

Abstract
FOXP3 is a necessary transcription factor for the development and function of CD4+ regulatory T-cells (Tregs). The role of Tregs in HIV-1 infection remains unclear. Here, we show expression of FOXP3 in primary human CD4 T-cells significantly inhibits HIV-1 infection. Since FOXP3 inhibits NFAT activity, and NFAT proteins contribute to HIV-1 transcription, we explore a transcriptional repressive function of HIV-1 LTR by FOXP3. Over-expression of FOXP3 in primary CD4 T-cells inhibits wild-type HIV-1 LTR reporter activity, and truncation mutants demonstrate repression of the LTR by FOXP3 requires the dual proximal NFκB/NFAT binding sites. Interestingly, FOXP3 decreases binding of NFAT2 to the HIV-1 LTR in vivo. Furthermore, FOXP3 does not inhibit infection of HIV-1 NL4-3 which is mutated to disrupt transcription factor binding at either proximal NFAT or NFκB binding sites. These data suggest resistance of Tregs to HIV-1 infection is due to inhibition of HIV-1 LTR transcription by FOXP3.
Keywords: T cells, Human, Transcription factors, Gene regulation, HIV-1, FOXP3, NFAT2, LTR
Introduction
The transcription factor, FOXP3, is highly expressed in CD4+CD25hi T-cells and is necessary for the development and function of regulatory T-cells (Tregs) (Fontenot, Gavin, and Rudensky, 2003; Hori, Nomura, and Sakaguchi, 2003; Khattri et al., 2003). FOXP3 has been shown to transactivate or to repress target genes (Zheng et al., 2007), and inhibits both NFAT- and NFκB-mediated transcriptional activation (Bettelli, Dastrange, and Oukka, 2005). FOXP3 inhibits transcriptional activation by interaction with forkhead binding sites located immediately adjacent to the cis-acting NFAT binding sites found in various cytokine promoters, including IL-2 (Marson et al., 2007; Wu et al., 2006). FOXP3 may also mediate transcriptional repression indirectly, without direct binding to DNA. Bettelli et al. showed that FOXP3 directly binds to NFAT and NFκB proteins and prevents the activation of a number of cytokine genes (Bettelli, Dastrange, and Oukka, 2005). Thus, there appear to be multiple mechanisms by which FOXP3 inhibits NFAT-responsive gene expression.
HIV/AIDS is characterized by the depletion of CD4 T-cells and progressive immune dysfunction, including HIV-1 specific T-cell responses. Recent studies suggest that Tregs play a major role in immune suppression. It has been postulated that Tregs are beneficial for HIV-1 infected individuals because detrimental immune activation may be controlled by Tregs (Sempere, Soriano, and Benito, 2007). In contrast, Kinter et al. showed that the HIV-1 specific CTL response is suppressed by Tregs and that this suppression continues throughout the course of HIV disease (Kinter et al., 2007a; Kinter et al., 2007b). Thus, Treg activity could impact the ability of HIV-1 infected individuals to control HIV-1 replication.
We show here that over-expression of FOXP3 inhibits HIV-1 infection of human primary CD4 T-cells, inhibiting infection of both FOXP3+ and FOXP3- cells. We further show that FOXP3 inhibits HIV-1 LTR activity and this transcriptional repression of the HIV-1 LTR requires the presence of the dual proximal NFκB/NFAT binding sites. Interestingly, FOXP3 expression decreases the binding of NFAT2 to the HIV-1 LTR. Based on these data, we hypothesize that FOXP3 makes CD4+ regulatory T-cells relatively resistant to HIV-1 infection via transcriptional inhibition of the LTR. Understanding the role of FOXP3 and Tregs in HIV-1 infection may lead to new therapeutic approaches to combat HIV-1 disease and has implications for the potential use of Treg therapy during HIV-1 infection.
Results and Discussion
Lentiviral mediated expression of FOXP3 in human primary CD4 T-cells
Human primary CD4 T-cells were activated with anti-CD3/CD28 beads and transduced with lentivirus expressing human FOXP3 or control (as described in the Materials and Methods). The level of expression of FOXP3 protein in transduced cells was determined by Western blot analysis. As shown in Fig. 1A, FOXP3-transduced cells expressed 3- to 4-fold more FOXP3 protein than the control transduced cells. Surprisingly, in some experiments significant levels of FOXP3 protein were noted in the control GFP-transduced cells. It is possible that the FOXP3 expression in control cells was due either to proliferation of a Treg population, or to prolonged expression of FOXP3 in recently activated T-cells, in response to exogenous IL-2 (Allan et al., 2007; Burchill et al., 2007; Murawski et al., 2006; Zorn et al., 2006). Nevertheless, the FOXP3-transduced cells expressed significantly more FOXP3 than the control cells (Fig. 1A).
Figure 1.
