

Abstract
In recent years, dihydrodipicolinate synthase (DHDPS, E.C. 4.2.1.52) has received considerable attention from a mechanistic and structural viewpoint. DHDPS catalyzes the reaction of (S)-aspartate-β-semialdehyde with pyruvate, which is bound via a Schiff base to a conserved active-site lysine (Lys161 in the enzyme from Escherichia coli). To probe the mechanism of DHDPS, we have studied the inhibition of E. coli DHDPS by the substrate analog, β-hydroxypyruvate. The K i was determined to be 0.21 (±0.02) mM, similar to that of the allosteric inhibitor, (S)-lysine, and β-hydroxypyruvate was observed to cause time-dependent inhibition. The inhibitory reaction with β-hydroxypyruvate could be qualitatively followed by mass spectrometry, which showed initial noncovalent adduct formation, followed by the slow formation of the covalent adduct. It is unclear whether β-hydroxypyruvate plays a role in regulating the biosynthesis of meso-diaminopimelate and (S)-lysine in E. coli, although we note that it is present in vivo. The crystal structure of DHDPS complexed with β-hydroxypyruvate was solved. The active site clearly showed the presence of the inhibitor covalently bound to the Lys161. Interestingly, the hydroxyl group of β-hydroxypyruvate was hydrogen-bonded to the main-chain carbonyl of Ile203. This provides insight into the possible catalytic role played by this peptide unit, which has a highly strained torsion angle (ω ∼201°). A survey of the known DHDPS structures from other organisms shows this distortion to be a highly conserved feature of the DHDPS active site, and we propose that this peptide unit plays a critical role in catalysis.
Keywords: dihydrodipicolinate synthase, lysine biosynthesis, conserved peptide distortion, X-ray crystallography
In the biosynthetic pathway leading to meso-diaminopimelate (DAP) and (S)-lysine in plants and bacteria, dihydrodipicolinate synthase (DHDPS, E.C. 4.2.1.52) catalyzes the branch point reaction: an aldol-like condensation of (S)-aspartate-β-semialdehyde [(S)-ASA] and pyruvate, to form (4S)-4-hydroxy-2,3,4,5-tetrahydro-(2S)-dipicolinic acid (HTPA, Scheme 1). Since (S)-lysine biosynthesis does not occur in animals, pathway members, such as DHDPS, are attractive targets for rational antibiotic and herbicide design (Coulter et al. 1999; Hutton et al. 2003, 2007). However, a potent inhibitor of DHDPS has yet to be characterized. As the purported control point in the (S)-lysine biosynthetic pathway, DHDPS is also of interest to those aiming to engineer plants rich in (S)-lysine, often the limiting nutrient in staple crops (Miflin et al. 1999).
Scheme 1.

Reaction catalyzed by DHDPS.
DHDPS from Escherichia coli has been extensively studied in the past decade from a mechanistic viewpoint. A combination of mutagenic and structural studies of the E. coli enzyme have both highlighted the role of the proton-relay and identified the residues responsible for binding the substrates and the allosteric effector, (S)-lysine (Mirwaldt et al. 1995; Blickling et al. 1997; Lawrence et al. 1997; Dobson et al. 2004b). (S)-Lysine inhibition of DHDPS [and the regulation of (S)-lysine biosynthesis] has been characterized in a number of plant species and Gram-negative bacterial species, but appears to be absent in the surveyed Gram-positive bacterial species (Patte 1996). A mode of inhibition has been proposed for DHDPS from Nicotiana sylvestris DHDPS, but thus far an inhibition mechanism for the E. coli enzyme is elusive. A number of structural studies have focused on DHDPS enzymes from a variety of other organisms (Blagova et al. 2006; Pearce et al. 2006; Kefala et al. 2008). With few exceptions, e.g., Thermotoga maritima DHDPS (Pearce et al. 2006), their equally important biochemical characterizations have been slow to follow.
In terms of structural features, the E. coli DHDPS enzyme consists of a homotetramer, which is made up of a dimer of tight dimers (Fig. 1), where there are many interactions between monomers “a” and “b” (or “c” and “d,” Fig. 1), but relatively few between the two tight-dimers “a/b” and “c/d” (Dobson et al. 2005). The active site is situated in the center of the (β/α)8-barrel in each monomer, while the allosteric lysine-binding site is situated in the cleft at the tight-dimer interface (Dobson et al. 2005). The structure of DHDPS from a number of organisms has been determined. Apart from the dimeric DHDPS from Staphylococcus aureus (Burgess et al. 2008) and the structure of a putative DHDPS from Agrobacterium tumefaciens (2HMC), which shows a hexameric structure, all other native DHDPS structures are tetrameric (Griffin et al. 2008).
Figure 1.

Structure of DHDPS from E. coli. The tetrameric structure is shown in A, while the monomeric (α/β)-barrel is shown in B. (C) Topology diagram of the monomeric structure of E. coli DHDPS showing the secondary structure and highlighting the relative positions of selected active-site residues. α-Helices are solid red circles and the β-strands are blue triangles. The residue Tyr107b contributes to the active site of the adjacent monomer of the tight dimer.
The proposed catalytic mechanism facilitated by DHDPS is outlined in Scheme 2, where it is the hydrate form of (S)-ASA that is considered to interact with the enzyme (Blickling et al. 1997). In the first step of the reaction, the ε-amino group of the active-site lysine (Lys161 in E. coli; Fig. 1C) stages a nucleophilic attack on the keto group of pyruvate, resulting in the formation of a Schiff base. Tyr133 is well placed to form a hydrogen bond with the keto oxygen of pyruvate, which accelerates the dehydration of a tetrahedral intermediate during Schiff-base formation. The enamine tautomer of the Schiff base adds to the dehydrated (S)-ASA, and this species then undergoes cyclization to form HTPA. Although Scheme 2 indicates formation of the enamine species prior to the arrival of (S)-ASA in the active site, there is, in fact, no evidence to support this; tautomerization may well be concomitant with binding and/or dehydration of (S)-ASA.
Scheme 2.

Currently accepted catalytic mechanism mediated by DHDPS. (S)-ASA is thought to be in the hydrate form.
