [DOI] [PubMed] [Google Scholar]
- 125.Kuo WN, Kocis JM, Mewar M. J Biochem Mol Biol Biophys. 2002;6(2):143–146. doi: 10.1080/10258140290027298. [DOI] [PubMed] [Google Scholar]
- 126.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. J Biol Chem. 2005;280(51):42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coles B, Bloodsworth A, Eiserich JP, Coffey MJ, McLoughlin RM, Giddings JC, Lewis MJ, Haslam RJ, Freeman BA, O’Donnell VB. J Biol Chem. 2002;277(8):5832–5840. doi: 10.1074/jbc.M105209200. [DOI] [PubMed] [Google Scholar]
- 128.Lim DG, Sweeney S, Bloodsworth A, White CR, Chumley PH, Krishna NR, Schopfer F, O’Donnell VB, Eiserich JP, Freeman BA. Proc Natl Acad Sci U S A. 2002;99(25):15941–15946. doi: 10.1073/pnas.232409599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wright MM, Schopfer FJ, Baker PR, Vidyasagar V, Powell P, Chumley P, Iles KE, Freeman BA, Agarwal A. Proc Natl Acad Sci U S A. 2006;103(11):4299–4304. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Trostchansky A, Souza JM, Ferreira A, Ferrari M, Blanco F, Trujillo M, Castro D, Cerecetto H, Baker PR, O’Donnell VB, Rubbo H. Biochemistry. 2007;46(15):4645–4653. doi: 10.1021/bi602652j. [DOI] [PubMed] [Google Scholar]
- 131.O’Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Chem Res Toxicol. 1999;12(1):83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]
- 132.Lima ES, Bonini MG, Augusto O, Barbeiro HV, Souza HP, Abdalla DS. Free Radic Biol Med. 2005;39(4):532–539. doi: 10.1016/j.freeradbiomed.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 133.Schopfer FJ, Baker PR, Giles G, Chumley P, Batthyany C, Crawford J, Patel RP, Hogg N, Branchaud BP, Lancaster JR, Jr, Freeman BA. J Biol Chem. 2005;280(19):19289–19297. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- 134.Cooper CE. Trends Biochem Sci. 2002;27(1):33–39. doi: 10.1016/s0968-0004(01)02035-7. [DOI] [PubMed] [Google Scholar]
- 135.Shiva S, Oh JY, Landar AL, Ulasova E, Venkatraman A, Bailey SM, Darley-Usmar VM. Free Radic Biol Med. 2005;38(3):297–306. doi: 10.1016/j.freeradbiomed.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 136.Metzen E, Ratcliffe PJ. Biol Chem. 2004;385(3–4):223–230. doi: 10.1515/BC.2004.016. [DOI] [PubMed] [Google Scholar]
- 137.Siddiq A, Aminova LR, Ratan RR. Neurochem Res. 2007;32(4–5):931–946. doi: 10.1007/s11064-006-9268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B. Mol Biol Cell. 2003;14(8):3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hagen T, Taylor CT, Lam F, Moncada S. Science. 2003;302(5652):1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 140.Doyle MP, Hoekstra JW. J Inorg Biochem. 1981;14(4):351–358. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 141.Ford PC, Wink DA, Stanbury DM. FEBS Lett. 1993;326(1–3):1–3. doi: 10.1016/0014-5793(93)81748-o. [DOI] [PubMed] [Google Scholar]
- 142.Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, Scheeren T, Godecke A, Schrader J, Schulz R, Heusch G, Schaub GA, Bryan NS, Feelisch M, Kelm M. Free Radic Biol Med. 2003;35(7):790–796. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 143.Lundberg JO, Weitzberg E, Cole JA, Benjamin N. Nat Rev Microbiol. 2004;2(7):593–602. doi: 10.1038/nrmicro929. [DOI] [PubMed] [Google Scholar]
- 144.Modin A, Bjorne H, Herulf M, Alving K, Weitzberg E, Lundberg JO. Acta physiologica Scandinavica. 2001;171(1):9–16. doi: 10.1046/j.1365-201X.2001.00771.x. [DOI] [PubMed] [Google Scholar]
