

Summary
Mycolic acids are long chain α-alkyl branched, β-hydroxy fatty acids that represent a characteristic component of the Mycobacterium tuberculosis cell wall. Through their covalent attachment to peptidoglycan via an arabinogalactan polysaccharide, they provide the basis for an essential outer envelope membrane. Mycobacteria possess two fatty acid synthases (FAS); FAS-I carries out de novo synthesis of fatty acids while FAS-II is considered to elongate medium chain length fatty acyl primers to provide long chain (C56) precursors of mycolic acids. Here we report the crystal structure of Mycobacterium tuberculosis β-ketoacyl acyl carrier protein synthase (ACP) II mtKasB, a mycobacterial elongation condensing enzyme involved in FAS-II. This enzyme, along with the M. tuberculosis β-ketoacyl ACP synthase I mtKasA, catalyzes the Claisen-type condensation reaction responsible for fatty acyl elongation in FAS-II and are potential targets for development of novel anti-tubercular drugs. The crystal structure refined to 2.4 Å resolution revealed that, like other KAS-II enzymes, mtKasB adopts a thiolase fold but contains unique structural features in the capping region that may be crucial to its preference for longer fatty acyl chains than its counterparts from other bacteria. Modeling of mtKasA using the mtKasB structure as a template predicts the overall structures to be almost identical, but a larger entrance to the active site tunnel is envisaged that might contribute to the greater sensitivity of mtKasA to the inhibitor thiolactomycin (TLM). Modeling of TLM binding in mtKasB shows that the drug fits the active site poorly and results of enzyme inhibition assays using TLM analogues are wholly consistent with our structural observations. Consequently, the structure described here further highlights the potential of TLM as an anti-tubercular lead compound and will aid further exploration of the TLM scaffold towards the design of novel compounds which inhibit mycobacterial KAS enzymes more effectively.
Keywords: fatty acid biosynthesis, FAS-II, mtKasB, thiolactomycin, drug target
Introduction
Mycobacterium tuberculosis, the aetiological agent of tuberculosis (TB), causes more human deaths than any other single infectious pathogen, with an estimated 8 million new cases and 2 million fatalities per annum worldwide 1. TB control has been made difficult in recent years by the apparent synergy between AIDS and TB 2; 3; 4. In addition, the length and complexity of current TB treatment regimens result in poor patient compliance, a major factor in emergence of multidrug resistant TB (MDR-TB) 5. Given the multiple problems in fighting TB, effective control of the disease requires the identification of new drug targets and discovery of novel drugs.
M. tuberculosis has an unusually complex and lipid-rich cell wall which is responsible for the pathogen's intrinsic resistance to many common antibiotics 6. The essentiality of the mycobacterial cell wall to the survival of the pathogen implies that the pathway to its biosynthesis is an attractive target for novel antimycobacterial chemotherapy 7. The most distinct feature of the mycobacterial cell wall is the presence of the α-alkyl, β-hydroxy fatty acids termed mycolic acids. These are esterified to the non-reducing termini of the arabinogalactan-peptidoglycan cell wall core forming the inner leaflet of the cell wall permeability barrier 7; 8. Although the longer meromycolate chain (up to 56 carbon atoms) of the mycolic acids are modified 9, its length is essentially provided via conventional fatty acid biosynthesis. Upon completion it is condensed with a C24-C26 fatty acid, which provides the α-alkyl branch of the mature mycolic acid 8; 10; 11. Mycobacteria are unusual in that they possess both known types of fatty acid synthase (FAS) 9; a mammalian-type FAS-I, which carries all of the necessary enzymatic activities and carrier functions on a single polypeptide 12, and a bacterial type FAS-II, in which separate enzymes interact with a discrete acyl carrier protein (ACP) that carries the growing fatty acyl chain between their active sites 13. In mycobacteria and related genera, FAS-I conducts the de novo synthesis of intermediate length (principally C16 and C24) fatty acids. Mycobacterial FAS-II, however, is incapable of de novo fatty acid synthesis 14; 15; 16; 17; 18. Its ability to elongate C14- and C16-CoA primers has been demonstrated 10; 18; 19; 20; 21 and current hypotheses suggest that FAS-I products are elongated via FAS-II to form meromycolates 8.
In both FAS-I and FAS-II, fatty acyl elongation is initiated by a condensation reaction in which malonyl-ACP is decarboxylated and the resulting carbanion attacks an enzyme-linked acyl thioester derived from an acyl-CoA primer 10; 12; 19. In FAS-II the initial condensation reaction is carried out by β-ketoacyl ACP synthase (KAS)-III, also referred to as FabH 22, however subsequent FAS-II cycles are initiated by acyl-ACP primed β-ketoacyl ACP synthases, KAS-I and KAS-II (KasA and KasB respectively in M. tuberculosis, FabB and FabF repectively in E. coli) 10; 16; 22; 23.
In E. coli, the temperature dependent regulation of fatty acid composition is modulated through KAS-II (FabF) 24; 25. E. coli mutants which lack KAS-II activity are deficient in elongation of palmitoleate to cis-vaccenate but show normal growth characteristics under standard culture conditions 24; 26; 27. Overexpression of mtkasB produced longer chain polyunsaturated hydrocarbons averaging 54 carbons in length 28. M. marinum kasB mutants grew poorly in macrophages and showed an altered mycolate profile and cell wall permeability 29. These results suggest the importance of KasB in mycobacteria and show that it represents an attractive target for novel drug development against M. tuberculosis. Thiolactomycin (TLM) has been shown to be an inhibitor of mycobacterial β-ketoacyl ACP synthases 10; 20; 23 and hence acts as a potent anti-tuberculous agent by inhibiting both fatty acid and mycolic acid biosynthesis 18.
In this work, the X-ray crystal structure of mtKasB has been determined using the molecular replacement method. The structure has been described and shows distinct features when compared to other known KAS-II structures 30; 31; 32; 33. To gain further knowledge of possible interactions between TLM and mtKasB, modeling of the complex has been attempted based on the known crystal structure of a TLM complex with KAS-I (ecFabB). The sequences of mtKasA and mtKasB are 69.5 % identical allowing the modeling of the structure of mtKasA based on mtKasB and thus comparisons with the mycobacterial KAS-I.