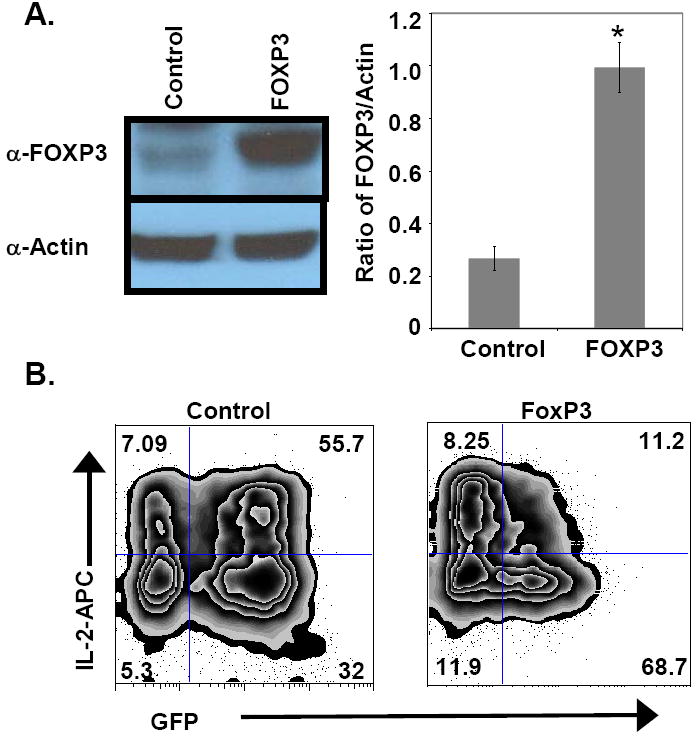
Lentiviral transduction of T-cells. FOXP3 or control GFP lentiviral vectors were transduced as described in the Material and Methods. Lentiviral transduction efficiency was between 50-85% measured by GFP expression (data not shown). More FOXP3 protein is expressed in transduced cells. (A) Western blotting was done to determine FOXP3 expression in FOXP3 lentiviral-transduced and control-transduced primary human CD4 T cells. The blot shown is from one representative experiment of 3. The graph to the right depicts ratios of FOXP3 to actin (control) optical density measurements for control and FOXP3 transduced CD4 T cells. The data shown are means ± SEM from 3 experiments (*p=0.002). These data show that FOXP3-transduced cells have substantially more FOXP3 protein, relative to actin controls, than control cells. (B) Intracellular staining of IL-2 in polyclonally activated (6 hrs) control- (left) or FOXP3- (right) transduced CD4 T cells. GFP expression (marking transduced cells) is depicted on the x-axis and IL-2 expression is portrayed on the y-axis. GFP+ FOXP3-transduced cells produced notably less IL-2 than GFP+ control cells. One representative of 3 experiments is shown.
Next, the FOXP3-transduced CD4 T-cells were tested for the ability to suppress IL-2 production [a well established function of FOXP3, (Bettelli, Dastrange, and Oukka, 2005)]. Lentiviral-transduced cells were activated again with PMA and ionomycin for 6 hrs and analyzed for intracellular IL-2. As expected, FOXP3 over-expression markedly suppressed IL-2 production compared to control cells (Fig. 1B). In this brief assay, the non-transduced (GFP-) cells did not lose IL-2 production, arguing that FOXP3 was responsible for the decreased IL-2 production in the GFP+ cells (Fig. 1B). Taken together, these data suggest that the lentiviral-transduced cells express functional FOXP3 protein and can be used to study the role of FOXP3 in HIV-1 infection of primary CD4 T-cells.
FOXP3 inhibits HIV-1 infection
Presently, the role of FOXP3-expressing Tregs during HIV-1 infection remains unclear. We investigated the effect of FOXP3 expression in an in vitro model of HIV-1 infection of primary CD4 T-cells. FOXP3- or control GFP-transduced cells were infected in vitro with the HIV-1 X4 viruses, NL4-3, or IIIB strains, and monitored for infection by assessing p24-gag expression by using flow cytometry and ELISA. Transduction of FOXP3 inhibited HIV-1 infection (Fig. 2); time course analyses showed that FOXP3 inhibited HIV-1 production at all time points tested post-infection (Fig. 2C and 2D). Interestingly, further analysis of FOXP3-transduced cell cultures showed that the GFP- (FOXP3-) population of cells also had fewer p24-gag+ cells (Fig. 2A, left upper quadrant). In comparison to control transduced cells, in FOXP3 transduced cells, NL4-3 p24 expression is reduced by 64%, and for the GFP–negative cells in the same culture, p24 expression is reduced by 49%. Similarly, for the IIIB virus, GFP+ (FOXP3+) cells had a 78% reduction in p24 expression, whereas GFP-negative cells in the same culture had a 54% reduction. Thus, the direct inhibitory effect in the FOXP3+ cells appears to be more substantial than the effect on neighboring FOXP3-negative cells. These comparisons are valid in that roughly equal percentages of cells were transduced with control versus FOXP3-expressing viruses (~50% for each virus). These data suggest that FOXP3+ cells could have suppressive effects on bystander (FOXP3-) cells within the same culture. Further experiments are needed to assess the trans-suppressive function of Tregs during HIV-1 infection. For example, is this phenomenon cell contact dependent? Our preliminary data reveal no change in CXCR4 expression arguing against a viral entry issue. Nevertheless, collectively, these data suggest that FOXP3 inhibits HIV-1 infection and viral particle production.