Importantly for this work, it is not known which residue(s) are responsible for facilitating proton abstraction during tautomerization to form an enamine (Scheme 2, step 2). As pointed out by Blickling et al. (1997), Tyr133 is unlikely to participate in this proton abstraction, since it is more than 4 Å from the methyl carbon (C3) in the Schiff base and its acute angle would not promote proton transfer. Another candidate is the main-chain oxygen of Ile203 (∼3.4 Å) (Blickling et al. 1997). Here we present evidence that provides a deeper understanding of the DHDPS “catalytic toolkit” (Gutteridge and Thornton 2005), using structural and kinetic data to implicate a highly conserved distorted peptide bond, here Ile203–Ser204, and an associated hydrogen-bonding network in the catalytic cycle of DHDPS.
Results and Discussion
β-Hydroxypyruvate is a time-dependent inhibitor of DHDPS
A number of pyruvate analogs have been found to inhibit DHDPS from E. coli, and these data both provide insights into the catalytic mechanism and reveal avenues for the design of novel antibacterial agents. Karsten (1997) showed that 3-fluoropyruvate, 2-ketobutyrate, and glyoxylate all show inhibition in the submillimolar range, and used this information to elucidate aspects of the kinetic mechanism displayed by DHDPS. We investigated inhibition of DHDPS by another pyruvate analog, β-hydroxypyruvate.
β-Hydroxypyruvate caused little inhibition when added directly to the assay, but appeared to alter the shape of the initial rate progress curve, compared to that observed without β-hydroxypyruvate, a symptom of time-dependent inhibition (Fig. 2). Further experiments revealed the mechanism to be that of the slow-binding variety described by Equation 1. The kinetic parameters were estimated by simultaneous fitting to Equations 2 and 3: the K i was 0.21 (±0.02) mM; k 3 was 0.007 (±0.001) min−1; and k 4 was 0.0021 (±0.0002) min−1. Interestingly, the K i is similar to that of (S)-lysine (Karsten 1997; Dobson et al. 2004a).
Figure 2.

Kinetic inhibition of E. coli DHDPS by β-hydroxypyruvate. E. coli DHDPS was exposed to β-hydroxypyruvate (0.01 mM [solid squares], 0.05 mM [open squares], 0.1 mM [solid circles], 0.5 mM [open circles], 1 mM [solid triangles], 5 mM [open triangles], 10 mM [solid stars], and 20 mM [open stars]) and incubated at 30°C. The initial rate of catalysis was measured at the indicated time points (refer to the Materials and Methods section for details). The ratio of the initial rate in the absence of inhibitor (V o) to that in the presence of inhibitor at each time point (V i) is plotted as a function of time. The V i/V o plot in the presence of both 5 mM β-hydroxypyruvate and 5 mM pyruvate is plotted with cross symbols and a dashed line.
Pyruvate protects against inhibition, suggesting that β-hydroxypyruvate inhibits the enzyme by binding to the active site (Fig. 2, dashed line). Protection by pyruvate is not complete, given that the enzyme–pyruvate complex is in equilibrium with populations of pyruvate and free enzyme, and therefore the enzyme–inhibitor complex will persist. However, from these data, we cannot preclude the binding of β-hydroxypyruvate to the enzyme at locations other than at the active site (see the following data from mass spectrometry and the X-ray model). Another pyruvate analog, 3-bromopyruvate, is a known irreversible inhibitor of DHDPS (Laber et al. 1992), although it does not specifically alkylate residues at the active site (Borthwick et al. 1995).
Presumably, the binding of β-hydroxypyruvate in a noncovalent manner constitutes the fast step (k 1/k 2, Equation 1) and Schiff-base formation (i.e., C–N bond formation) constitutes the slow step (k 3/k 4, Equation 1). In the case of irreversible inhibition of N-acetylneuraminate lyase (NAL) by β-hydroxypyruvate, the adduct is thought to proceed through to an aldehyde, which interacts with the NAL equivalent of Tyr133 in DHDPS, and appears to be essentially irreversible (Lawrence et al. 1997). Whether such an adduct is formed upon coordination with DHDPS was unclear. Accordingly, mass spectrometry data with DHDPS in the presence of β-hydroxypyruvate were sought.
Mass spectrometry analysis of DHDPS in the presence of β-hydroxypyruvate
It was possible to follow the interaction of β-hydroxypyruvate and DHDPS with respect to time using ESI-Q/TOF mass spectrometry at 0, 5, 12, and 25 min following the addition of inhibitor (0.5 mM). The raw data at each time point showed a charge distribution envelope and indicated the presence of adducts, some that were present in the enzyme sample alone (Supplemental Fig. S1A), and others that appeared as a function of time (Supplemental Fig. S1B–E). These data were subsequently deconvoluted using the Mass Analyzer software to yield the mass spectra shown in Figure 3. The spectrum quality and intensity were degraded with the addition of β-hydroxypyruvate; however, it is apparent from Figure 3 that the native monomer (31,270.0 Da) is decreasing in abundance with time, while the adduct at 31,356.3 Da, which represents the native enzyme mass +86.3 Da, increases over time. This adduct is consistent with the covalently attached β-hydroxypyruvate species (Scheme 3). In addition, a peak at 31,373.1 Da (native mass +103.1 Da) is observed, which decreases in relative abundance with time. This is consistent with the noncovalent adduct (Scheme 3) that binds to the active site rapidly (relative to the timescale of the MS experiment). With respect to the kinetic mechanism described in Equation 1, the noncovalent adduct represents the EI complex. The covalent adduct (EI* complex) then forms more slowly, as judged by the loss of the native and noncovalent adducts and the increasing peak at 31,356.3 Da (Fig. 3). This is consistent with the kinetic data and model obtained. It appears that only one inhibitor molecule binds to each monomer of the tetramer. Several other peaks are observed in Figure 3 that are likely to represent buffer or chemical adducts. Two of these peaks are abundant following the addition of β-hydroxypyruvate at 31,311 Da (+41 Da) and 31,332 Da (+62 Da). Neither of these adducts change as a function of time, and may therefore represent low-level contaminants in the commercial β-hydroxypyruvate preparation or an artifact of the method used to collect the data. Nevertheless, the only time-dependent adducts observed by ESI-Q/TOF mass spectrometry are those of β-hydroxypyruvate, which is consistent with the kinetic results (Fig. 2). To support these findings, the structure of DHDPS complexed β-hydroxypyruvate was obtained.