Results and discussion
Quality of the model
The mtKasB structure has been solved using a molecular replacement strategy to a resolution of 2.4 Å. The final model has been refined to twinned R/R-free of 23.6%/18.1%. The model consists of two molecules of mtKasB and 229 water molecules per asymmetric unit. The electron density was clear for the both molecules in the asymmetric unit except a stretch of residues 128 to 138 and the sidechains of residues 38, 48, 51, 55, 78, 144 and 213 of chain A and the stretch of residues 50 to 58 and 130 to 138 and the sidechains of residues 73, 121, 123, 203, 211, 212 and 213 of chain B. Although C16-CoA was present in the crystallization drop, electron density for the substrate was not observed consistent with mtKasB being an acyl-ACP-primed KAS-II. Data collection and refinement statistics are given in Table 1.
Table 1.
Data collection and refinement statistics for mtKasB structure determination
| Data collection: | |
|---|---|
| Space group | R3 |
| Cell dimensions (a, b, c in Å, α°, β°, γ°) | 198.7, 198.7, 71.8, 90, 90, 120 |
| Resolution (Å) | 50-2.39 |
| Completeness % (last shell) | 98.6 (88.4) |
| I/σ(I) (last shell) | 31.7 (2.5) |
| No. of total reflections | 230006 |
| No. of unique reflections | 41266 |
| Rsym % (last shell) | 5.3 (43.1) |
| Refinement: | |
| Resolution (Å) | 50-2.4 |
| No. of reflections in test set | 1911 |
| No. of reflections in working set | 35874 |
| No. of protein molecules in asu | 2 |
| No. of protein atoms in asu | 6192 |
| No. of water molecules in asu | 229 |
| Twinned R-factor (%) | 23.6 |
| Twinned Rfree factor (%) | 18.1 |
| RMS deviations: | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.15 |
| Average B-factors (Å2): | |
| Main chain | 53.9 |
| Side chain and waters | 54.4 |
| RMS B main chain | 0.75 |
| RMS B side chain | 0.89 |
Rsym = Σ |I - <I>| / Σ I, where <I> is the average intensity over symmetry equivalents.
Subunit structure
mtKasB shows a homodimeric assembly in the crystal structure. Each subunit has a conserved thiolase fold 34 with a five layered αβαβα core structure similar to the structures of other elongation β-ketoacyl ACP synthases 30; 31; 32; 33; 35 as shown by structural alignments of the elongation condensing enzymes (Figures 1 and 2). Each subunit is composed of 12 α helices and 15 β strands. The subunit structure consists of two domains – one defining the highly-structured core and the other forming a capping region. Residues 1-260 and 261-415 form topologically equivalent N- and C-terminal βαβαβαββ motifs. The secondary structural elements of the two substructures contribute to similar parts of the core region. This structural aspect suggests that these parts might have arisen as a result of gene duplication although the sequence identity between these parts is only 7%. Although the N-term substructure is more than 100 residues longer than the C-term substructure, the motifs can be superposed with a rmsd of 1.99 Å over 80 Cα atoms. The two substructures have different number of insertions between the helices and strand forming the core. The capping region is primarily composed of some of these additional helical segments (α4, α6, α7 and α10).
Figure 1.

Ribbon representation of mtKasB dimer. The two subunits are shown in magenta and cyan. The positions of the active site residues are shown as red spheres and labeled. The segments shown in black and labeled 1 and 2 correspond to the regions showing large deviations when compared to KAS-I/II structures. These segments are also refered to in Figure 2.
Figure 2.

Structure based alignment of KAS-I/II sequences. A structure based alignment was generated for KAS-I/II sequences using Protein structure comparison service SSM at European Bioinformatics institute59 (http://www.ebi.ac.uk/msd-srv/ssm) and Boxshade (http://www.ch.embnet.org/software/BOX_form.html). The red stars mark the active site residues. The stretches of sequence marked by the black line above the mtKasB sequence and labeled 1 and 2 refer to the segments colored in black in Figure 1. Ssp_KASII - Synechocystis sp. KAS II (PDB code of the structure used in alignment: 1E5M, rmsd of alignment with mtKasB reported by the SSM service: 1.6 Å), Tth_KASII - T. thermophilus KAS II (1J3N, 1.5 Å), Eco_KASII - E.coli FabF (1KAS, 1.6 Å), Spn_KASII - S. pneumoniae KAS II (1OX0, 1.7 Å), Eco_KASI - E.coli FabB (1DD8, 1.7 Å), mtKasA - M. tuberculosis KasA (0.6 Å), Ath_KAS - A. thaliana mitochondrial KAS (1W0I, 1.7 Å).
Structure of Dimer
The homodimer observed in the asymmetric unit is the functional assembly of this enzyme, based on the similarity to the E. coli FabF dimer that has been shown to be dimeric both in solution and in the crystal structure 30; 36. In the crystal structure the two subunits are related by a pseudo-two-fold axis (Figure 1). The solvent accessible surface area buried by the dimer is about 6433 Å2, 17.7% of the entire surface area of the dimer. Extensive interactions are formed between the subunits and these involve elements from both the core region and the additional elements that are not part of the core. Strand β5 of the subunits come together in the center of the dimer assembly effectively extending one of the core β-sheets to a 10-stranded twisted β-sheet. The insertion between β4 and α8 of the N-term substructure contains the additional elements α6 and α7. These elements contribute significantly to both the formation of the dimer and the channel leading to the active site. The residues Ala116, Leu119, Val120 and Tyr123 of α6 of the capping region contribute significantly to the van der Waals interactions with the helical counterpart from the partner subunit. The residues of the helix α7 from one subunit line the channel leading to the active site of the partner subunit. Other interactions contributing to the dimer formation include van der Waals interactions between residues that form the loop regions between β5 and β6 which includes α6 and α7, between β10 and α12 and the helix α10.
Architecture of the active site
The Cys-His-His active site triad is located approximately 15 Å from the surface of the enzyme at the bottom of a molecular tunnel lined by primarily hydrophobic groups from the protein. The mouth of this tunnel measures about 12 Å at its widest part. The residues Phe237, Pro280, Thr313, Thr315, and Phe405 lining the tunnel are conserved in the four other KAS-II structures determined to date (Synechocystis sp, 1E5M 31; Thermus thermophilus, 1J3N33; Streptococcus pneumoniae, 1OX0 32 and E. coli 1KAS 30) and A. thaliana mitochondrial KAS (1W0I37).