- 145.Rodriguez J, Maloney RE, Rassaf T, Bryan NS, Feelisch M. Proc Natl Acad Sci U S A. 2003;100(1):336–341. doi: 10.1073/pnas.0234600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Nat Med. 1995;1(8):804–809. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 147.Fukuto JM, Cho JY, Switzer CH. The Chemical Properties of Nitric Oxide and Realted Nitrogen Oxides. In: Ignarro L, editor. Nitric Oxide. Academic Press; San Diego: 2000. [Google Scholar]
- 148.Furchgott RF, Bhadrakom S. J Pharmacol Exp Ther. 1953;108(2):129–143. [PubMed] [Google Scholar]
- 149.Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, Kelm M. Proc Natl Acad Sci U S A. 2001;98(22):12814–12819. doi: 10.1073/pnas.221381098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dykhuizen RS, Frazer R, Duncan C, Smith CC, Golden M, Benjamin N, Leifert C. Antimicrob Agents Chemother. 1996;40(6):1422–1425. doi: 10.1128/aac.40.6.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kozlov AV, Staniek K, Nohl H. FEBS Lett. 1999;454(1–2):127–130. doi: 10.1016/s0014-5793(99)00788-7. [DOI] [PubMed] [Google Scholar]
- 152.Nohl H, Staniek K, Sobhian B, Bahrami S, Redl H, Kozlov AV. Acta Biochim Pol. 2000;47(4):913–921. [PubMed] [Google Scholar]
- 153.Alikulov ZA, L’Vov NP, Kretovich VL. Biokhimiia. 1980;45(9):1714–1718. [PubMed] [Google Scholar]
- 154.Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, Harrison R. J Biol Chem. 2000;275(11):7757–7763. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- 155.Millar TM, Stevens CR, Benjamin N, Eisenthal R, Harrison R, Blake DR. FEBS Lett. 1998;427(2):225–228. doi: 10.1016/s0014-5793(98)00430-x. [DOI] [PubMed] [Google Scholar]
- 156.Zhang Z, Naughton D, Winyard PG, Benjamin N, Blake DR, Symons MC. Biochem Biophys Res Commun. 1998;249(3):767–772. doi: 10.1006/bbrc.1998.9226. [DOI] [PubMed] [Google Scholar]
- 157.Zhang Z, Naughton DP, Blake DR, Benjamin N, Stevens CR, Winyard PG, Symons MC, Harrison R. Biochem Soc Trans. 1997;25(3):524S. doi: 10.1042/bst025524s. [DOI] [PubMed] [Google Scholar]
- 158.Li H, Samouilov A, Liu X, Zweier JL. J Biol Chem. 2001;276(27):24482–24489. doi: 10.1074/jbc.M011648200. [DOI] [PubMed] [Google Scholar]
- 159.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, 3rd, Gladwin MT. Nat Med. 2003;9(12):1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 160.Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. J Biol Chem. 2003;278(47):46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 161.Kim-Shapiro DB, Gladwin MT, Patel RP, Hogg N. J Inorg Biochem. 2005;99(1):237–246. doi: 10.1016/j.jinorgbio.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 162.Cooper CE. Biochim Biophys Acta. 1999;1411(2–3):290–309. doi: 10.1016/s0005-2728(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 163.Poli G, Albano E, Dianzani MU. Chemistry and physics of lipids. 1987;45(2–4):117–142. doi: 10.1016/0009-3084(87)90063-6. [DOI] [PubMed] [Google Scholar]
- 164.Esterbauer H, Schaur RJ, Zollner H. Free Radical Biology & Medicine. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 165.Strohmaier H, Hinghofer-Szalkay H, Schaur RJ. J Lipid Mediat Cell Signal. 1995;11(1):51–61. doi: 10.1016/0929-7855(94)00027-a. [DOI] [PubMed] [Google Scholar]
- 166.Schwarzer E, Muller O, Arese P, Siems WG, Grune T. FEBS Lett. 1996;388(2–3):119–122. doi: 10.1016/0014-5793(96)00523-6. [DOI] [PubMed] [Google Scholar]