Figure 2.
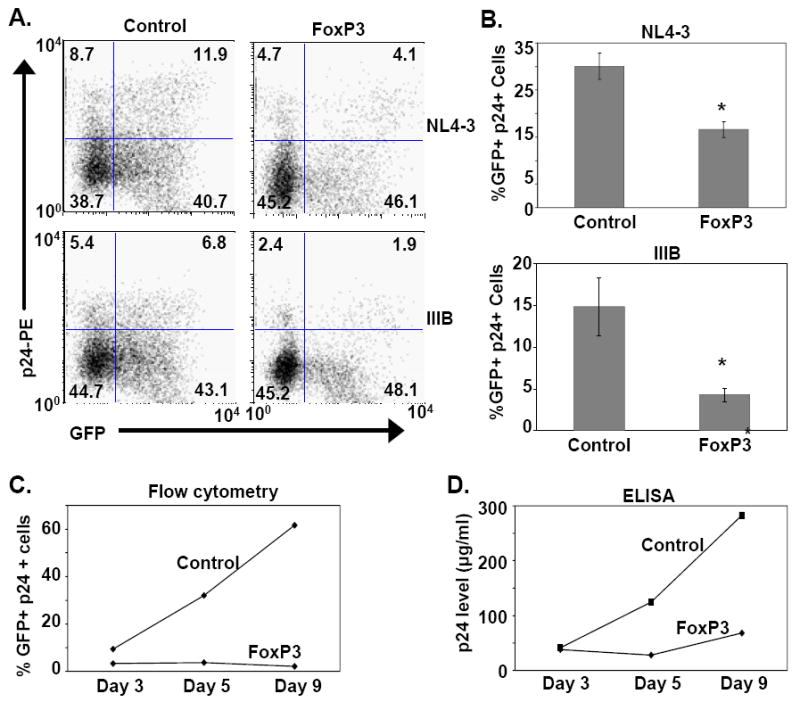
FOXP3 expression inhibits HIV-1 infection in T-cells. (A) Primary human CD4 T-cells were activated with anti-CD3/CD28 antibody conjugated beads and transduced with FOXP3 or control lentivirus as described in the Materials and Methods. Transduced T-cells were infected with HIV-1 NL4-3 or IIIB and monitored for p24-gag expression by flow cytometry. Lentivirus-transduced cells were monitored by their expression of GFP. Dot plot analysis of one representative experiment of 3 is shown. GFP expression (transduced cells) is noted along the x-axis, and p24-gag expression is depicted on the y-axis. The percentages of cells for each quadrant are noted in the corners of the respective quadrants. (B) The percentages of p24+ cells in transduced (control or FOXP3) T-cell cultures are shown in the bar graphs for the NL4-3 and IIIB infected cells, respectively. The mean ± SEM of 3 experiments performed is shown and compared between control and FOXP3-transduced cells (p=0.015 for NL4-3, and p=0.06 for IIIB). (C) The time course of HIV-1 IIIB infection of the control- and FOXP3-transduced CD4 T-cells is shown as % p24+ cells (y-axis) versus days of infection (x-axis). One representative of 3 experiments is shown. (D) p24 levels (y-axis) in the supernatant of the IIIB infected CD4 T-cells was measured by ELISA and is shown over time in days post-infection (x-axis). One representative of 3 experiments is presented.
In contrast, there are at least three published manuscripts reporting that FOXP3 increases or augments HIV-1 infection (Antons et al., 2008; Holmes et al., 2007; Oswald-Richter et al., 2004). Why such dichotomous results exist remains unclear but there are differences in the experimental design of these studies. Specifically, Oswald-Richter et al. utilized a VSV-G pseudotyped CCR5-tropic (R5) GFP virus (nef gene deleted) in their experiments (Oswald-Richter et al., 2004). The authors did not report on the effect of FOXP3 on an HIV-1 R4 virus in natural Tregs or FOXP3-transduced cells. Although we have not analyzed natural Tregs directly, our data with VSV-G pseudotyped R5-Luc (luciferase) virus (nef gene deleted) show that FOXP3 similarly increases HIV-1 infection by 38.5 ± 5.7% (mean ± SEM of 4 experiments). Thus, it is possible that X4 and R5 viruses respond differently to FOXP3. Interestingly, a second published report also utilized an X4 reporter-virus (NL4-Luc, nef gene deleted) in their experiments and showed that FOXP3 increased HIV-1 infection (Holmes et al., 2007). Both of these reporter viruses are deleted in the nef gene, so, theoretically, the Nef protein could play an important role in the inhibition of HIV-1 infection by FOXP3. However, our initial experiments show that FOXP3 also inhibits an HIV-1 NL4-3 virus deleted in the nef gene (data not shown). Another possible difference explaining the discrepant results is the VSV-G envelope used with the NL4-3 virus, although our data show that FOXP3 still inhibits VSV-G pseudotyped wild-type NL4-3 T-cell infection (data not shown). Thus, the type of virus cannot fully explain the disparate results.