Alignment of the available structures of the elongation condensing enzymes clearly indicates that the residues Cys170, His311 and His346 of mtKasB are highly conserved and positioned appropriately to function as the catalytic residues. Biochemical and mutagenesis studies of condensing enzymes 38; 39; 40; 41 also show that these residues are the catalytic residues. Cys170 is located at the N-terminus of helix α9 similar to other KAS-II structures. This location has been proposed to promote activation of its sulfhydryl group. Currently, it is accepted that the dipole moment of this helix enhances the nucleophilicity of the sulfur atom by lowering its pKa 42. In subunit A a water molecule (Wat4) is hydrogen bonded to the backbone carbonyl oxygens of Cys170 (2.7 Å) and Val348 (2.5 Å) (Figure 3a). Wat4 is also within hydrogen-bonding distance from the backbone carbonyl oxygen of Gly349 (2.8 Å) and backbone amide nitrogens of Ser172 (3.5 Å) Gly173 (2.9 Å), and Gly352 (3.0 Å). A water molecule is conserved in this position in S. pneumoniae, T. thermophilus and Synechocystis sp. KAS-II, apo and acyl-bound ecFabB (KAS-I) structures (1DD8 35 and 1EK4 43 respectively) and A. thaliana mitochondrial KAS37 suggesting that it may play a structural role such as orienting or maintaining the position of Cys170 in the active site. Incidentally, this water molecule is seen only in subunit A of mtKasB and not in subunit B.
Figure 3.


a) active site in subunit A of mtKasB. For clarity some of the residues that hydrogen bond with Wat4 and described in the text are not shown. b) active site in subunit B of mtKasB. In both a and b electron density contoured at 1.0σ level is shown in blue.
Interestingly, His311 and Ala312 were found to be in two different conformations in subunits A and B of mtKasB (Figure 3b). Ala312 is unique to mtKasB with a glycine occupying the position in the other KAS-II structures. His311 has phi/psi values of −104.6°/55.0° in subunit A and −86.4°/−13.2° in subunit B. Similarly, Ala312 has phi/psi values of −91.9°/118.3° in subunit A and −30.6°/116.3° in subunit B. Comparison of the environment around His311 in the two subunits showed that the difference in the Ala312 conformations is caused by a change in the conformation of Lys341. The conserved Lys341/Glu355 pair is assumed to control the electronic state of His311 32; 42. Despite the differing backbone conformations, the Nδ of His311 interacts with Lys341 in both the subunits (2.5 Å and 3.4 Å in subunits A and B respectively). Hence, this change in the backbone conformation might not affect the catalytic role of His311. The Lys341-Glu355 salt-bridge interaction is not seen in subunit A of mtKasB (Nζ of Lys341 is at distances of 3.8 Å and 3.4 Å from the sidechain carboxylate oygens of Glu355).
There is also significant difference in orientation of the imidazole ring of the other catalytic histidine residue His346 in the two subunits of mtKasB. The plane of the ring in subunint B is about 50° rotated about the Cβ-Cγ bond from that seen in subunit A. Also there are variations in the orientation of the imidazole plane when comparing mtKasB and other KAS-II structures (for eg. rotations of about 75° in the S. pneumoniae KAS-II structure and about 45° in the T. thermophilus structure when comparing with the orientation in subunit A). In all the other KAS-II structures, Nε of this His residue interacts with the sulfur of the catalytic Cys residue with an average distance of 3.27 Å between these two atoms. However, in mtKasB it is farther away and does not interact with the sulfur (3.6 Å in subunit A and 4.0 Å in subunit B). Nδ of His346 is hydrogen bonded to the backbone amide of the conserved Leu348 in mtKasB hence orienting the sidechain so that the Nε points toward the active site.
The role of the conserved Lys341 in the catalysis of the KAS enzymes has been analyzed by biochemical and structural studies44; 45. Based on the biochemical studies it was suggested that the interaction between Lys341 and His311 may not be structural in chracter and that Lys341 may play a more important role in the transacylation half reaction than in the decarboxylation half reaction of the catalysis of the KAS enzymes with Cys-His-His catalytic triad. In this work, we observed different conformations of the His311 and Ala312 due to slight changes in the Lys341 sidechain conformation in mtKasB. Wang et al observed significant conformational changes of the catalytic histidines and lack of plastensimycin binding to ecFabF (KAS-II) Lys335Ala mutant. These observations suggest that a structural role for this conserved at this position cannot be ruled out. It now seems clear that the conserved lysine plays a critical stereochemical role in orienting the sidechain of the other catalytic histidine His34644.
Comparison of KAS II structures
Alignment of mtKasB, KAS-I/II, and A. thaliana mitochondrial KAS structures revealed interesting structural rearrangements of the capping region of mtKasB (rmsd of alignments are given in the legend to Figure 2). Most of the deviations are seen in the insertion segments and the capping region. There is significant rearrangement of the segment containing the helices α6, α7 and α10, all of which are part of the capping region. The α6 helices of the partner subunits contribute to the dimer interface in all of the structures. Analysis of crystal packing of mtKasB dimers showed that α4 of subunit B of a dimer lies approximately parallel to α6 of subunit A and prevents a closer approach of α6 of partner subunits in a symmetry-related dimer. This kind of crystal packing is not observed in any of the other KAS-I/II structures.
The apparent rearrangement of the α10 of mtKasB involves a shift along its axis towards the surface of the protein. mtKasb has a unique Arg residue at position 200 in the short 4-residue stretch connecting β6 and α10. Ser or Ala is found at the equivalent position in othe KAS-II structures. These shorter sidechains point toward the acyl-binding channels of the corresponding KAS-II structures. The long Arg sidechain in mtKasB forces this residue to take a β-conformation exposing the sidechain to solvent. mtKasB also has an Ala at position 203 compared to a conserved Pro at the equivalent position in all the other KAS-II structures. A further analysis of α10 showed that the expected backbone hydrogen bonds of an α-helix are formed only in the segment between residues 203 and 210 and not seen in that between residues 207 and 213 in mtKasB. The electron density for the residues of α10 of mtKasB is clear and thus the elongation of this helix cannot be attributed to errors in building the structural model. Thus, the unique Arg200 residue and the the observed hydrogen bonding pattern might explain the difference in arrangement of α10 in mtKasB compared to that in other KAS-II structures. Arg213 located at the loop following α10 is one of the residues at the mouth of the active site tunnel. The unique arrangement of α10 described above “pushes” the loop containing Arg213 further towards the surface of the molecule. This makes the mouth of the active site tunnel appear constricted in mtKasB in comparison with the other KAS-I/II structures.
It should be noted that α10 spans both the active site tunnel and the acyl binding channel thus forming a structural link between these two cavities. Hence any change in its structure could affect both the channels at the same time. mtKasB elongates acyl chains longer than 16 carbons in length. The distal end of the acyl binding channel defined by the ecFabB-acyl intermediate structures is closed by α6. Hence, it is intriguing how longer acyl chains could be accommodated in mtKasB in this short channel. Attempts to model a C12 acyl chain in the defined channel in mtKasB shows that it is not of sufficient length to accommodate longer acyl chains.