- 167.Uchida K. Prog Lipid Res. 2003;42(4):318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 168.Zarkovic N. Mol Aspects Med. 2003;24(4–5):281–291. doi: 10.1016/s0098-2997(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 169.Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Thorpe SR. Nephrol Dial Transplant. 1996;11(Suppl 5):48–53. doi: 10.1093/ndt/11.supp5.48. [DOI] [PubMed] [Google Scholar]
- 170.Floyd RA, Hensley K. Neurobiol Aging. 2002;23(5):795–807. doi: 10.1016/s0197-4580(02)00019-2. [DOI] [PubMed] [Google Scholar]
- 171.Leonarduzzi G, Scavazza A, Biasi F, Chiarpotto E, Camandola S, Vogl S, Dargel R, Poli G. FASEB Journal. 1997;11:851–857. doi: 10.1096/fasebj.11.11.9285483. [DOI] [PubMed] [Google Scholar]
- 172.Srivastava S, Chandra A, Ansari NH, Srivastava SK, Bhatnagar A. Biochem J. 1998;329(Pt 3):469–475. doi: 10.1042/bj3290469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Reichard JF, Vasiliou V, Petersen DR. Biochim Biophys Acta. 2000;1487(2–3):222–232. doi: 10.1016/s1388-1981(00)00095-0. [DOI] [PubMed] [Google Scholar]
- 174.Alary J, Gueraud F, Cravedi JP. Mol Aspects Med. 2003;24(4–5):177–187. doi: 10.1016/s0098-2997(03)00012-8. [DOI] [PubMed] [Google Scholar]
- 175.Siems W, Grune T. Mol Aspects Med. 2003;24(4–5):167–175. doi: 10.1016/s0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- 176.Srivastava SK, Singhal SS, Bajpai KK, Chaubey M, Ansari NH, Awasthi YC. Exp Eye Res. 1994;59(2):151–159. doi: 10.1006/exer.1994.1093. [DOI] [PubMed] [Google Scholar]
- 177.Singhal SS, Zimniak P, Awasthi S, Piper JT, He NG, Teng JI, Petersen DR, Awasthi YC. Archives of Biochemistry and Biophysics. 1994;311:242–250. doi: 10.1006/abbi.1994.1233. [DOI] [PubMed] [Google Scholar]
- 178.Singhal SS, Awasthi S, Srivastava SK, Zimniak P, Ansari NH, Awasthi YC. Invest Ophthalmol Vis Sci. 1995;36(1):142–150. [PubMed] [Google Scholar]
- 179.Forman HJ, Dickinson DA, Iles KE. Mol Aspects Med. 2003;24(4–5):189–194. doi: 10.1016/s0098-2997(03)00013-x. [DOI] [PubMed] [Google Scholar]
- 180.Cheng JZ, Sharma R, Yang Y, Singhal SS, Sharma A, Saini MK, Singh SV, Zimniak P, Awasthi S, Awasthi YC. Journal of Biological Chemistry. 2001;276(44):41213–41223. doi: 10.1074/jbc.M106838200. [DOI] [PubMed] [Google Scholar]
- 181.Spycher S, Tabataba-Vakili S, O’Donnell VB, Palomba L, Azzi A. Biochemical and Biophysical Research Communications. 1996;226(2):512–516. doi: 10.1006/bbrc.1996.1386. [DOI] [PubMed] [Google Scholar]
- 182.Petersen DR, Doorn JA. Free Radic Biol Med. 2004;37(7):937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 183.Schaur RJ. Mol Aspects Med. 2003;24(4–5):149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 184.Eckl PM. Mol Aspects Med. 2003;24(4–5):161–165. doi: 10.1016/s0098-2997(03)00010-4. [DOI] [PubMed] [Google Scholar]
- 185.Friguet B, Szweda LI. FEBS Lett. 1997;405(1):21–25. doi: 10.1016/s0014-5793(97)00148-8. [DOI] [PubMed] [Google Scholar]
- 186.Okada K, Wangpoengtrakul C, Osawa T, Toyokuni S, Tanaka K, Uchida K. J Biol Chem. 1999;274(34):23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 187.Dianzani MU, Barrera G, Parola M. Acta Biochim Pol. 1999;46(1):61–75. [PubMed] [Google Scholar]
- 188.Cambiaggi C, Dominici S, Comporti M, Pompella A. Life Sci. 1997;61(8):777–785. doi: 10.1016/s0024-3205(97)00559-6. [DOI] [PubMed] [Google Scholar]
- 189.Semlitsch T, Tillian HM, Zarkovic N, Borovic S, Purtscher M, Hohenwarter O, Schaur RJ. Anticancer Res. 2002;22(3):1689–1697. [PubMed] [Google Scholar]
- 190.Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC. Acta Biochim Pol. 2003;50(2):319–336. [PubMed] [Google Scholar]