Alternatively, rather than due to the virus, the differences between our data and previously published reports may have been due to differences in the method of T-cell activation and expansion. In both of the previously published manuscripts, T-cells were activated with anti-mouse IgG coated plates and soluble anti-CD3 and anti-CD28 antibodies. It is likely that the T-cells were more completely activated by the anti-CD3/CD28 coated beads used in our experiments (Levine et al., 1998). In addition, we added 100 U/ml of IL-2 to expand the T-cells and to maintain their viability. It is possible, for example, that our activation protocol maintained FOXP3 expression in the transduced cells for a longer period of time. In sum, while our data using intact HIV-1 virions demonstrate that over-expression of FOXP3 inhibits HIV-1 infection, the mechanism remains unclear.
FOXP3 inhibits HIV-1 LTR transcriptional activity
FOXP3 is a well-known transcriptional repressor of many of its target genes (Marson et al., 2007). In particular, FOXP3 has been proposed to inhibit both NFAT and NFκB transcriptional activity (Bettelli, Dastrange, and Oukka, 2005). Interestingly, the HIV-1 LTR possesses dual proximal NFAT/NFκB binding sites, which are critical to optimal transcriptional activity of the promoter (Cron, 2001; Giffin et al., 2003; Robichaud et al., 2002; Romanchikova et al., 2003). In order to explore a potential transcriptional repressive function of the HIV-1 LTR by FOXP3, unstimulated primary human CD4 T-cells were transfected with an HIV-1 LTR-driven luciferase reporter construct, with or without co-transfection of a wild-type FOXP3 mammalian expression vector. The CD4 T-cells were activated with PMA and ionomycin for 6 hrs and luciferase activity was determined by luminometry as described in the Materials and Methods. Expression of FOXP3 significantly inhibited intact HIV-1 LTR transcriptional activity (Fig. 3). Truncation of the distal LTR up to -122bp upstream of the transcriptional start site still revealed substantial inhibition by FOXP3 (Fig. 3), but transcriptional inhibition was lost with further deletion up to -97bp. Comparing the HIV-1 LTR sequences between -122 and -97 with published consensus sequences for FOXP3 binding did not reveal a good match or theoretical binding site for FOXP3 within this stretch of nucleotides. Interestingly, deletion up to -97bp disrupts the tandem proximal NFAT/NFκB binding sites, suggesting that FOXP3-mediated transcriptional inhibition of the HIV-1 LTR occurs via NFAT and/or NFκB pathways. With each truncation of the LTR, the transcriptional activity is reduced. However, even the -97 construct still has notable inducible activity (~40,000 arbitrary light units) above background (Fig. 3, bottom). In summary, our data show that FOXP3 over-expression inhibits HIV-1 LTR transcriptional activity in primary CD4 T-cells and this inhibition requires the presence of the dual proximal NFAT/NFκB binding sites (Fig. 3).
Figure 3.
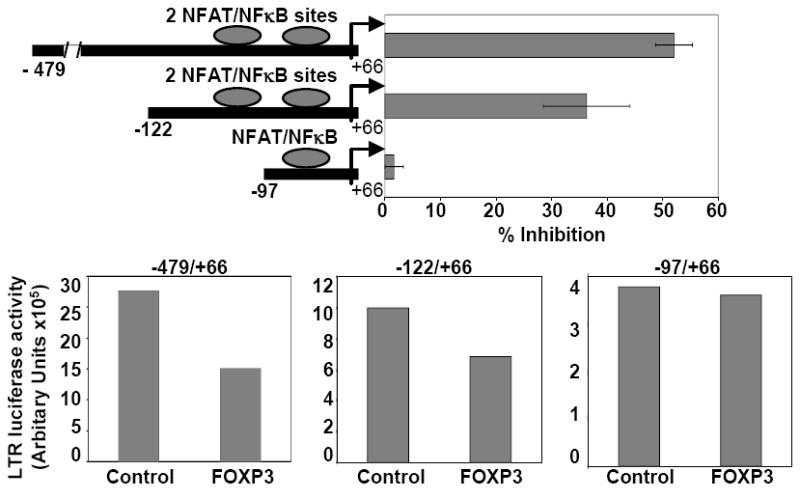
FOXP3 inhibits HIV-1 LTR activity. Primary CD4 T-cells were co-transfected with a FOXP3 (or control) expression vector, one of two different 5’ truncation deletions of HIV-1 LTR-responsive firefly-luciferase reporter genes, and a transfection efficiency control Renilla-luciferase construct. T-cells were activated with PMA and ionomycin for 6 hrs and corrected luciferase reporter gene activity was measured as a surrogate of HIV-1 LTR transcription rate. Data are represented as percent inhibition of LTR activity by FOXP3 compared to the pcDNA expression control vector. Comparisons of LTR activity between the -122/+66 and -97/+66 reporter constructs were made using the Student’s t-test. Data depicted are the mean ± SEM of 3 experiments performed (p=0.05). Graphs on the bottom represent LTR-luciferase activity for the different deletion constructs as indicated, with and without co-expression of FOXP3. One representative of 3 experiments is shown. With each truncation of the LTR, the transcriptional activity is reduced. However, even the -97/+66 construct still has notable inducible activity (~40,000 arbitrary light units) above background luminometry values (~1,000 ALU, data not shown).
