

Abstract
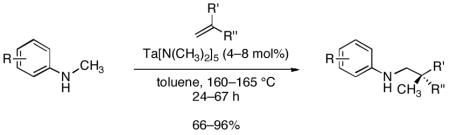
A tantalum-catalyzed addition of N-alkylarylamine α-C–H bonds across olefins is reported. These reactions occur with mono- and 2,2-disubstituted olefins to form the branched insertion products in high yield and regioselectivity. The reactions encompass additions of the α-C–H bonds of cyclic and acyclic amines, as well as intramolecular additions. NMR studies indicate that the starting homoleptic, Ta(NMe2)5precatalyst converts to bis- and tris(N-methylanilide) complexes (amongst others) in solution. Deuterium-labeling studies suggest that reversible ortho-metalation of the arene substituent occurs under the reaction conditions. However, several experiments imply that this ortho-metalation does not lie on the reaction pathway. Instead, these complexes are proposed to eliminate amine to form N-aryl imine complexes, which insert olefins into the Ta–C bond and undergo protonolysis to regenerate the active catalyst and eliminate the addition product.
Recent efforts to develop the synthesis of amines from olefins have led to improved catalytic procedures for the addition of N–H bonds across olefins (hydroamination)1 and for the tandem hydroformylation and reductive amination to form homologated amines (hydroaminomethylation)2 We describe a complementary metal-catalyzed strategy for olefin amination: the addition of amine α-C–H bonds across olefins3 to form branched alkylamines in a process that can be termed hydroaminoalkylation (eq 1). This reaction broadens the scope of α-functionalizations of amines that occur in the absence of typical coordinating directing groups4, 5 to include reactions with olefins, and it displays an unusual selectivity for functionalization of saturated over aromatic C–H bonds through organometallic intermediates.
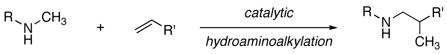 |
(1) |
Over 20 years ago, Maspero6 and Nugent7 reported the α-alkylation of dimethylamine with simple olefins in the presence of homoleptic dimethylamido complexes of tantalum, niobium, and tungsten. Although this transformation was new at the time, yields did not exceed 38 %, even after one week of reaction time, and no improved conditions have since been reported. These additions were suggested7 to occur by amine elimination to form an η2-imine complex,8 followed by olefin insertion into the resulting M–C bond, as shown in eq 2. Related stoichiometric transformations of methylzirconocene amido complexes9 and group V η2-imine complexes10 were subsequently developed. In parallel, Whitby and co-workers showed that the rate of formation of zirconocene-η2-imine complexes was faster from N-alkylanilide complexes than from dialkylamido complexes.11
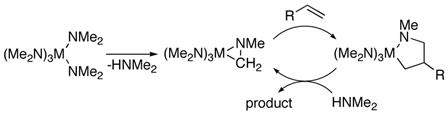 |
(2) |
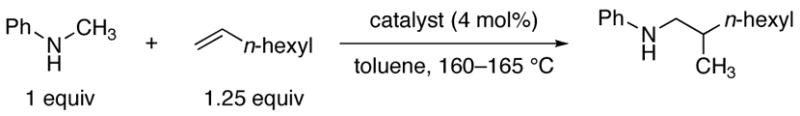
The last result suggested to us that the catalytic α-alkylation of amines might proceed more efficiently with N-arylalkylamines than with dialkylamines. Consistent with this hypothesis, the reaction of N-methylaniline with 1-octene formed the branched hydroaminoalkylation product in >95% yield within 24 h in the presence of 4 mol% Ta(NMe2)512a with only a moderate excess of olefin. This reaction is shown as entry 1 in Table 1.
Table 1.
Coupling of N-methylaniline and 1-octene by early metal dimethylamido complexes.
 | ||||
|---|---|---|---|---|
| % Yielda |
||||
| Catalyst Precursor | 1.3 h | 5.1 h | 24 h | |
| 1 | Ta[N(CH3)2]5 | 32 | 60 | 96 |
| 2 | Ta[N(CH2CH3)2]5 | 23 | 41 | 66 |
| 3 | Nb[N(CH3)2]5 | 20 | 29 | 35 |
| 4 | Cp2Zr[N(CH3)2]2 | 0.6 | 1.2 | 3.0 |
| 5 | Zr[N(CH3)2]4 | 0b | 0b | 0.1 |
| 6 | none | 0b | 0b | 0b |
Determined by GC using dodecane as an internal standard.