Acyl binding channel
The acyl binding channel of elongation condensing enzymes was defined with the structure of the KAS-I (ecFabB)-C12/C10 complexes (1EK4/1F91) 43. Comparison of KAS-I/II structures clearly indicates that Gly114, Ala169, Phe209 and Phe405 lining the channel are conserved. Gly114 of mtKasB appears to be conserved in its position as a bigger sidechain cannot be accommodated in the space available. It is clear that the phenyl ring of Phe405 (Phe392 in 1EK4) has to be rotated about the CB-CG bond in order accommodate the head part of acyl chains. Alignment of 1EK4 and mtKasB structures also showed that the β-turn between the strands β12 and β13 which contains Phe405 has shifted by about 2 Å, in turn moving the sidechain of this residue towards the active site tunnel. As a consequence, the active site tunnel in mtKasB appears more occluded than in the ecFabB-acyl intermediate. In the acyl-bound structure, the backbone amide nitrogens of Phe405 and Ala169 (Ala162 in 1EK4) interact with the oxo-group of the acyl chain. Phe209 is equivalent to Phe201 in 1EK4, the latter dictating the U-shaped conformation of the acyl chain in the acyl-bound structure.
Some interesting differences in the residues lining the acyl binding channel were observed when comparing the structures of ecFabB-acyl complex and mtKasB. By comparing the structures of complexes of ecFabB with C10 and C12 acyl chains (1EK4 and 1FJ1), it was suggested that Glu200 moves to accommodate a longer acyl chain 43. In mtKasB, the equivalent residue is Gly208 and in the other KAS-II structures, it is a glycine or an alanine. Gln113 in ecFabB was found to be in different conformations in the A and B subunits of the structure and the electron density for this residue was well-defined in the complex structure in comparison with the apoenzyme. The sidechain of Gln113 also interacts with the backbone carbonyl group of Gly107 in subunit A. In mtKasB, Gln113 is substituted by Leu121 and Gly107 by Leu115. In other KAS-II structures, Ile is found in the place of Gly107 of ecFabB. It is seen that the substitution of a Gly107 in ecFabB with a longer side chain in KAS-II necessitates a compensatory change in the residues equivalent to Met197 of ecFabB to one having a smaller sidechain to avoid a clash between the sidechains in these positions. Hence, in mtKasB Met197 is substituted by Pro205 and by Ser, Gly or Ala in the other KAS-II structures. In ecFabB, Met197A along with Met138B delineate opposite sides of the acyl binding channel. The latter residue is conserved between ecFabB and mtKasB but substituted by Ile or Leu in the other KAS-II structures. It should be noted that Pro205, Gly208 and Phe209 of mtKasB are located in α10. These substitutions may reflect the necessary changes required to bind substrates specific to the KAS-II enzymes of different organisms.
The structures of ecFabB/F-cerulenin complexes46; 47 were also compared with mtKasB structure. It is clear from this comparison that Leu115 of mtKasB has to be reoriented for CER binding. Olsen et al. observed that the equivalent residue Ile154 in A. thaliana mitochondrial KAS would require a similar reorientation to facilitate CER binding37. The structural alignment also clearly shows that Tyr144B of mtKasB (Ile183 in A. thaliana mitochondrial KAS, Ala137 in ecFabB and Thr137 in ecFabF) will not allow the orientation of CER seen in the above complexes in mtKasB. The energetic barrier to reorient Tyr144B in mtKasB to allow a similar CER binding mode as seen in the complexes is expected to be high. Hence we predict that beyond the C7 position CER in mtKasB would proceed in a direction different from that seen in the above complexes.
Modeling of mtKasB-TLM complex
TLM is a natural product that reversibly inhibits β-ketoacyl ACP synthases. The structure of an ecFabB-TLM complex 46 (PDB code: 1FJ4) has been determined and shows the mechanism of inhibition of this KAS-I enzyme. Although TLM is a poor inhibitor of condensing enzymes, it provides an avenue for rational design of analogues that could be better inhibitors of this class of enzyme. Crystallization of an mtKasB-TLM complex was attempted towards this end. The crystals did not diffract well enough to get useful data required to determine the structure of the complex, hence, modeling of TLM in the mtKasB structure was performed with the knowledge of ecFabB-TLM complex structure. Energy minimization was done using the program Sybyl and the Discover module of the program package Insight 2000.
The structural model shows that the isoprenoid branch of TLM is sandwiched between the peptide bonds 279-280 and 405-406 similar to that in the ecFabB-TLM structure (Figure 4). The model also shows that the active site tunnel in the model is significantly constricted in comparison with that in the ecFabB-TLM structure. The model suggests that the β-turn between β12 and β13 and the loop connecting β8 and α12 might have to undergo significant rearrangements to create an appropriate binding pocket for the TLM. This is in contrast to the ecFabB-TLM complex structure where only minimal distortion is required to fit the TLM 46. The movement of the main chain in the segment of residues from 403 to 409 spanning β12, β13 and the β12-β13 turn results in partial stacking of the phenyl ring of Phe405 with the thiolcatone ring of TLM (Figure 4). The distortion in the loop formed by residues 272-286 between β8 and α12 is partly due to a two residue (Pro282-Asn283) insertion in mtKasB in comparison with the loop in ecFabB and moves Pro280 to the appropriate position for TLM binding. This two residue insertion occurs in all the other KAS-II structures and A. thaliana mitochondrial KAS (Figure 2). It is also seen from the model that the conserved Asp273 in mtKasB has to swing away from its position in the apo structure to avoid a clash with the isoprenoid moiety of TLM (Figure 4)
Figure 4.

Superposition of TLM binding sites in ecFabB-TLM complex (1FJ4, green) and mtKasB-TLM model (magenta). The greem dotted lines indicate interactions of TLM O2 with active site histidines in ecFabB-TLM complex.
Kremer et al. demonstrated that hydrophobic chain substituents at the C5 position with varying chain length and saturation showed better in vivo inhibition activity than TLM 20. In contrast, Kim et al. 48 reported that their studies revealed very little tolerance for substitution at C5 of TLM in assays of condensing enzymes derived from both E. coli and M. tuberculosis. The significant modifications that seem to be required to the TLM binding site in mtKasB as suggested above by our model may explain the high IC50 values reported for TLM and its analogues 23, 48.