Figure 3.

Deconvoluted mass spectrum of E. coli DHDPS incubated with 0.5 mM β-hydroxypyruvate. Times are indicated on the plot. The asterisk indicates that the 0 time point was generated by addition of the inhibitor to enzyme followed by immediate injection into the mass spectrometer. After 25 min little change in the relative peak intensities was observed.
Scheme 3.

Proposed mechanism of inhibition. Both the noncovalent (A) and covalent (B and/or C) adducts are seen by mass spectrometry; however, only the imine (B) is observed in the crystal structure.
Structure of DHDPS soaked with β-hydroxypyruvate
The global structure of DHDPS complexed with β-hydroxypyruvate (solved to a resolution of 2.4 Å) showed no discernable difference when compared to the wild-type structure (RMSD was 0.13 Å for the overlay of the dimer to 1YXC) (Dobson et al. 2005). The enzyme remains homotetrameric, consisting of four (β/α)8-barrels, each with a C-terminal domain, for which there is no known function (Fig. 1B). The active site is situated within the β-barrel and is identified around the lysine residue at position 161 (Fig. 1C).
The structure of DHDPS soaked with β-hydroxypyruvate was solved to ascertain whether β-hydroxypyruvate binds to more than one site on the enzyme; to determine if the inhibitor binds to the active site and if the binding is covalent; and to resolve the structural form of the bound inhibitor. Defined and continuous electron density links the ε-nitrogen of Lys161 and C2 of β-hydroxypyruvate, which indicates that the inhibitor is covalently bound (Fig. 4A). Informed by the inhibition (Fig. 2) and mass-spectrometry results (Fig. 3), the occupancy of the covalent β-hydroxypyruvate adduct was set to 0.75. The carboxyl moiety was clearly distinguishable, hydrogen bonding to the main-chain N–H moieties of Thr44 and Thr45 and to the side-chain hydroxyl of Thr45 (Fig. 4B); a configuration reminiscent of the native pyruvate complex. An elevated B-factor (compared to atoms in the side chain of Lys161, B-factor of 37.3) for the hydroxyl of β-hydroxypyruvate suggests some mobility. The averaged B-factor values for the β-hydroxypyruvate moiety were: hydroxyl oxygen, 49.5 Å2; carboxyl moiety 39.4 Å2; C2, 42.8 Å2; and C3, 45.9 Å2. The average B-factor for the protein was 41.4 Å2. β-Hydroxypyruvate was modeled as the imine form (Scheme 3, second active site), given that the hydroxyl oxygen appears to form a hydrogen bond to the main-chain oxygen of Ile203 (3.0 Å) (Fig. 4B). The enol form (Scheme 3, third active site) is also possible, but, given that the hydroxyl and NZ are not in plane, this is likely to be a minor species. The connection between the hydroxyl and the main chain of Ile203 was of interest, since the hydrogen bond suggests that the main-chain carbonyl of Ile203, rather than the hydroxyl of Tyr133 or the main chain of Gly186, may be responsible for facilitating proton abstraction during enamine formation.
Figure 4.

Structure of the E. coli DHDPS active site with bound β-hydroxypyruvate. (A) An omit-map of the adduct bound to Lys161 in the active site of DHDPS and (B) the inhibitor modeled into this density. In both A and B, the electron density is contoured to 1 σ for the 2Fo-Fc map (blue) and 3 σ (green) and −3 σ (red, not observed in this region) for the Fo-Fc map. (C) Stereoplot of an overlay of the active site of DHDPS with, and without, β-hydroxypyruvate. The red line highlights the distance (7.9 Å) between Arg138 and Asp187.
An overlay with the unligated wild-type structure (1YXC) shows that the active-site residues hold the same position when β-hydroxypyruvate is bound (Fig. 4C). An interesting and thus far overlooked feature of the E. coli DHDPS structure (1YXC) is the significant distortion in the main-chain peptide torsion angle (ω) between Ile203 and Ser204 (Fig. 5A), which belongs to strand β8 (Fig. 1C). This angle is a measure of the planarity across the peptide bond. The ω distortion observed here (ω ∼201°) is a simple twist about the peptide N–C bond, and is a conserved feature of all structurally characterized DHDPS. This is highlighted in Table 1 and Figure 5B, where all DHDPS structures, apart from the recently posted A. tumefaciens model, which does not appear to possess all of the key catalytic residues of DHDPS, show significant distortion in the planarity of this peptide bond. Although both the Aquifex aeolicus and N. sylvestris enzymes show seemingly little distortion in this peptide bond, an examination of the mean ω angle of each structure (and its standard deviation, Table 1) suggests that bond angles in these models were very tightly restrained during refinement. Moreover, in each case where Ile203 is significantly distorted, it is the most distorted main-chain peptide torsion angle in the entire structure (the Tyr203 residue for the A. tumefaciens st. c58 model shows the second most strained ω value). Significantly, the greatest distortion in this peptide bond is found in the highest resolution DHDPS structure: the S. aureus DHDPS structure at 1.45 Å resolution. This structure has the weakest geometrical restraints, including no noncrystallographic symmetry restraints between the two crystallographically independent molecules in the asymmetry unit, and has the greatest weight placed on X-ray terms in refinement. Although not as pronounced, the ω value for the linkage before this (Val202–Ile203) is also somewhat strained (∼166°). Inspection of the higher resolution native E. coli model (1YXC) and the dimeric S. aureus model (3DAQ) shows no unusual hydrogen bonding directly associated with these linkages that might explain the strained conformation. However, the ω distortion of E. coli DHDPS could be caused by two hydrogen bonds: the first between the side-chain oxygen of Ser204 and the carboxylate group of the highly conserved residue Asp188 (2.6 Å), and the second between the main-chain N–H and the same carboxylate group of Asp188 (2.9 Å). The adjacent residue, Asp187, which is also conserved, has been suggested to be responsible for binding the amino moiety of (S)-ASA, given its proximity in the active site (Blickling et al. 1997). Both Asp187 and Asp188 sit in a loop region linking β7 and α7 (Fig. 1C).
Figure 5.