FOXP3 decreases the binding of NFAT2 to the HIV-1 LTR
To understand the mechanism of FOXP3 mediated inhibition of HIV-1 infection, we first determined whether FOXP3 binds along with NFAT2 to the HIV-1 LTR, as shown for the IL-2 promoter (Wu et al., 2006). For strength of manipulation, 293T cells were co-transfected with control or wild-type FOXP3 expression vector, an NFAT2 expression vector, and an HIV-1 LTR luciferase reporter plasmid. After 48 hrs of culture, cells were harvested and either ChIP or luciferase assays were performed. The luciferase transcriptional assay shows that FOXP3 inhibits LTR activity in 293T cells similar to primary CD4+ T-cells (Fig 4A). However, FOXP3 binding to HIV-1 LTR by ChIP was not detected (Fig. 4B). Interestingly, FOXP3 expression significantly decreases NFAT2 binding to the HIV-1 LTR (Fig. 4B). Our data show a different mechanism for FOXP3 mediated inhibition of transcription than reported for IL-2 (Wu et al., 2006). Nevertheless, Bettelli et al. showed that FOXP3 could bind to and functionally inhibit NFAT2 and NFκB (Bettelli, Dastrange, and Oukka, 2005). Therefore, it is possible that FOXP3 binds to NFAT2 and thus blocks the subsequent binding of NFAT2 to the HIV-1 LTR, required for optimal activation-dependent transcription of HIV-1 in primary CD4 T-cells. Alternatively, FOXP3 is known to inhibit induction of NFAT2 in T cells (Fontenot et al., 2005; Marson et al., 2007), so that less NFAT2 might be available to bind the LTR. Whatever the mechanism turns out to be, NFAT2 engagement of the LTR is decreased in the presence of FOXP3. Along these lines, our preliminary data suggest FOXP3 over-expression disrupts NFAT2 engagement of the HIV-1 LTR in vivo following activation of virally infected primary human CD4 T cells (data not shown).
Figure 4.
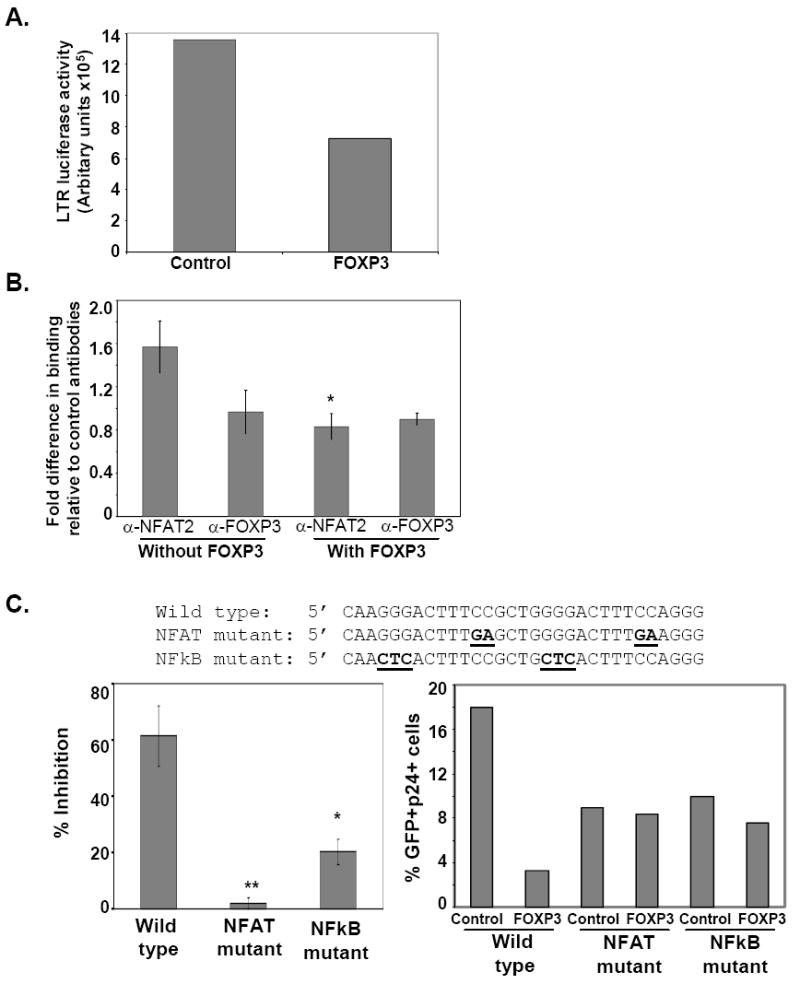
FOXP3 inhibits HIV-1 infection by interfering with NFAT and/or NFκB function on the HIV-1 LTR. (A) FOXP3 inhibits LTR activity in 293T cells. 293T cells were co-transfected with a control or wild-type FOXP3 expression vector, an NFAT2 expression vector, and an HIV-1 LTR luciferase reporter plasmid as described in the Materials and Methods. Data show that FOXP3 inhibits NFAT2 induced LTR activity in 293T cells. One representative of 3 experiments is presented. (B) FOXP3 decreases the binding of NFAT2 to the LTR. ChIP with anti-NFAT2 and anti-FOXP3 antibodies was performed using control (without FOXP3) and FOXP3 transfected 293 cells, which were co-transfected with an HIV-1 LTR reporter plasmid. NFAT2 and FOXP3 engagement in vivo of the HIV-1 LTR reporter sequence was analyzed using PCR amplification primers specific to the proximal HIV-1 LTR sequence as described in the Materials and Methods. ChIP analysis data are presented as fold differences (mean ± SEM) between specific antibody and isotype control from 3 independent experiments (*p=0.053, compared to NFAT2 without FOXP3). (C) FOXP3 does not substantially inhibit productive infection by NL4-3 viruses mutated at either the dual NFAT or NFκB binding sites. FOXP3 or control lentivirus transduced T-cells were infected with NL4-3 wild type, NFAT-mutant, or NFκB-mutant HIV-1 viruses and were monitored for p24-gag expression. The wild type and NFAT and NFκB mutant sequences are depicted above the bar graphs with the mutant sequences underlined and bolded. Data in the bar graph to the left are represented as % inhibition of infection in FOXP3 transduced cells compared to control cells for the wild type and each binding mutant virus. Data are the mean ± SEM of 3 experiments (**p=0.005; *p=0.024). Data shown on the right is a representative example of 1 of 3 experiments. Data are presented as %GFP+, p24+ cells for each virus and for both the control and FOXP3 transduced cells. The binding mutant viruses have reduced but not absent HIV-1 production in response to T cell activation.