Modeling of mtKasA structure
The high sequence identity between mtKasB and mtKasA has allowed modeling of the structure of the latter using the mtKasB structure determined in this work as a template and as expected, the two structures are almost identical (rmsd of 0.6 Å). The positions of the catalytic site residues of mtKasA (Cys167, His307 and His341) and the active site environment are conserved. The Glu355/Lys341 interaction of mtKasB is also conserved in mtKasA (Glu350/Lys336). The residues of α6 and α10 and those lining the acyl binding channel are mostly conserved in the mtKasA model.
The residues lining the active site tunnel are also conserved in the two structures and hence the conservation of the hydrophobic nature of the tunnel in mtKasA. Superposition of the mtKasA model over the mtKasB structure shows that the loop following α6 is shorter in mtKasA than in mtKasB. In mtKasA, a segment of this loop has the sequence 209MRAMST214 whereas in mtKasB the sequence is 212MRIVMST218. In mtKasB Ile214, Val215 and Phe239 form a hydrophobic core in the active site tunnel (Figure 5). The substitution of 214IV215 of mtKasB by single residue (Ala211) in mtKasA results in loss of this hydrophobic core in the latter structure.
Figure 5.

a) Superposition of mtKasA model (gold) and crystal structure of mtKasB (magenta). The structures are shown in worm representation. The Cα atoms of the active site residues Cys170, His311 and His341 are shown as spheres. The black frame encloses the region of a loop at the entrance to the active site tunnel where the structures are different. b) A magnified view of the region shown in the black frame in a) representing the difference in the structure of the loop between mtKasA and mtKasB. The residues of this loop that are different between the two structures are labeled. This loop region follows the helix α6.
Interestingly, IC50 of TLM for mtKasA is lower than that for mtKasB (20 μM vs. 90 μm) 23. Kim et al reported IC50's of two TLM analogues for mtKasA that are lower (8 and 16 fold) than that for mtKasB48. The MIC of TLM for M. bovis BCG strain overexpressing mtKasA is lower than that for mtKasB (80 μg/mL vs 100 μg/mL) 20. It would be interesting to investigate if an increased accessibility resulting from a more open active site tunnel in mtKasA as consequence of the shorter loop could account at least partly for these lower values.
Synthesis of TLM analogues and structural and biochemical studies will be required to determine structure activity relationships that could be used in further steps of development of inhibitors of mtKasB and mtKasA. In this regard, IC50 values against mtKasA were higher than TLM for analogues (Table 2) 1 (63 μM), 2 (86 μM), 3 (46 μM) and 4 (66 μM) prepared prior to our structural insight into mtKasB and mtKasA. Analogues 1-4 also possessed IC50 values greater than 150 μM against mtKasB. Although, compound 3 possessed a weaker IC50 value against mtKasA, it is much more potent in vivo possessing a minimum inhibitory concentration (MIC) of 34 μM, in comparison to TLM, which possessed an MIC value of 142 μM against Mycobacterium bovis BCG. These results suggest that analogues with improved activities may now be synthesized in a more rationale way aided by the use of the crystal structure of mtKasB and modelling studies of mtKasA presented in this current report.
Table 2.
Structures and IC50 values against mtKasA of TLM analogues 1-4
| No. | R-group | IC50 (μM) |
|---|---|---|
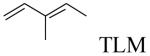 |
||
| 1 | 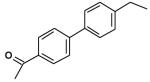 |
63 |
| 2 | 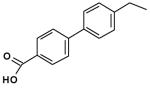 |
86 |
| 3 | 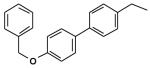 |
46 |
| 4 | 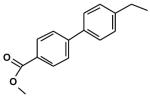 |
66 |
Methods
Cloning, overexpression and purification of mtKasB
The mtkasB gene (Rv2246) was cloned from M. tuberculosis H37Rv genomic DNA using the upstream primer 5′-aagaagacatatggttaccgggaaagcctttccc-3′ with a NdeI restriction site (bold letters) and the downstream primer 5′-aagctcgagtcagtaccgtccgaaggcgattgc-3′ with a XhoI restriction site. The amplified product was then digested with the restriction enzymes and ligated with similarly cut pET28b plasmid DNA (Novagen) to form pET28-mtkasB; the construct was verified by nucleotide sequencing. For overexpression of mtkasB, Rosetta (DE3) cells (Novagen) were transformed with pET28-mtkasB. Transformants were selected on Luria-Bertani (LB) agar medium containing kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL) at 37° C. A single colony was picked and used to inoculate 5 mL of LB medium containing 50 μg/mL kanamycin and 34 μg/mL chloramphenicol. This culture was grown to an OD600nm of 0.6 Units at 37° C and used to inoculate a 100 mL starter culture. This starter culture was grown to an OD600nm of 0.6 Units at 37° C and used to inoculate 4 liters of LB medium. After this culture reached an OD600nm of 0.8 Units, it was cooled to 20° C. IPTG was added to a concentration of 1 mM to induce mtKasB overexpression and the medium was further incubated for 12 hrs at 20° C.
The cells were harvested by centrifuging at 3000 × g for 30 minutes and the pelleted cells were resuspended in buffer A (20 mM Tris pH 7.5, 0.5 M NaCl, 2 mM β-mercaptoethanol) at 4° C. DNase I at a final concentration of 10 μg/mL and protease inhibitor cocktail (Calbiochem) were added to the suspension before lysis using a French pressure cell at 1000 psi. The crude lysate was clarified by centrifugation at 10,000 × g for 60 minutes maintaining the temperature at 4° C. The clarified lysate was filtered (0.22 microns) before subsequent chromatographic purification steps performed at 4° C.
mtKasB was purified to near homogeneity using nickel affinity and gel filtration chromatography techniques with performance being monitored using SDS-PAGE. The clarified lysate was passed through the Ni2+-charged HiTrap chelating agarose columns equilibrated with buffer A. The columns were washed with 5 column volumes of buffer A to remove unbound and loosely bound contaminant proteins. mtKasB bound to the matrix in the columns was eluted using a stepwise gradient of imidazole concentration using 20 mM Tris pH 7.5, 0.5 M NaCl, 2 mM β-mercaptoethanol, 0.5 mM imidazole as buffer B. The bound protein was found to elute with 150 mM imidazole. The eluted protein was dialyzed against gel filtration buffer (20 mM Tris pH 7.5, 10 mM NaCl, 10% glycerol, 1 mM EDTA, 1mM DTT) to remove imidazole. mtKasB was concentrated and further purified using a Superdex 200 gel filtration column (Amersham Pharmacia).
mtKasB eluted in two peaks from the gel filtration column. The fractions of the higher molecular weight peak contained a small fraction of mtKasB and contaminating proteins and were not used any further. Most of the mtKasB eluted in the fractions of the second peak, corresponding to the expected elution volume of mtKasB dimer (m.wt. of approximately 88 kDa). These fractions were pooled, concentrated to 30 mg/mL and stored at −80° C in the gel filtration buffer or used directly for crystallization purposes. TLM analogues (Table 2) previously reported by Senior et al. 49 were assayed against mtKasB and mtKasA using methods previously described by Schaeffer et al. 50.