The distortion between Ile203 and Ser204 (A). This view is through the oxygen–carbon bond. Electron density (2Fo-Fc, contoured to 1.5 σ) across both residues shows the backbone to be well defined. (B) A stereoplot showing an overlay of current DHDPS models (confirmed and putative) with E. coli complexed with β-hydroxypyruvate (yellow). This view looks down the oxygen–carbon bond of the peptide joining I203 and S204, highlighting the distortion in the peptide. The DHDPS models are: Thermatoga maritima (green, RMSD 0.9 Å, dimer); Bacillus anthracis (light blue, RMSD 1.0 Å, dimer); Mycobacterium tuberculosis (pink, RMSD 1.1 Å, dimer); Aquifex aeolicus, (peach, RMSD 0.9 Å, dimer); Agrobacterium tumefaciens, (silver, RMSD 2.8 Å, monomer); Agrobacterium tumefaciens st. c58, (black, RMSD 2.9 Å, dimer); Hahella chejuensis, (sand, RMSD 1.1 Å, dimer); Staphylococcus aureus, (marine, RMSD 0.9 Å, dimer); and Methanocaldococcus jannaschii (chocolate, RMSD 1.0 Å, dimer). The plot was generated by aligning a monomer from each structure with chain A from E. coli (1YXC) using PyMOL (DeLano Scientific). Residue numbering refers to the E. coli structure.
Table 1.
Analysis of ω values in DHDPS structures

Mechanistic consequences of this distortion
A systematic survey by MacArthur and Thornton (1996), based on a sample of protein structures modeled from X-ray data, estimates the mean planarity for peptide bonds in proteins to be 179.6°, with a standard deviation of 4.5°. Given the distortion across the N–C peptide bond associated with Ile203, it is reasonable to conclude that there is somewhat less than the commonly estimated 40% double-bond character associated with this bond and thus more double-bond character in the C–O bond. MacArthur and Thornton (1996) estimate the energetic cost for such a distortion (20°) in a protein to be ∼3.5 kcal/mol. This represents a considerable energetic expense to the folded enzyme. Recent theoretical calculations confirm a similar substantial energy penalty for a simple 20° twist (Mannfors et al. 2003).
Herzberg and Moult (1991) conclude that polypeptide backbone strain is located “overwhelmingly in regions concerned with function” and reason that such strain may be “necessary for ligand binding and catalysis, compared with the requirements of satisfactory folding.” In this regard, the mechanistic consequences of the main-chain strain observed in the DHDPS structures are perplexing. If the main chain of Ile203 were indeed involved in facilitating proton abstraction from the pyruvate C3 group during enamine formation, it would then make more sense for the peptide oxygen to be primarily anionic, as would be the case in a standard planar peptide bond (MacArthur and Thornton 1996), but this is not the case here. The Ile203 peptide distortion would be expected to decrease the ability of the carbonyl oxygen to promote tautomerization. Given that the peptide twist is not associated with enhanced basicity that would facilitate Schiff-base tautomerism, the distorted ω angle may be playing a different role.
However, the C3 carbon of pyruvate interacts with the main chain of Ile203. The structure of E. coli DHDPS with pyruvate bound at the active site shows the carbonyl oxygen of Ile203 <3.4 Å from the C3 atom of pyruvate (Blickling et al. 1997), indicating that Ile203 may contribute to polarizing the C2–C3 bond. In addition, DHDPS from T. maritima has been solved with pyruvate in the active site, although it was not added to the crystallization conditions (Pearce et al. 2006). The active site of this structure (Fig. 6) shows that the C3 atom of pyruvate is in close proximity to an ordered water molecule, which is hydrogen bonded to the carbonyl of Ile203 (C3–H2O, 2.7 Å, H2O–CO[Ile203] 2.8 Å). The structure of E. coli DHDPS bound to β-hydroxypyruvate shows that one of these waters is conserved (Fig. 6, cyan water), while the other is likely displaced by the presence of β-hydroxypyruvate. Significantly, the C3 atom of pyruvate is also close to two ordered waters that are hydrogen bonded to Asp187 (E. coli numbering). Thus, Asp187 may be involved in proton abstraction. Although the standard reaction mechanism (Scheme 2) presumes tautomerization to the enamine prior to arrival of (S)-ASA, this has not been proven. Thus, tautomerization may well be concomitant with positioning of (S)-ASA in the active site, which in bulk solvent exists predominantly in the hydrated form.
Figure 6.

The DHDPS from Thermatoga maritima (1O5K, yellow) showing the relationship of pyruvate bound at the active site Lys161 with Ile203 and Asp188. This is overlaid with E. coli DHDPS bound to β-hydroxypyruvate (cyan), showing that one water is conserved (cyan sphere, 2.8 Å from the carboxyl of Asp188) while the other is likely displaced by the presence of β-hydroxypyruvate in the active site.
Barbosa et al. (2000) and Blickling et al. (1997) suggest that the amine and carboxylate moieties of (S)-ASA interact with the side chains of Asp187 and Arg138, respectively. As observed in this and other DHDPS structures, the side chain to side chain distance between Asp187 and Arg138 is ∼9 Å (Fig. 4B), which is too great for both the amine and carboxylate moieties of (S)-ASA to be in hydrogen-bonding contact with these side chains simultaneously, unless the side chains are reoriented. With the conformation of Arg138 already fully extended toward Asp187, only Asp187 can move to optimally accommodate the amine of (S)-ASA. In order for Asp187 to occupy an allowed rotamer following this movement, reorientation of the peptide backbone of Asp187–Asp188 is necessary, which is likely to lead to changes in the orientation of Asp188. If the connection between Asp188 and Ser204 is responsible for the torsional strain in the peptide bond, then changes in orientation of Asp188 would disrupt the Asp188–Ser204 interaction, thereby triggering relaxation of the Ile203–Ser204 peptide link. In essence, we propose that the binding of (S)-ASA triggers conformational changes that lead to relaxation of the torsionally strained Ile203–Ser204 peptide bond. Concomitant with change in position of this group, there will be a repositioning of the water network. The carboxyl of Asp187 (via ordered water molecules) in conjunction with a more basic Ile203 main-chain carbonyl (due to relaxation of the distorted peptide) can now effect proton abstraction.