None was observed under conditions where >0.05% could be detected.
Reactions catalyzed by other d0 homoleptic dialkylamido complexes, such as Ta(NEt2)512 (entry 2), Nb(NMe2)513 (entry 3), and Zr(NMe2)414 (entry 4), occurred to lower conversions. Cp2Zr(NMe2)215 (entry 5) did not catalyze this transformation in substantial yields, although it is similar to the complexes that have been used for stoichiometric reactions of η2-imine complexes with olefins.9 Under otherwise identical conditions, N-methyl-trimethylacetamide, N-methyl-trifluoroacetamide, N-methyl-p-toluenesulfonamide, and N-methyl-methanesulfonamide did not form detectable levels of the analogous addition products, as determined by GC/MS and 1H NMR spectroscopic analysis.
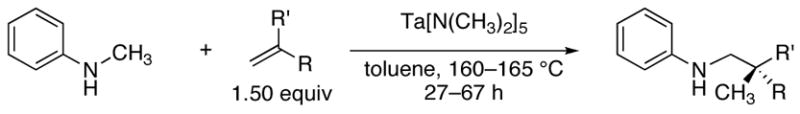
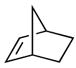
The scope of the olefin that reacts with N-methylaniline is summarized in Table 2. Mono- and 2,2-disubstituted olefins reacted in high yields (entries 1–6) using 4–8 mol% Ta(NMe2)5. Norbomene also added N-methylaniline in high yield (entry 7), but unstrained, 1,2-disubstituted olefins, such as trans-2-octene or cyclohexene, have not yet formed detectable levels of the expected alkylation products.
Table 2.
Coupling of N-methylaniline with terminal olefins.
 | ||||
|---|---|---|---|---|
| Entry | Olefin | Mol% Ta | Products(s) | Yielda |
| 1 |
|
4 |
|
88% |
|
|
50% | |||
| 2 |
|
4 |
|
28% |
| 3 |
|
4 |
|
77% |
| 4b |
|
8 |
|
76% |
| 5b |
|
4 |
|
71% |
| 6b |
|
8 |
|
66% |
| 7 |
|
4 |
|
96% |
Isolated yield after purification by flash-column chromatography.
Reaction conducted neat.
The selectivities of this process are notable. The branched alkylation products were formed as the sole detectable regioisomer, with the exception of the alkylaniline products shown in entry 2. Products arising from multiple alkylations were not observed. Many reactions of α-olefins are complicated by competitive isomerization processes. 1H NMR analysis of these crude hydroaminoalkylation reaction mixtures revealed that only 10–20% of the unreacted olefin consisted of internal isomers. Finally, in contrast to most C–H bond functionalizations that occur through organometallic intermediates, the reactions occur preferentially at sp3 C–H bonds over sp2 C–H bonds.
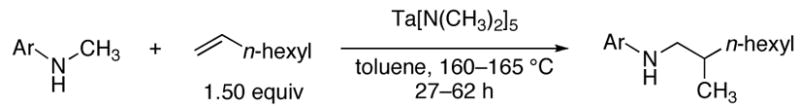
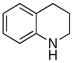
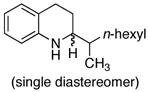
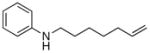
The scope of the amine component is shown in Table 3. A variety of substituted alkylaniline derivatives added to 1-octene in high yield. For example, N-methyl-3,5-dimethylaniline, N-methyl-3,5-di-t-butylaniline, N-methyl-3,5-di-fluoroaniline, and N-methyl-4-fluoroaniline formed the branched addition products in high yields (entries 1, 2, 3, and 5, respectively). N-Methyl-4-methoxyaniline also reacted with 1-octene in high yield, and this aryl group can be cleaved from nitrogen by oxidation (entry 4).16 The alkylation of 1,2,3,4-tetrahydroquinoline (entry 6) and N-(6-heptenyl)aniline (entry 7) illustrate the ability of this aminoalkylation process to form products that cannot be generated by aminomethylation with CO and H2.