Crystallization and data collection
Initial screening for crystallization of mtKasB was done using the hanging drop vapor diffusion method. Crystallization conditions obtained commercially from Hampton Research (Crystal Screen and Crystal Screen II) and Emerald Biostructures (Wizard Screens I and II) were screened. Wizard screen II condition 39 (100 mM CAPS pH 10.5, 20 % w/v PEG 8000, 200 mM NaCl) produced crystals that diffracted poorly to 8 Å resolution. Optimization of the above crystallization condition was carried out with additives and detergents (from Hampton research) using sitting drop plates in the presence of C16-CoA. Finally, addition of 5 mM C16-CoA, 0.3 μL Spermine HCl as additive and 0.1 μL Foscholine-9 as detergent with the above crystallization buffer produced crystals that diffracted to 3 Å resolution.
X-ray intensity data was collected at the synchrotron facility at the Center for Advanced Microstructures and Devices (CAMD), Louisiana using a MarCCD system under cryogenic conditions. Several crystals were screened to find one that diffracted better than 3 Å resolution. One of the crystals diffracted to 2.4 Å resolution and was used to collect a total of 180° of data using 1° oscillation range per frame.
Structure determination
The diffraction data was indexed using HKL2000 51 which showed that mtKasB crystallized in the space group R3 with unit cell dimensions of a=b=198.68 Å, c=71.83 Å, α= β =90°, γ =120°. The data were further integrated and scaled using the program HKL2000. The crystal structure of mtKasB was solved using molecular replacement method. First, an initial structural model of mtKasB was generated by submitting the protein sequence to and using the first approach mode of the SwissModel software server (http://swissmodel.expasy.org/SWISS-MODEL.html) 52. In this mode, the software automatically chooses the structural templates from the Protein Data Bank (PDB) to build a model for the sequence submitted. This structure was used as the search model to obtain initial phases. Molecular replacement rotation and translation searches to determine the correct orientation of the search model in the unit cell were done using the MolRep program 53 of the CCP4 suite 54. Although a clear solution was obtained, initial electron density maps (2mFo-DFc) obtained after a few cycles of coordinate and B-factor refinement, carried out using the Refmac 55 program of the CCP4 suite, was poor with many discontinuities along the main chain of the electron density and poor or non-existent density for the sidechains of many residues. However, model building using XtalView 56 was attempted to fit as many residues as possible in the available map. After a few rounds of iterative model building and refinement there was no improvement in the quality of the electron density map. Further, the R-factor/R-free factor remained high at 36%/25% after several steps of refinement without further improvement. At this point, data twinning was suspected and the data was analyzed for twinning, using Yeates' Twinning Server (http://nihserver.mbi.ucla.edu/Twinning/) 57, CCP4, and the Crystallography and NMR System (CNS) 58 suites. Analysis of these results indicated that the diffraction data was merohedrally twinned with a twin fraction of 41%. When the structure was refined using scripts in the CNS suite that take data twinning into account (the detwin_partial.inp script was used which incorporates the twin fraction directly into the refinement target) the structure and electron density maps improved dramatically, During the final few rounds of model building, water molecules were also added to the model. Iterative refinement and model building yielded the final model of mtKasB with an R-factor of 18.1% and an R-free of 23.6%. and good stereo chemistry. The data and structure are deposited in the Protein Data Bank (PDB code 2GP6).
The structural model of mtKasA was generated in SwissModel 52 using the structure of mtKasB as the template. The sequence of mtKasA and the structure of mtKasB were submitted to the server and the first approach mode was used to generate the mtKasA structural model.
ACKNOWLEDGEMENTS
GSB was supported by a Lister-Jenner Institute Research Fellowship and by the Medical Research Council (UK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. Jama. 1999;282:677–86. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 2.Wallis RS, Ellner JJ, Shiratsuchi H. Macrophages, mycobacteria and HIV: the role of cytokines in determining mycobacterial virulence and regulating viral replication. Res Microbiol. 1992;143:398–405. doi: 10.1016/0923-2508(92)90053-q. [DOI] [PubMed] [Google Scholar]
- 3.Toossi Z, Mayanja-Kizza H, Hirsch CS, Edmonds KL, Spahlinger T, Hom DL, Aung H, Mugyenyi P, Ellner JJ, Whalen CW. Impact of tuberculosis (TB) on HIV-1 activity in dually infected patients. Clin Exp Immunol. 2001;123:233–8. doi: 10.1046/j.1365-2249.2001.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paolo J, William F, Nosanchuk JD. Tuberculosis in New York city: recent lessons and a look ahead. The Lancet Infectious Diseases. 2004;4:287–293. doi: 10.1016/S1473-3099(04)01004-7. [DOI] [PubMed] [Google Scholar]
- 5.Kaye K, Frieden TR. Tuberculosis control: the relevance of classic principles in an era of acquired immunodeficiency syndrome and multidrug resistance. Epidemiol Rev. 1996;18:52–63. doi: 10.1093/oxfordjournals.epirev.a017916. [DOI] [PubMed] [Google Scholar]
- 6.Hong X, Hopfinger AJ. Molecular modeling and simulation of Mycobacterium tuberculosis cell wall permeability. Biomacromolecules. 2004;5:1066–77. doi: 10.1021/bm0345155. [DOI] [PubMed] [Google Scholar]
- 7.Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 8.Dover LG, Cerdeno-Tarraga AM, Pallen MJ, Parkhill J, Besra GS. Comparative cell wall core biosynthesis in the mycolated pathogens, Mycobacterium tuberculosis and Corynebacterium diphtheriae. FEMS Microbiol Rev. 2004;28:225–50. doi: 10.1016/j.femsre.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Kremer L, Baulard A, Besra GS. In: Molecular Genetics of Mycobacteria. Hatfull GF, Jacobs WR Jr., editors. ASM; Washington D.C.: 2000. [Google Scholar]