Blickling et al. (1997) reasoned that the product cyclization step (Scheme 2) was mediated by Tyr133. Here, relaxation of conformational strain in the Ile203 peptide bond may provide free energy to lower the activation-energy barrier to cyclization. However, inspection of the active site indicates that there is very little space, if any, to achieve cyclization of this intermediate species, consistent with observations that putative cyclic analogs (such as piperidine ester [Mitsakos et al. 2008]) inhibit DHDPS very poorly. If cyclization occurs off-enzyme, then water is the likely nucleophile to break the pyruvyl–Lys161 bond. A relaxation of the Ile203 peptide group on binding (S)-ASA may facilitate the repositioning and polarization of a nucleophilic water.
Whether the free energy of strain relief contributes just to (S)-ASA binding or is coupled to facilitating a specific catalytic step, such as the condensation of (S)-ASA with the pyruvyl enamine species, remains to be elucidated. We propose further mutagenic studies to test these hypotheses; specifically, mutating Ser204 to Ala to break the link between the loop bearing the distorted peptide bond Ile203 and the loop bearing the putative hydrogen-bonding partner for (S)-ASA, Asp187.
N-Acetylneuraminate lyase enzymes, which are closely related to DHDPS in structure and function (Lawrence et al. 1997), mediating essentially the reverse reaction, do not show the same ω distortion in the equivalent position. Here, a glycine residue separates the NAL equivalents of Eco-DHDPS Ile203 and Ser204, which likely alleviates the strain in this motif. Furthermore, it is unclear whether the main chain of Ile203 plays a role in NAL catalysis, although it is similarly placed in the NAL active site when compared to DHDPS.
β-Hydroxypyruvate as a regulator of DHDPS
The degree of inhibition of DHDPS by β-hydroxypyruvate (K i = 0.21 [±0.02] mM) is similar to that of (S)-lysine (K i ∼20.3 mM) (Dobson et al. 2004a). Yugari and Gilvarg (1962), in one of their seminal works with E. coli DHDPS, showed that (S)-lysine inhibited DHDPS activity and that a number of other DAP pathway metabolites including meso-diaminopimelate, (S)-diaminopimelate, ornithine, and homoserine did not. Accordingly, they concluded that (S)-lysine regulates the DAP pathway via DHDPS. Nevertheless, there may be other bacterial metabolites that regulate the activity of DHDPS in vivo. The results of this study suggest that β-hydroxypyruvate, which is a known biomolecule in E. coli, may also be an important regulatory metabolite. β-Hydroxypyruvate is found in glyoxylate and dicarboxylate metabolism via glyoxylate reductase (NADP+) (E.C. 1.1.1.79) (Nunez et al. 2001), and hydroxypyruvate aldose-ketose-isomerase (E.C. 5.3.1.22) (Ashiuchi and Misono 1999).
The (S)-lysine/DAP biosynthetic pathway is feedback-regulated via inhibition of DHDPS and aspartokinase III (lysA) by (S)-lysine (Patte 1996), and it has recently been shown that the dapA gene is regulated by the level of diaminopimelate (Acord and Masters 2004). The fact that DHDPS is inhibited by β-hydroxypyruvate hints that (S)-lysine biosynthesis is not only feedback-regulated by (S)-lysine, but may be sensitive to metabolic flux through the glyoxylate and dicarboxylate pathways. We note that E. coli is able to grow on glycolate or glyoxylate as a sole carbon source; thus, global carbon flux may influence (S)-lysine biosynthesis. To our knowledge, this is the first time such a regulatory link to DHDPS has been suggested. Moreover, it is tempting to speculate that β-hydroxypyruvate may be a regulatory agent for those DHDPS known to be insensitive to lysine, particularly Gram-positive bacteria. This opens up the possibility of conducting metabolomic studies to compare the flux of metabolites into the DAP pathway of Gram-positive and Gram-negative bacteria fed on different carbon sources.
Conclusions
We present data regarding the interaction of E. coli DHDPS and a hitherto unidentified inhibitor, β-hydroxypyruvate. Inhibition is time dependent and reversible, as was found in previous studies characterizing the interaction of NAL and β-hydroxypyruvate. Given that β-hydroxypyruvate is a known bacterial biomolecule, and that the level of inhibition is equivalent to (S)-lysine, we posit that β-hydroxypyruvate may be involved in regulating DAP and (S)-lysine biosynthesis in vivo. We also describe the crystal structure E. coli DHDPS complexed with β-hydroxypyruvate. The hydroxyl of β-hydroxypyruvate hydrogen bonds to the main-chain oxygen of Ile203. In addition, we note that the C3 atom of pyruvate is close to the Ile203 peptide carbonyl oxygen atom in the pyruvate-bound structures (and 15OK) (Blickling et al. 1997). Closer examination of the Ile103–Ser204 linkage revealed that the ω value for the peptide bond was greater than 200°, which is in excess of four standard deviations from the mean ω value observed generally in proteins, with an estimated energy cost of ∼3.5 kcal/mol. Furthermore, strain in this peptide bond is conserved in all structurally and biochemically characterized DHDPS structures. Given its position in the active site, we speculate that this strained peptide bond plays two roles: first, in the polarization of both the pyruvate moiety, for enhanced reactivity, and a possible nucleophilic water that hydrolyzes the Lys161–substrate bond; and second, in the release of strain energy, which is triggered by conformational changes on binding (S)-ASA and may be coupled to the condensation of (S)-ASA with the protein-bound pyruvate. This work highlights the importance of examining structures for the functional significance of conserved, small, but energy-rich conformational distortions that may be easily overlooked.
Materials and Methods
Materials
Unless otherwise stated, all chemicals were obtained from Sigma Chemical Company, GE Healthcare, or Invitrogen Ltd. Protein concentration was measured by the method of Bradford (1976) using BSA as a standard. Enzymes were manipulated at 4°C, or on ice, and were stored in Tris·HCl buffer (20 mM, pH 8.0) at −20°C. (S)-ASA was synthesized using the methods of Roberts et al. (2003), and was of the highest quality (>95% purity), as judged by 1H NMR and titration using the coupled assay. Stock solutions of (S)-ASA and NADPH were prepared fresh for each experiment. Dihydrodipicolinate reductase (DHDPR) from E. coli, required for the coupled assay, was purified by previously reported methods (Dobson et al. 2004b).