- 191.Rinaldi M, Barrera G, Spinsanti P, Pizzimenti S, Ciafre SA, Parella P, Farace MG, Signori E, Dianzani MU, Fazio VM. Free Radic Res. 2001;34(6):629–637. doi: 10.1080/10715760100300521. [DOI] [PubMed] [Google Scholar]
- 192.Fukuda A, Nakamura Y, Ohigashi H, Osawa T, Uchida K. Biochemical and Biophysical Research Communications. 1997;236:505–509. doi: 10.1006/bbrc.1997.6585. [DOI] [PubMed] [Google Scholar]
- 193.Singhal SS, Godley BF, Chandra A, Pandya U, Jin GF, Saini MK, Awasthi S, Awasthi YC. Invest Ophthalmol Vis Sci. 1999;40(11):2652–2659. [PubMed] [Google Scholar]
- 194.Raza H, Robin MA, Fang JK, Avadhani NG. Biochem J. 2002;366(Pt 1):45–55. doi: 10.1042/BJ20020533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, Kimura H. Biochem J. 2004;381(Pt 1):113–123. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang H, Dickinson DA, Liu RM, Forman HJ. Free Radic Biol Med. 2005;38:463–471. doi: 10.1016/j.freeradbiomed.2004.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. Free Radic Biol Med. 2002;33(7):974–987. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 198.Zhang H, Court N, Forman HJ. Redox Rep. 2007;12(1):101–106. doi: 10.1179/135100007X162266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chiarpotto E, Domenicotti C, Paola D, Vitali A, Nitti M, Pronzato MA, Biasi F, Cottalasso D, Marinari UM, Dragonetti A, Cesaro P, Isidoro C, Poli G. Hepatology. 1999;29(5):1565–1572. doi: 10.1002/hep.510290510. [DOI] [PubMed] [Google Scholar]
- 200.Nitti M, Domenicotti C, d’Abramo C, Assereto S, Cottalasso D, Melloni E, Poli G, Biasi F, Marinari UM, Pronzato MA. Biochem Biophys Res Commun. 2002;294(3):547–552. doi: 10.1016/S0006-291X(02)00512-0. [DOI] [PubMed] [Google Scholar]
- 201.Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Am J Physiol Cell Physiol. 2003;285(2):C334–342. doi: 10.1152/ajpcell.00043.2003. [DOI] [PubMed] [Google Scholar]
- 202.Gopalakrishna R, Jaken S. Free Radic Biol Med. 2000;28(9):1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 203.Torres M, Forman HJ. Biofactors. 2003;17(1–4):287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 204.Kakishita H, Hattori Y. Life Sci. 2001;69(6):689–697. doi: 10.1016/s0024-3205(01)01166-3. [DOI] [PubMed] [Google Scholar]
- 205.Soh Y, Jeong KS, Lee IJ, Bae MA, Kim YC, Song BJ. Mol Pharmacol. 2000;58(3):535–541. doi: 10.1124/mol.58.3.535. [DOI] [PubMed] [Google Scholar]
- 206.Camandola S, Poli G, Mattson MP. Journal of Neurochemistry. 2000;74(1):159–168. doi: 10.1046/j.1471-4159.2000.0740159.x. [DOI] [PubMed] [Google Scholar]
- 207.Tamagno E, Robino G, Obbili A, Bardini P, Aragno M, Parola M, Danni O. Exp Neurol. 2003;180(2):144–155. doi: 10.1016/s0014-4886(02)00059-6. [DOI] [PubMed] [Google Scholar]
- 208.Bruckner SR, Estus S. J Neurosci Res. 2002;70(5):665–670. doi: 10.1002/jnr.10437. [DOI] [PubMed] [Google Scholar]
- 209.Song BJ, Soh Y, Bae M, Pie J, Wan J, Jeong K. Chem Biol Interact. 2001;130–132(1–3):943–954. doi: 10.1016/s0009-2797(00)00247-7. [DOI] [PubMed] [Google Scholar]
- 210.Kumagai T, Nakamura Y, Osawa T, Uchida K. Arch Biochem Biophys. 2002;397(2):240–245. doi: 10.1006/abbi.2001.2601. [DOI] [PubMed] [Google Scholar]
- 211.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, Gentilini P, Dianzani MU. J Clin Invest. 1998;102(11):1942–1950. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rinna A, Forman HJ. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2007-0371OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Shi Q, Vaillancourt F, Cote V, Fahmi H, Lavigne P, Afif H, Di Battista JA, Fernandes JC, Benderdour M. Arthritis research & therapy. 2006;8(6):R159. doi: 10.1186/ar2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Fiaschi T, Buricchi F, Cozzi G, Matthias S, Parri M, Raugei G, Ramponi G, Chiarugi P. Hepatology. 2007;46(1):130–139. doi: 10.1002/hep.21643. [DOI] [PubMed] [Google Scholar]