- 10.Kremer L, Dover LG, Carrere S, Nampoothiri KM, Lesjean S, Brown AK, Brennan PJ, Minnikin DE, Locht C, Besra GS. Mycolic acid biosynthesis and enzymic characterization of the beta-ketoacyl-ACP synthase A-condensing enzyme from Mycobacterium tuberculosis. Biochem J. 2002;364:423–30. doi: 10.1042/BJ20011628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Portevin D, De Sousa-D'Auria C, Houssin C, Grimaldi C, Chami M, Daffe M, Guilhot C. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc Natl Acad Sci U S A. 2004;101:314–9. doi: 10.1073/pnas.0305439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith S, Witkowski A, Joshi AK. Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res. 2003;42:289–317. doi: 10.1016/s0163-7827(02)00067-x. [DOI] [PubMed] [Google Scholar]
- 13.Lu YJ, Zhang YM, Rock CO. Product diversity and regulation of type II fatty acid synthases. Biochem Cell Biol. 2004;82:145–55. doi: 10.1139/o03-076. [DOI] [PubMed] [Google Scholar]
- 14.Bloch K. Control mechanisms for fatty acid synthesis in Mycobacterium smegmatis. Adv Enzymol Relat Areas Mol Biol. 1977;45:1–84. doi: 10.1002/9780470122907.ch1. [DOI] [PubMed] [Google Scholar]
- 15.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 16.Mdluli K, Slayden RA, Zhu Y, Ramaswamy S, Pan X, Mead D, Crane DD, Musser JM, Barry CE., 3rd Inhibition of a Mycobacterium tuberculosis beta-ketoacyl ACP synthase by isoniazid. Science. 1998;280:1607–10. doi: 10.1126/science.280.5369.1607. [DOI] [PubMed] [Google Scholar]
- 17.Peterson DO, Bloch K. Mycobacterium smegmatis fatty acid synthetase. Long chain transacylase chain length specificity. J Biol Chem. 1977;252:5735–9. [PubMed] [Google Scholar]
- 18.Slayden RA, Lee RE, Armour JW, Cooper AM, Orme IM, Brennan PJ, Besra GS. Antimycobacterial action of thiolactomycin: an inhibitor of fatty acid and mycolic acid synthesis. Antimicrob Agents Chemother. 1996;40:2813–9. doi: 10.1128/aac.40.12.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi KH, Kremer L, Besra GS, Rock CO. Identification and substrate specificity of beta -ketoacyl (acyl carrier protein) synthase III (mtFabH) from Mycobacterium tuberculosis. J Biol Chem. 2000;275:28201–7. doi: 10.1074/jbc.M003241200. [DOI] [PubMed] [Google Scholar]
- 20.Kremer L, Douglas JD, Baulard AR, Morehouse C, Guy MR, Alland D, Dover LG, Lakey JH, Jacobs WR, Jr., Brennan PJ, Minnikin DE, Besra GS. Thiolactomycin and related analogues as novel anti-mycobacterial agents targeting KasA and KasB condensing enzymes in Mycobacterium tuberculosis. J Biol Chem. 2000;275:16857–64. doi: 10.1074/jbc.M000569200. [DOI] [PubMed] [Google Scholar]
- 21.Brown AK, Sridharan S, Kremer L, Lindenberg S, Dover LG, Sacchettini JC, Besra GS. Probing the mechanism of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthase III mtFabH: factors influencing catalysis and substrate specificity. J Biol Chem. 2005;280:32539–47. doi: 10.1074/jbc.M413216200. [DOI] [PubMed] [Google Scholar]
- 22.Heath RJ, Rock CO. The Claisen condensation in biology. Nat Prod Rep. 2002;19:581–96. doi: 10.1039/b110221b. [DOI] [PubMed] [Google Scholar]
- 23.Schaeffer ML, Agnihotri G, Volker C, Kallender H, Brennan PJ, Lonsdale JT. Purification and biochemical characterization of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthases KasA and KasB. J Biol Chem. 2001;276:47029–37. doi: 10.1074/jbc.M108903200. [DOI] [PubMed] [Google Scholar]
- 24.Garwin JL, Klages AL, Cronan JE., Jr. Beta-ketoacyl-acyl carrier protein synthase II of Escherichia coli. Evidence for function in the thermal regulation of fatty acid synthesis. J Biol Chem. 1980;255:3263–5. [PubMed] [Google Scholar]
- 25.de Mendoza D, Klages Ulrich A, Cronan JE., Jr. Thermal regulation of membrane fluidity in Escherichia coli. Effects of overproduction of beta-ketoacyl-acyl carrier protein synthase I. J Biol Chem. 1983;258:2098–101. [PubMed] [Google Scholar]
- 26.Gelmann EP, Cronan JE., Jr. Mutant of Escherichia coli deficient in the synthesis of cis-vaccenic acid. J Bacteriol. 1972;112:381–7. doi: 10.1128/jb.112.1.381-387.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garwin JL, Klages AL, Cronan JE., Jr. Structural, enzymatic, and genetic studies of beta-ketoacyl-acyl carrier protein synthases I and II of Escherichia coli. J Biol Chem. 1980;255:11949–56. [PubMed] [Google Scholar]
- 28.Slayden RA, Barry CE., 3rd The role of KasA and KasB in the biosynthesis of meromycolic acids and isoniazid resistance in Mycobacterium tuberculosis. Tuberculosis (Edinb) 2002;82:149–60. doi: 10.1054/tube.2002.0333. [DOI] [PubMed] [Google Scholar]
- 29.Gao LY, Laval F, Lawson EH, Groger RK, Woodruff A, Morisaki JH, Cox JS, Daffe M, Brown EJ. Requirement for kasB in Mycobacterium mycolic acid biosynthesis, cell wall impermeability and intracellular survival: implications for therapy. Mol Microbiol. 2003;49:1547–63. doi: 10.1046/j.1365-2958.2003.03667.x. [DOI] [PubMed] [Google Scholar]
- 30.Huang W, Jia J, Edwards P, Dehesh K, Schneider G, Lindqvist Y. Crystal structure of beta-ketoacyl-acyl carrier protein synthase II from E.coli reveals the molecular architecture of condensing enzymes. Embo J. 1998;17:1183–91. doi: 10.1093/emboj/17.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moche M, Dehesh K, Edwards P, Lindqvist Y. The crystal structure of beta-ketoacyl-acyl carrier protein synthase II from Synechocystis sp. at 1.54 A resolution and its relationship to other condensing enzymes. J Mol Biol. 2001;305:491–503. doi: 10.1006/jmbi.2000.4272. [DOI] [PubMed] [Google Scholar]