Overexpression and purification
Wild-type DHDPS enzyme, cloned into pBluescript, was expressed in E. coli XL1-blue. The recombinant product was purified as described previously (Dobson et al. 2004b).
Kinetics
Wild-type DHDPS (1.2 μM) was incubated at 30°C in the presence of HEPES (50 mM, pH 8.0, adjusted with NaOH) with various concentrations of β-hydroxypyruvate. Aliquots (10 μL) were removed at various times and the activity was measured using a coupled assay (800 μL) with DHDPR, as previously described (Coulter et al. 1999). Briefly, the reaction was initiated with DHDPS (final concentration was 15 nM) after the cuvette had been preincubated for at least 10 min with DHDPR, pyruvate, and NADPH, followed by a 2-min incubation with (S)-ASA. Assays were performed in HEPES buffer (100 mM, pH 8.0, adjusted with NaOH) at 30°C, kept constant using a circulating water bath. Initial rates were measured as the change in NADPH concentration, monitored by absorbance at 340 nm. Since DHDPR activity was kept well in excess of DHDPS activity, the rate of oxidation of NADPH by DHDPR corresponds to the rate of formation of the DHDPS product. Care was taken to ensure the enzymes and substrates were stable over the course of the assay (usually 2 min), an excess of DHDPR (125 μg per assay, final concentration of 5.4 μM) was used, and that the DHDPS concentration was proportional to initial rate. Initial velocities were reproducible to within 10% error for duplicate measurements. Data were analyzed using OriginLab and fitted simultaneously to the “slow-binding” type mechanism (Equations 1–3), as described for Rubisco (Pearce and Andrews 2003).
The slow-binding mechanism is characterized according to Equation 1 and models a two-step process, whereby a fast-binding equilibrium between inhibitor and enzyme is established (EI), followed by a slow reaction to a tighter enzyme–inhibitor complex (EI*). Here, E represents the free enzyme, I is the inhibitor, EI is the initial enzyme–inhibitor complex, EI* is the tight enzyme–inhibitor complex in equilibrium with EI, and k 1–k 4 are the individual rate constants for the forward and reverse steps.
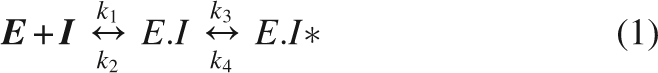 |
The equations that model this process (using initial rate data from the enzyme incubated with various inhibitor concentrations) are:
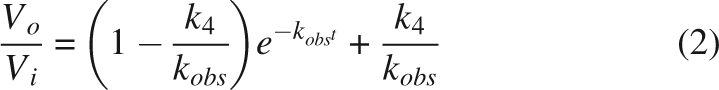 |
where
 |
Here, V i is the rate at time t, V o is the rate at time zero, I is the inhibitor concentration, K i = k 2/k 1, and k obs is the rate at which the final equilibrium is reached. We make the assumption that k 3 and k 4 are much smaller than k 1 and k 2, and that [I] >> [E].
Mass spectrometry
A stock solution of β-hydroxypyruvate was freshly prepared in water and added to the enzyme (final inhibitor concentration of 0.5 mM; final DHDPS concentration of ∼0.5 mg mL−1 in 20 mM Tris.HCl, pH 8.0). The enzyme inhibitor mix was incubated at room temperature, and directly injected (5 μL) into the mass spectrometer at various time points with 25% v/v acetonitrile in water with 0.1% v/v formic acid at 0.25 mL min−1. Mass spectrometric data were collected using an Agilent 6510 LC/Q-TOF mass spectrometer with an electrospray ionizing (ESI) source coupled to an Agilent 1100 LC system. All data were acquired and reference mass corrected via a dual-spray ESI source. Each scan or data point on the total ion chromatogram was an average of 10,000 transients, producing a scan every second. Spectra were created by averaging the scans across each peak. Data for the enzyme without inhibitor were also collected. The conditions for the mass spectrometer were: positive mode; drying gas flow was 7 L min−1; nebulizer was 30 psi; drying gas temp was 300°C; Vcap was set to 3000 V; the fragmentor was set to 225 V; the skimmer was set to 60 V; the OCT RFV was 750 V; and the scan range acquired was 100–2500 m/z. The quality and intensity of the spectra were noticeably decreased upon addition of β-hydroxypyruvate.
Crystallization, soaking with β-hydroxypyruvate, and X-ray data collection
Crystallization experiments were undertaken as described by Mirwaldt et al. (1995) using the hanging-drop vapor-diffusion method at 12°C. Drops contained: protein solution (∼12 mg mL−1 in Tris·HCl, 20 mM, pH 8.0, 2.5 μL), precipitant (K2HPO4 1.8 M, pH 10.0, 1.2 μL), and N-octyl-®-R-glucopyranoside (6% w/v, 0.6 μL). Crystals appeared after 3–5 d and grew to dimensions of up to 0.3 mm. Crystals were then soaked in the mother liquor containing β-hydroxypyruvate (20 mM) for 2 d. For X-ray data collection, the crystals were briefly soaked in cryo-protectant solution (K2HPO4, 2.0 M, pH 10.0, 20% glycerol [v/v], b-hydroxypyruvate 20 mM) and directly flash-frozen in liquid nitrogen. Intensity data were collected at 110 K using an RAxisIV++ image-plate detector coupled to a Rigaku Micromax 007 X-ray generator operating at 40 kV and 20 mA. The crystals belong to space group P3121. Diffraction data sets were processed and scaled using the package CRYSTALCLEAR (Pflugrath 1999).
Structure determination and refinement of wild-type DHDPS soaked with β-hydroxypyruvate
DATAMAN was used to transfer the R Free flags from the wild-type data set (Dobson et al. 2005) to the β-hydroxypyruvate-soaked data set. The orientation and location of DHDPS was determined using AMORE (Navaza and Saludjian 1997), where the search model was the E. coli DHDPS monomer (PDB code 1YXC). Refinement was achieved using REFMAC5 (Collaborative Computational Project Number 4 1994) with manual model corrections using the program O (Jones and Kjeldgaard 1997). Informed by the kinetic analysis, the occupancy of the β-hydroxypyruvate adduct was set to 0.75. The final refinement rounds involved the placement of solvent molecules using the program Arp (Lamzin and Wilson 1997). PROCHECK (Laskowski et al. 1993) was used to examine the quality of the final structure and assess other DHDPS models. Structural alignments were performed using the program O (Jones and Kjeldgaard 1997).