Based on the in vivo binding studies (Fig. 4B) and our mapping studies (Fig. 3), it is likely that FOXP3 disrupts NFAT binding at the dual proximal NFAT/NFκB binding sites (Cron et al., 2000). However, it is formally possible, yet unlikely, that FOXP3 inhibits LTR activity via disruption of NFAT binding in the upstream negative regulatory element (Gaynor, 1992), or in the NFAT binding site in the 5’ untranslated region (Romanchikova et al., 2003). To directly address the role of the dual proximal NFAT/NFκB sites in FOXP3-mediatied inhibition of LTR transcription, the effect of mutations in the proximal NFAT sites on the ability of FOXP3 to inhibit HIV-1 expression was studied.
Specifically, the importance of NFAT and/or NFκB binding to the proximal LTR during FOXP3 mediated inhibition of HIV-1 infection was tested. NL4-3 viruses mutagenized to disrupt binding of transcription factors to the dual proximal NFAT or NFκB binding sites were analyzed. FOXP3- or control GFP-transduced cells were infected in vitro with NL4-3 wild-type, dual NFAT mutant, or dual NFκB mutant HIV-1 viruses and monitored for infection by assessing p24-gag expression. Interestingly, FOXP3 did not substantially inhibit productive infection by the mutant HIV-1 viruses, with remarkable reversal of inhibition seen particularly with the NFAT mutant virus (Fig. 4C). Neither the NFAT or NFκB mutant viruses are inactive but their expression is reduced (Fig. 4C), consistent with our proposed mechanism of how FOXP3 inhibits viral replication. Similarly, others have shown that mutations in this region of the LTR leading to reduced binding of NFAT or NFκB can be found in different HIV clades. These viruses have reduced but not absent transcription in response to T cell activation (Lemieux et al., 2004). In addition, we and others have previously shown that specific mutations used to generate the viruses in Fig. 4C lead to specific inhibition of binding of NFAT or NFκB factors to the proximal HIV-1 LTR (Cron et al., 2000; Kinoshita et al., 1997). Collectively, these data suggest that FOXP3 mediates inhibition of HIV-1 infection primarily via interference with NFAT and/or NFκB transcriptional activation of the proximal HIV-1 LTR.
Taken together, our studies suggest that FOXP3-expressing Tregs may be relatively resistant to HIV-1 infection. Moreover, FOXP3-expressing T-cells may also inhibit HIV-1 infection of neighboring CD4 T-cells (Fig. 2A). We speculate that FOXP3 suppresses HIV-1 transcription and could, thereby, convert these cells to latent infection. Currently, standard latency protocols are performed on resting T-cells after positive selection of CD4+CD25- T-cells from HIV+ individuals. It is possible that by removing CD25+ cells, potential Tregs (FOXP3+ cells) are also removed; this could result in underestimation of the number of HIV-1 latently infected T-cells in patients. In addition, recent publications suggest that Tregs in HIV-1+ individuals suppress HIV-1 specific CTLs (Kinter et al., 2007a; Kinter et al., 2007b; Sempere, Soriano, and Benito, 2007), further confounding the benefit or lack thereof of Tregs during HIV-1 infection. Immune control of viral replication represents a balance between the cell-mediated immune response and the Treg mediated counter-regulation of such responses. Given the complexity of Treg biology in HIV-1 disease, treatment of HIV-1 disease with Tregs may be more complicated than the proposed use of Tregs in cancer or autoimmunity.