Table 3.
Coupling of substituted anilines with 1-octene.
 | ||||
|---|---|---|---|---|
| Entry | Arylamine | Product | Yielda | |
| 1 | Ar = m-(CH3)2-C6H3 | 88<& | ||
| 2 |
|
Ar = m-t-Bu2-C6H3 |
|
93% |
| 3 | Ar = m-F2-C6H3 | 84% | ||
| 4 | Ar = p-(CH3O)-C6H4 | 90% | ||
| 5 | Ar = p-F-C6H4 | 78% | ||
| 6 |
|
|
72%b | |
| 7 |
|

|
88%c | |
Isolated yield after purification by flash-column chromatography.
Reaction conducted in neat 1-octene (2.50 equiv) using 8 mol % Ta(Nme2)5; relative stereochemistry not assigned.
8 mol% Ta(NMe2)5.
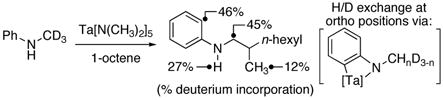
The higher yields from reactions of N-aryl alkylamines than from reactions of dialkylamines,6, 7 along with the usual trend that activation of aryl C–H bonds occurs faster than activation of aliphatic C–H bonds, led us to investigate whether metalation of the aryl ring occurred during the catalytic process. Indeed, the product from reactions of N-(methyl-d3)aniline contained 46% deuterium incorporation into the ortho position on the arene (eq 3). In addition, identical amounts of deuterium in the ortho position of the arene (15%) were contained in the reactant and product at 25% conversion (1H NMR spectral analysis) Thus, the formation of ortho-metalated intermediates, as depicted in eq 3, appeals to occur faster than the overall catalytic process. However, the reaction of N-(methyl-d3)-3,5-di-tert-butylaniline formed the expected alkylation product in 78% yield and with comparable rates to the reaction of N-(methyl-d3)aniline, with only 16% deuterium incorporation into the ortho positions of the arene. This experiment implies that the amine α-alkylation occurs in a similar fashion when ortho-metalation is faster or slower than the overall process and that the ortho-metalation lies off of the reaction pathway. We suggest that the N-aryl substituents facilitate generation of an η2-imine complex by serving as an electron-withdrawing group,9a,11 without deactivating the catalyst by formation of a stable chelate.
 |
(3) |
To gain insight into the identity of the tantalum complexes in the catalytic system, we analyzed reactions of N-methyl-p-toludine by 1H NMR spectroscopy. After heating this substrate with Ta[N(CH3)2]5 and 1-octene in toluene at 160 °C for 3 h, the bis(anilide) and tris(anilide) complexes [(p-tol)(CH3)N]2Ta[N(CH3)2]3 (1) and [(p-tol)(CH3)N]3Ta[N(CH3)2]2 (2) (~ 1:1 ratio) accounted for >75% of the tantalum-amido species in solution (1H NMR analysis).17 Studies on the chemistry of these complexes relevant to this catalytic process are ongoing.
In summary, we have described an efficient C–H bond functionalization process that constitutes a hydroaminoalkylation of alkenes. The selectivity of this reaction appears to be controlled by the electronic properties of the amine, but this reaction does not require functionality on nitrogen, such as a pyridyl, iminyl, or carbamoyl groups, that directly coordinate to the metal.
Supplementary Material
Detailed experimental procedures and spectral data of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the NIH (NRSA fellowship to S.B.H and NIGMS GM-55382 to JFH) for support of this work.
References
- 1.(a) Muller TE, Beller M. Chem Rev. 1998;98:675. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]; (b) Nobis M, Driessen-Holscher B. Angew Chem, Int Ed Engl. 2001;40:3983. doi: 10.1002/1521-3773(20011105)40:21<3983::aid-anie3983>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (c) Hong S, Marks TJ. Acc Chem Res. 2004;37:673. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 2.(a) Eilbracht P, Baerfacker L, Buss C, Hollmann C, Kitsos-Rzychon BE, Kranemann CL, Rische T, Roggenbuck R, Schmidt A. Chem Rev. 1999;99:3329. doi: 10.1021/cr970413r. [DOI] [PubMed] [Google Scholar]; (b) Beller M, Seayad J, Tillack A, Jiao H. Angew Chem, Int Ed Engl. 2004;43:3368. doi: 10.1002/anie.200300616. [DOI] [PubMed] [Google Scholar]
- 3.For the coupling of methane and propene, see: Sadow AD, Tilley TD. J Am Chem Soc. 2003;125:7971. doi: 10.1021/ja021341a.