The final model contained 292 amino acid residues in each chain, with two chains in each asymmetric unit, from which the tetramer can be generated by the crystallographic twofold symmetry. The R Cryst and R Free values were 0.174 and 0.237 (Table 2). PROCHECK showed that 92.5% of the residues (within the asymmetric unit) fell into the favored regions of the Ramachandran plot (Ramachandran and Sasisekharan 1968), 7.1% were in the additionally allowed regions, and 0.4% (Tyr107) were in the disallowed region. For position 107, the structure showed well-defined electron density and the dihedral angles were similar to those of the wild-type structure. Data collection and model refinement statistics are summarized in Table 2. The coordinate and structure factor files were deposited with the RCS Protein Data Bank (3C0J). Images in this work were produced using PyMOL (deLano Scientific).
Table 2.
Data collection and refinement statistics

Acknowledgments
J.A.G. would like to acknowledge the Royal Society of New Zealand Marsden Fund (contract UOC303). M.A.P., R.C.J.D., and J.A.G. would like to acknowledge the Defense Threat Reduction Agency (project W911NF-07-1-0105) and the Australian Research Council (DP0770888). We thank Professor Robert Huber (Max-Planck-Institut für Biochemie, DE) for supplying the coordinates of the N. sylvestris DHDPS enzyme.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Renwick C.J. Dobson, Bio21 Molecular Science and Biotechnology Institute, 30 Flemington Road, University of Melbourne, Parkville, Victoria 3010, Australia; e-mail: rdobson@unimelb.edu.au; fax: 61-3-9348-1421.
Abbreviations: HTPA, (4S)-4-hydroxy-2,3,4,5-tetrahydro-(2S)-dipicolinic acid; DHDPS, dihydrodipicolinate synthase; DHDPR, dihydrodipicolinate reductase; (S)-ASA, (S)-aspartate-β-semialdehyde; DAP, meso-diaminopimelate; MS, mass spectrometry.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.037440.108.
References
- Acord J., Masters M. Expression from the Escherichia coli dapA promoter is regulated by intracellular levels of diaminopimelic acid. FEMS Microbiol. Lett. 2004;235:131–137. doi: 10.1016/j.femsle.2004.04.022. [DOI] [PubMed] [Google Scholar]
- Ashiuchi M., Misono H. Biochemical evidence that Escherichia coli hyi (orf b0508, gip) gene encodes hydroxypyruvate isomerase. Biochim. Biophys. Acta. 1999;1435:153–159. doi: 10.1016/s0167-4838(99)00216-2. [DOI] [PubMed] [Google Scholar]
- Barbosa J.A., Smith B.J., DeGori R., Ooi H.C., Marcuccio S.M., Campi E.M., Jackson W.R., Brossmer R., Sommer M., Lawrence M.C. Active site modulation in the N-acetylneuraminate lyase sub-family as revealed by the structure of the inhibitor-complexed Haemophilus influenzae enzyme. J. Mol. Biol. 2000;303:405–421. doi: 10.1006/jmbi.2000.4138. [DOI] [PubMed] [Google Scholar]
- Blagova E., Levdikov V., Milioti N., Fogg M.J., Kalliomaa A.K., Brannigan J.A., Wilson K.S., Wilkinson A.J. Crystal structure of dihydrodipicolinate synthase (BA3935) from Bacillus anthracis at 1.94 Å resolution. Proteins. 2006;62:297–301. doi: 10.1002/prot.20684. [DOI] [PubMed] [Google Scholar]
- Blickling S., Renner C., Laber B., Pohlenz H.-D., Holak T.A., Huber R. Reaction mechanism of Escherichia coli dihydrodipicolinate synthase investigated by X-ray crystallography and NMR spectroscopy. Biochemistry. 1997;36:24–33. doi: 10.1021/bi962272d. [DOI] [PubMed] [Google Scholar]
- Borthwick E.B., Connel S.J., Tudor D.W., Robins D.J., Shneier A., Abell C., Coggins J.R. Escherichia coli dihydrodipicolinate synthase: Characterisation of the imine intermediate and the product of bromopyruvate treatment by electrospray mass spectrometry. Biochem. J. 1995;305:521–524. doi: 10.1042/bj3050521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Burgess B.R., Dobson R.C.J., Griffin M.D.W., Jameson G.B., Parker M.W., Gerrard J.A., Perugini M.A. Structure and evolution of a novel dimeric enzyme from a clinically-important bacterial pathogen. J. Biol. Chem. 2008 doi: 10.1074/jbc.M804231200. (in press) [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Coulter C.V., Gerrard J.A., Kraunsoe J.A.E., Pratt A.J. Escherichia coli dihydrodipicolinate synthase and dihydrodipicolinate reductase: Kinetic and inhibition studies of two putative herbicide targets. Pestic. Sci. 1999;55:887–895. [Google Scholar]
- Dobson R.C.J., Griffin M.D.W., Roberts S.J., Gerrard J.A. Dihydrodipicolinate synthase (DHDPS) from Escherichia coli displays partial mixed inhibition with respect to its first substrate, pyruvate. Biochimie. 2004a;86:311–315. doi: 10.1016/j.biochi.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Dobson R.C.J., Valegård K., Gerrard J.A. The crystal structure of three site-directed mutants of Escherichia coli dihydrodipicolinate synthase: Further evidence for a catalytic triad. J. Mol. Biol. 2004b;338:329–339. doi: 10.1016/j.jmb.2004.02.060. [DOI] [PubMed] [Google Scholar]
- Dobson R.C.J., Griffin M.D.W., Jameson G.B., Gerrard J.A. The crystal structures of native and (S)-lysine-bound dihydrodipicolinate synthase from Escherichia coli with improved resolution show new features of biological significance. Acta Crystallogr. D Biol. Crystallogr. 2005;61:1116–1124. doi: 10.1107/S0907444905016318. [DOI] [PubMed] [Google Scholar]