Materials and Methods
Isolation of CD4 T-cells
CD4 T-cells were isolated by negative selection (RosetteSep; StemCell Technologies, Vancouver, British Columbia) from heparinized venous blood of healthy adult human donors as described (Hamilton et al., 2003; Selliah and Finkel, 2001). Isolated cells were 90-95% CD3+CD4+ as detected by flow cytometry.
Lentiviral Transduction and HIV-1 Infection
CD4 T-cells were activated with anti-CD3/CD28 coated beads [Torcylated Dynal beads (Invitrogen, Carlsbad, CA). were conjugated with anti-CD3 mAb (OKT3, Ortho Biotech Products, L.P., Bridgewater, NJ) and anti-CD28 mAb (clone 9.3, Ortho Biotech), as per the manufacturer’s recommendations] for 24 hrs in RPMI containing 10% human serum (Gemini Bio-Products, Sacramento, CA), penicillin/streptomycin, L-glutamine and IL-2 (100 U/ml, NIH AIDS Research and Reference Reagent Program, Bethesda, MD). T-cells were transduced with lentivirus expressing human FOXP3 co-expressed with GFP, or control GFP alone, in the presence of Polybrene (8 μg/ml, Sigma, St. Louis, MO) at TU=40 and cultured at 5 × 105 cells/ml (Humeau et al., 2004). Transduced T-cells were washed after 3 days and cultured for another two days. Beads were removed using a magnet (Invitrogen), and T-cells were infected with HIV-1 NL4-3 or IIIB strains (2 μg of p24-gag by ELISA per 1 × 106 cells, with 20 μg/ml DEAE-dextran) and cultured with IL-2 (100 U/ml) for 3 days (Rapaport et al., 1998; Selliah et al., 2006). Infection of T-cells was monitored by p24-gag staining by flow cytometry and by measuring p24-gag in the supernatant by ELISA, as described (Rapaport et al., 1998; Selliah et al., 2006). In some experiments, NL4-3 virus mutated at NFAT or NFκB proximal binding sites were used for the infection. Mutations of NFAT or NFκB sites were performed by PCR site-directed mutagenesis (kit from Stratagene, La Jolla, CA). Briefly, wild type HIV-1 LTR sequence (from the Xho I site to the Nco I site) was first subcloned by conventional methods from the pUC-based vector, pNL4-3 (kindly supplied by Dr. Frederic Bushman, University of Pennsylvania, Philadelphia, PA), into pMIGR1 (kindly provided by Dr. Warren Pear, University of Pennsylvania) for manipulation purposes. PCR primer pairs containing the proximal HIV-1 LTR mutant sequences listed below (coding strands only shown) were used to generate transcription factor binding site variant sequences using the Site-Directed Mutagenesis Kit. Underlined sequences correspond to the dual proximal NFAT/NFκB overlapping binding sites, and the bolded/underlined nucleotides are the altered sequences used to disrupt NFAT and NFκB binding, respectively.
Wild type: 5’CAAGGGACTTTCCGCTGGGGACTTTCCAGGG
NFAT mutant: 5’CAAGGGACTTTGAGCTGGGGACTTTGAAGGG
NFκB mutant: 5’CAACTCACTTTCCGCTGCTCACTTTCCAGGG
The resultant variant LTR sequences were then subsequently religated into pNL4-3 prior to transfection into 293T cell lines for generation of mutant virions. All mutant sequences were confirmed by DNA sequencing of coding and non-coding strands. Infection of T-cells was monitored by p24-gag staining by flow cytometry and by measuring p24-gag in the supernatant by ELISA, as described (Selliah et al., 2006).
Western Blotting and IL-2 Detection
FOXP3 or control GFP lentiviral transduced cells were checked for protein expression by Western blotting, as described previously (Selliah and Finkel, 2001). PVDF membrane was blotted with anti-FOXP3 antibody (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA) or anti-actin control mAb (Sigma), and developed with goat anti-rabbit IgG-HRP or goat anti-mouse IgG–HRP, respectively, and super signal substrate (Pierce, Rockford, IL). Optical density of the bands was measured with a BioRad Quantity One imaging system (BioRad, Hercules, CA).