- 4.(a) Murahashi SI. Angew Chem, Int Ed Engl. 1995;34:2443. [Google Scholar]; (b) Davies HML, Venkataramani C, Hansen T, Hopper DW. J Am Chem Soc. 2003;125:6462. doi: 10.1021/ja0290072. [DOI] [PubMed] [Google Scholar]; (c) Catino AJ, Nichols JM, Nettles BJ, Doyle MP. J Am Chem Soc. 2006;128:5648. doi: 10.1021/ja061146m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li ZP, Bohle DS, Li CJ. Proc Natl Acad Sci USA. 2006;103:8928. doi: 10.1073/pnas.0601687103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For examples of directed activation of C–H bonds adjacent to nitrogen, see: Jordan RF, Taylor DF. J Am Chem Soc. 1989;111:778.Jun CH, Hwang DC, Na SJ. Chem Commun. 1998:1405.Chatani N, Asaumi T, Ikeda T, Yorimitsu S, Ishii Y, Kakiuchi F, Murai S. J Am Chem Soc. 2000;122:12882. doi: 10.1021/ja011540e.Wang DH, Hao XS, Wu DF, Yu JQ. Org Lett. 2006;8:3387. doi: 10.1021/ol061384m.Pastine SJ, Gribkov DV, Sames D. J Am Chem Soc. 2006;128:14220. doi: 10.1021/ja064481j.Lewis JC, Bergman RG, Ellman JA. [accessed April 6, 2007];J Am Chem Soc. doi: 10.1021/ja070388z. [Online early access], Published online: April 6,2007. http://pubs3.acs.org/acs/journals/doilookup?in_doi=10.1021/ja070388z.
- 6.Clerici MG, Maspero F. Synthesis. 1980:305. [Google Scholar]
- 7.Nugent WA, Ovenall DW, Holmes SJ. Organometallics. 1983;2:161. [Google Scholar]
- 8.Formation of η2-imine complexes by amine elimination: Takahashi Y, Onoyama N, Ishikawa Y, Motojima S, Sugiyama K. Chem Lett. 1978:525.
- Airoldi C, Bradley DC, Vuru G. Transition Met Chem (London) 1979;4:64. [Google Scholar]
- 9.(a) Buchwald SL, Watson BT, Wannamaker MW, Dewan JC. J Am Chem Soc. 1989;111:4486. [Google Scholar]; (b) Barluenga 1, Rodriguez F, Alvarez-Rodrigo L, Fananas FJ. Chem Soc Rev. 2005;34:762. doi: 10.1039/b504557f. [DOI] [PubMed] [Google Scholar]; (c) Cummings SA, Tunge JA, Norton JR. Top Organomet Chem. 2005;10:1. [Google Scholar]
- 10.Roskamp EJ, Pedersen SF. J Am Chem Soc. 1987;109:6551. [Google Scholar]
- 11.Coles N, Harris MCJ, Whitby RJ, Blagg J. Organometallics. 1994;13:190. [Google Scholar]
- 12.(a) Bradley DC, Thomas IM. Can J Chem. 1962;40:1355. [Google Scholar]; (b) Davies HO, Jones AC, McKinnell EA, Raftery J, Muryn CA, Afzaal M, O’Brien P. J Mater Chem. 2006:2226. [Google Scholar]
- 13.Bradley DC, Thomas IM. Can J Chem. 1962;40:449. [Google Scholar]
- 14.Bradley DC, Thomas IM. J Chem Soc. 1960:3857. [Google Scholar]
- 15.Chandra G, Lappert MF. J Chem Soc A. 1968:1940. [Google Scholar]
- 16.Kronenthal DR, Han CY, Taylor MK. J Org Chem. 1982;47:2765. [Google Scholar]
- 17.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures and spectral data of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.