- Griffin M.D.W., Dobson R.C.J., Antonio L., Pearce F.G., Whitten A.E., Liew C.K., Mackay J.P., Trewhella J., Perugini M.A., Jameson G.B., et al. Evolution of quaternary structure in a homotetrameric protein. J. Mol. Biol. 2008;380:691–703. doi: 10.1016/j.jmb.2008.05.038. [DOI] [PubMed] [Google Scholar]
- Gutteridge A., Thornton J.M. Understanding nature's catalytic toolkit. Trends Biochem. Sci. 2005;30:622–629. doi: 10.1016/j.tibs.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Herzberg O., Moult J. Analysis of the steric strain in the polypeptide backbone of protein molecules. Proteins. 1991;11:223–229. doi: 10.1002/prot.340110307. [DOI] [PubMed] [Google Scholar]
- Hutton C.A., Southwood T.J., Turner J.J. Inhibitors of lysine biosynthesis as antibacterial agents. Mini Rev. Med. Chem. 2003;3:115–127. doi: 10.2174/1389557033405359. [DOI] [PubMed] [Google Scholar]
- Hutton C.A., Perugini M.A., Gerrard J.A. Inhibition of lysine biosynthesis: An evolving antibiotic strategy. Mol. Biosyst. 2007;3:458–465. doi: 10.1039/b705624a. [DOI] [PubMed] [Google Scholar]
- Jones T., Kjeldgaard M. Electron density map interpretation. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- Karsten W.E. Dihydrodipicolinate synthase from Escherichia coli: pH-Dependent changes in the kinetic mechanism and kinetic mechanism of allosteric inhibition by L-lysine. Biochemistry. 1997;36:1730–1739. doi: 10.1021/bi962264x. [DOI] [PubMed] [Google Scholar]
- Kefala G., Evans G., Griffin M.D., Devenish S.R., Pearce F.G., Perugini M.A., Gerrard J.A., Weiss M.S., Dobson R.C. Crystal structure and kinetic study of dihydrodipicolinate synthase from Mycobacterium tuberculosis . Biochem. J. 2008;411:351–360. doi: 10.1042/BJ20071360. [DOI] [PubMed] [Google Scholar]
- Laber B., Gomis-Rüth F.-X., Româo M.J., Huber R. Escherichia coli dihydrodipicolinate synthase. Identification of the active site and crystallization. Biochem. J. 1992;288:691–695. doi: 10.1042/bj2880691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamzin V.S., Wilson K.S. Automated refinement for protein crystallography. Methods Enzymol. 1997;277:269–305. doi: 10.1016/s0076-6879(97)77016-2. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- Lawrence M.C., Barbosa J.A.R.G., Smith B.J., Hall N.E., Pilling P.A., Ooi H.C., Marcuccio S.M. Structure and mechanism of a sub-family of enzymes related to N-acetylneuraminate lyase. J. Mol. Biol. 1997;266:381–399. doi: 10.1006/jmbi.1996.0769. [DOI] [PubMed] [Google Scholar]
- MacArthur M.W., Thornton J.M. Deviations from planarity of the peptide bond in peptides and proteins. J. Mol. Biol. 1996;264:1180–1195. doi: 10.1006/jmbi.1996.0705. [DOI] [PubMed] [Google Scholar]
- Mannfors B.E., Mirkin N.G., Palmo K., Krimm S. Analysis of the pyramidalization of the peptide group nitrogen: Implications for molecular mechanics energy functions. J. Phys. Chem. A. 2003;107:1825–1832. [Google Scholar]
- Miflin B.J., Napier J., Shewry P.R. Improving plant product quality. Nat. Biotechnol. 1999;17:13–14. [Google Scholar]
- Mirwaldt C., Korndorfer I., Huber R. The crystal structure of dihydrodipicolinate synthase from Escherichia coli at 2.5 Å resolution. J. Mol. Biol. 1995;246:227–239. doi: 10.1006/jmbi.1994.0078. [DOI] [PubMed] [Google Scholar]
- Mitsakos V., Dobson R.C., Pearce F.G., Devenish S.R., Evans G.L., Burgess B.R., Perugini M.A., Gerrard J.A., Hutton C.A. Inhibiting dihydrodipicolinate synthase across species: Towards specificity for pathogens? Bioorg. Med. Chem. Lett. 2008;8:842–844. doi: 10.1016/j.bmcl.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Navaza J., Saludjian P. AMoRe: An automated molecular replacement program package. Methods Enzymol. 1997;276:581–594. doi: 10.1016/S0076-6879(97)76079-8. [DOI] [PubMed] [Google Scholar]
- Nunez M.F., Pellicer M.T., Badia J., Aguilar J., Baldoma L. Biochemical characterization of the 2-ketoacid reductases encoded by ycdW and yiaE genes in Escherichia coli . Biochem. J. 2001;354:707–715. doi: 10.1042/0264-6021:3540707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patte J.C. Biosynthesis of threonine and lysine. In: Neidhart F.C., editor. Escherichia coli and Salmonella typhimurium: Cellular and molecular biology. ASM Press; Washington DC: 1996. pp. 528–541. [Google Scholar]
- Pearce F.G., Andrews T.J. The relationship between side reactions and slow inhibition of ribulose-bisphosphate carboxylase revealed by a loop 6 mutant of the tobacco enzyme. J. Biol. Chem. 2003;278:32526–32536. doi: 10.1074/jbc.M305493200. [DOI] [PubMed] [Google Scholar]
- Pearce F.G., Perugini M.A., McKerchar H.J., Gerrard J.A. Dihydrodipicolinate synthase from Thermotoga maritima . Biochem. J. 2006;400:359–366. doi: 10.1042/BJ20060771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugrath J.W. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- Ramachandran G.N., Sasisekharan V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- Roberts S.J., Morris J.C., Dobson R.C.J., Gerrard J.A. The preparation of (S)-aspartate semi-aldehyde appropriate for use in biochemical studies. Bioorg. Med. Chem. Lett. 2003;13:265–267. doi: 10.1016/s0960-894x(02)00923-x. [DOI] [PubMed] [Google Scholar]
- Yugari Y., Gilvarg C. Coordinate end-product inhibition in lysine synthesis in Escherichia coli . Biochim. Biophys. Acta. 1962;62:612–614. doi: 10.1016/0006-3002(62)90256-1. [DOI] [PubMed] [Google Scholar]