- 32.Price AC, Rock CO, White SW. The 1.3-Angstrom-resolution crystal structure of beta-ketoacyl-acyl carrier protein synthase II from Streptococcus pneumoniae. J Bacteriol. 2003;185:4136–43. doi: 10.1128/JB.185.14.4136-4143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bagautdinov B, Miyano M, Tahirov TH. Crystal Structure of 3-Oxoacyl-(Acyl-Carrier Protein) Synthase II from Thermus Thermophilus. 2003 doi: 10.1107/S1744309108010336. to be published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathieu M, Zeelen JP, Pauptit RA, Erdmann R, Kunau WH, Wierenga RK. The 2.8 A crystal structure of peroxisomal 3-ketoacyl-CoA thiolase of Saccharomyces cerevisiae: a five-layered alpha beta alpha beta alpha structure constructed from two core domains of identical topology. Structure. 1994;2:797–808. doi: 10.1016/s0969-2126(94)00081-6. [DOI] [PubMed] [Google Scholar]
- 35.Olsen JG, Kadziola A, von Wettstein-Knowles P, Siggaard-Andersen M, Lindquist Y, Larsen S. The X-ray crystal structure of beta-ketoacyl [acyl carrier protein] synthase I. FEBS Lett. 1999;460:46–52. doi: 10.1016/s0014-5793(99)01303-4. [DOI] [PubMed] [Google Scholar]
- 36.Edwards P, Nelsen JS, Metz JG, Dehesh K. Cloning of the fabF gene in an expression vector and in vitro characterization of recombinant fabF and fabB encoded enzymes from Escherichia coli. FEBS Lett. 1997;402:62–6. doi: 10.1016/s0014-5793(96)01437-8. [DOI] [PubMed] [Google Scholar]
- 37.Olsen JG, Rasmussen AV, von Wettstein-Knowles P, Henriksen A. Structure of the mitochondrial beta-ketoacyl-[acyl carrier protein] synthase from Arabidopsis and its role in fatty acid synthesis. FEBS Lett. 2004;577:170–4. doi: 10.1016/j.febslet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Davies C, Heath RJ, White SW, Rock CO. The 1.8 A crystal structure and active-site architecture of beta-ketoacyl-acyl carrier protein synthase III (FabH) from escherichia coli. Structure Fold Des. 2000;8:185–95. doi: 10.1016/s0969-2126(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 39.Funabashi H, Kawaguchi A, Tomoda H, Omura S, Okuda S, Iwasaki S. Binding site of cerulenin in fatty acid synthetase. J Biochem (Tokyo) 1989;105:751–5. doi: 10.1093/oxfordjournals.jbchem.a122739. [DOI] [PubMed] [Google Scholar]
- 40.Kauppinen S, Siggaard-Andersen M, von Wettstein-Knowles P. beta-Ketoacyl-ACP synthase I of Escherichia coli: nucleotide sequence of the fabB gene and identification of the cerulenin binding residue. Carlsberg Res Commun. 1988;53:357–70. doi: 10.1007/BF02983311. [DOI] [PubMed] [Google Scholar]
- 41.McGuire KA, Siggaard-Andersen M, Bangera MG, Olsen JG, von Wettstein-Knowles P. beta-Ketoacyl-[acyl carrier protein] synthase I of Escherichia coli: aspects of the condensation mechanism revealed by analyses of mutations in the active site pocket. Biochemistry. 2001;40:9836–45. doi: 10.1021/bi0105577. [DOI] [PubMed] [Google Scholar]
- 42.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 43.Olsen JG, Kadziola A, von Wettstein-Knowles P, Siggaard-Andersen M, Larsen S. Structures of beta-ketoacyl-acyl carrier protein synthase I complexed with fatty acids elucidate its catalytic machinery. Structure (Camb) 2001;9:233–43. doi: 10.1016/s0969-2126(01)00583-4. [DOI] [PubMed] [Google Scholar]
- 44.von Wettstein-Knowles P, Olsen JG, McGuire KA, Henriksen A. Fatty acid synthesis. Role of active site histidines and lysine in Cys-His-His-type beta-ketoacyl-acyl carrier protein synthases. Febs J. 2006;273:695–710. doi: 10.1111/j.1742-4658.2005.05101.x. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358–61. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 46.Price AC, Choi KH, Heath RJ, Li Z, White SW, Rock CO. Inhibition of beta-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin. Structure and mechanism. J Biol Chem. 2001;276:6551–9. doi: 10.1074/jbc.M007101200. [DOI] [PubMed] [Google Scholar]
- 47.Moche M, Schneider G, Edwards P, Dehesh K, Lindqvist Y. Structure of the complex between the antibiotic cerulenin and its target, beta-ketoacyl-acyl carrier protein synthase. J Biol Chem. 1999;274:6031–4. doi: 10.1074/jbc.274.10.6031. [DOI] [PubMed] [Google Scholar]
- 48.Kim P, Zhang YM, Shenoy G, Nguyen QA, Boshoff HI, Manjunatha UH, Goodwin MB, Lonsdale J, Price AC, Miller DJ, Duncan K, White SW, Rock CO, Barry CE, 3rd, Dowd CS. Structure-activity relationships at the 5-position of thiolactomycin: an intact (5R)-isoprene unit is required for activity against the condensing enzymes from Mycobacterium tuberculosis and Escherichia coli. J Med Chem. 2006;49:159–71. doi: 10.1021/jm050825p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Senior SJ, Illarionov PA, Gurcha SS, Campbell IB, Schaeffer ML, Minnikin DE, Besra GS. Biphenyl-based analogues of thiolactomycin, active against Mycobacterium tuberculosis mtFabH fatty acid condensing enzyme. Bioorg Med Chem Lett. 2003;13:3685–8. doi: 10.1016/j.bmcl.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 50.Schaeffer ML, Carson JD, Kallender H, Lonsdale JT. Development of a scintillation proximity assay for the Mycobacterium tuberculosis KasA and KasB enzymes involved in mycolic acid biosynthesis. Tuberculosis (Edinb) 2004;84:353–60. doi: 10.1016/j.tube.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Otwinowski Z, Minor W, Carter CWJ, Sweet RM, editors. Methods Enzymol. Vol. 276. Academic Press; 1997. Processing of X-ray diffraction data collected in oscillation mode. [DOI] [PubMed] [Google Scholar]
- 52.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vagin AA, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–25. [Google Scholar]
- 54.CCP4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 55.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 56.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–65. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 57.Yeates TO. Detecting and overcoming crystal twinning. Methods Enzymol. 1997;276:344–58. [PubMed] [Google Scholar]
- 58.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, T. S, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1999;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 59.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–68. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]