- 215.Burlando B, Magnelli V, Panfoli I, Berti E, Viarengo A. Cell Physiol Biochem. 2003;13(3):147–154. doi: 10.1159/000071865. [DOI] [PubMed] [Google Scholar]
- 216.Saito S, Frank GD, Mifune M, Ohba M, Utsunomiya H, Motley ED, Inagami T, Eguchi S. J Biol Chem. 2002;277(47):44695–44700. doi: 10.1074/jbc.M208332200. [DOI] [PubMed] [Google Scholar]
- 217.Meves A, Stock SN, Beyerle A, Pittelkow MR, Peus D. Toxicol Lett. 2001;122(3):205–214. doi: 10.1016/s0378-4274(01)00359-9. [DOI] [PubMed] [Google Scholar]
- 218.Liu W, Akhand AA, Kato M, Yokoyama I, Miyata T, Kurokawa K, Uchida K, Nakashima I. J Cell Sci. 1999;112(Pt 14):2409–2417. doi: 10.1242/jcs.112.14.2409. [DOI] [PubMed] [Google Scholar]
- 219.Negre-Salvayre A, Vieira O, Escargueil-Blanc I, Salvayre R. Mol Aspects Med. 2003;24(4–5):251–261. doi: 10.1016/s0098-2997(03)00020-7. [DOI] [PubMed] [Google Scholar]
- 220.Escargueil-Blanc I, Salvayre R, Vacaresse N, Jurgens G, Darblade B, Arnal JF, Parthasarathy S, Negre-Salvayre A. Circulation. 2001;104(15):1814–1821. doi: 10.1161/hc4001.097179. [DOI] [PubMed] [Google Scholar]
- 221.Hool LC, Corry B. Antioxid Redox Signal. 2007;9(4):409–435. doi: 10.1089/ars.2006.1446. [DOI] [PubMed] [Google Scholar]
- 222.Carini R, Bellomo G, Paradisi L, Dianzani MU, Albano E. Biochemical and Biophysical Research Communications. 1996;218:772–776. doi: 10.1006/bbrc.1996.0137. [DOI] [PubMed] [Google Scholar]
- 223.Malecki A, Garrido R, Mattson MP, Hennig B, Toborek M. J Neurochem. 2000;74(6):2278–2287. doi: 10.1046/j.1471-4159.2000.0742278.x. [DOI] [PubMed] [Google Scholar]
- 224.Raess BU, Keenan CE, McConnell EJ. Biochem Biophys Res Commun. 1997;235(3):451–454. doi: 10.1006/bbrc.1997.6819. [DOI] [PubMed] [Google Scholar]
- 225.Siems W, Capuozzo E, Lucano A, Salerno C, Crifo C. Life Sci. 2003;73(20):2583–2590. doi: 10.1016/s0024-3205(03)00661-1. [DOI] [PubMed] [Google Scholar]
- 226.Lu C, Chan SL, Fu W, Mattson MP. J Biol Chem. 2002;277(27):24368–24375. doi: 10.1074/jbc.M201924200. [DOI] [PubMed] [Google Scholar]
- 227.Akaishi T, Nakazawa K, Sato K, Saito H, Ohno Y, Ito Y. Biol Pharm Bull. 2004;27(2):174–179. doi: 10.1248/bpb.27.174. [DOI] [PubMed] [Google Scholar]
- 228.Natarajan V, Scribner WM, Taher MM. Free Radical Biology & Medicine. 1993;15:365–375. doi: 10.1016/0891-5849(93)90036-t. [DOI] [PubMed] [Google Scholar]
- 229.Rossi MA, Garramone A, Dianzani MU. International journal of tissue reactions. 1988;10(5):321–325. [PubMed] [Google Scholar]
- 230.Dozza B, Smith MA, Perry G, Tabaton M, Strocchi P. J Neurochem. 2004;89(5):1224–1232. doi: 10.1111/j.1471-4159.2004.02413.x. [DOI] [PubMed] [Google Scholar]
- 231.Nakashima I, Liu W, Akhand AA, Takeda K, Kawamoto Y, Kato M, Suzuki H. Mol Aspects Med. 2003;24(4–5):231–238. doi: 10.1016/s0098-2997(03)00018-9. [DOI] [PubMed] [Google Scholar]
- 232.Dianzani C, Parrini M, Ferrara C, Fantozzi R. Cell Biochem Funct. 1996;14(3):193–200. doi: 10.1002/cbf.683. [DOI] [PubMed] [Google Scholar]
- 233.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N. Free Radic Biol Med. 2004;37(6):755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 234.Echtay KS. Free Radic Biol Med. 2007;43(10):1351–1371. doi: 10.1016/j.freeradbiomed.2007.08.011. [DOI] [PubMed] [Google Scholar]