IL-2 production was evaluated in lentiviral transduced T-cells by intracellular staining. FOXP3 or control GFP transduced T-cells were activated with PMA (10 ng/ml, Sigma) and ionomycin (1 μM, MP Biomedicals, Solon, OH) for 6 hrs without exogenous IL-2. Two μl of Golgi STOP (BD Biosciences, San Jose, CA) was added for the final 4 hrs and cells were fixed in 1% paraformaldehyde (Sigma). T-cells were washed with PBS and resuspended in 100 μl of PBS containing 0.1% saponin and 10% FCS. 2.5 μl of anti-human IL-2-APC (BD Biosciences) was added and incubated at room temperature for 30 min. T-cells were then washed two times in PBS and analyzed by flow cytometry (FACS Calibur, BD). Data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Luciferase assays
CD4 T-cells were co-transfected, as described (Cron et al., 2000) using AMAXA technology (AMAXA® Biosystems, Cologne, Germany), with pcDNA3 (empty control) or pcDNA3-FOXP3 expression vector, (1.5 μg, a kind gift from Dr. Troy Torgerson, University of Washington, Seattle, WA) (Lopes et al., 2006), an HIV-1 LTR-firefly luciferase reporter gene (1 μg) (Cron et al., 2000), and a Renilla luciferase-expressing transfection control plasmid (pRL-null, 0.5 μg) (Promega, Madison, WI). HIV-1 LTR-luciferase deletion constructs were generated as previously described (Chipitsyna et al., 2006). T-cells were activated with PMA (10 ng/ml) and ionomycin (1 μM) for 6 hrs and lysed with passive lysis buffer to measure luciferase activity, as per the manufacturer’s recommendations (Pierce) (Cron et al., 2000). Cell viability was determined and samples were normalized to equal numbers of viable cells prior to lysis.
Chromatin Immunoprecipitation (ChIP) Assays
293T cells were co-transfected, using Fugene 6 reagent (Roche, Indianapolis, IN), with pcDNA3 (empty control) or pcDNA3-FOXP3 expression vector, along with an NFAT2 expression vector, and an HIV-1 LTR-luciferase transcriptional reporter plasmid. After 48 hrs of culture, cells were harvested and ChIP assay was performed with anti-NFAT2 antibody (Santa Cruz) or anti-FOXP3 antibody (Santa Cruz) or control Rabbit IgG antibody (Santa Cruz), as described (Selliah et al., 2006), using the EZ ChIP assay kit (Upstate Biotechnology, Billerica, MA). Specifically, cells were cross-linked with 1% formaldehyde, washed, pelleted, and resuspended in lysis buffer. Each lysate was diluted to 1 ml using Tris/EDTA and sonicated. Sonicates were precleared with protein G Sepharose beads and incubated in dilution buffer overnight at 4°C with 10 μl of anti-FOXP3, anti-NFAT2, or IgG isotype control antibodies. Samples were immunoprecipitated with protein G-Sepharose for 1 h. The beads were washed and eluted, and supernatants were reverse cross-linked by addition of NaCl and heat for 6 h at 65°C. Immunoprecipitated samples were digested with proteinase K for 1 h at 45°C, and DNA was extracted with phenol/chloroform/isoamyl alcohol. The DNA was then treated with RNase for 30 min at 37°C, and equal volumes of immunoprecipitated DNA were analyzed by real-time PCR. Real-time quantitative PCR was done with an ABI Prism 7500 Sequence Detection System (PerkinElmer Life Sciences, Waltham, MA) using PCR primers specific to the proximal HIV-1 LTR sequence (Selliah et al., 2006). Fold differences (between specific antibody and isotype control antibody) were calculated using the formula: 2-ΔCt, where ΔCt=Ct(sample)-Ct(control), and Ct corresponds to the number of amplification cycles needed to reach a specified threshold value.
Acknowledgments
Supported by the National Institutes of Health (NIH) P30 AI45008, NIH R01 AI35513, the Joseph L. Hollander Chair (T.H.F.), the Mary L. Smith Charitable Trust, the Elizabeth Glaser Pediatric AIDS Foundation (R.Q.C.), the Joseph Stokes Jr. Research Institute, the Penn Cancer Center, the NIH AIDS Research and Reference Reagent Program, and the University of Pennsylvania Center for AIDS Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, Roncarolo MG, Levings MK. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19(4):345–54. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- Antons AK, Wang R, Oswald-Richter K, Tseng M, Arendt CW, Kalams SA, Unutmaz D. Naive precursors of human regulatory T cells require FoxP3 for suppression and are susceptible to HIV infection. J Immunol. 2008;180(2):764–73. doi: 10.4049/jimmunol.180.2.764. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A. 2005;102(14):5138–43. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol. 2007;178(1):280–90. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- Chipitsyna G, Sawaya BE, Khalili K, Amini S. Cooperativity between Rad51 and C/EBP family transcription factors modulates basal and Tat-induced activation of the HIV-1 LTR in astrocytes. J Cell Physiol. 2006;207(3):605–13. doi: 10.1002/jcp.20612. [DOI] [PubMed] [Google Scholar]
- Cron RQ. HIV-1, NFAT, and cyclosporin: immunosuppression for the immunosuppressed? DNA Cell Biol. 2001;20(12):761–7. doi: 10.1089/104454901753438570. [DOI] [PubMed] [Google Scholar]
- Cron RQ, Bartz SR, Clausell A, Bort SJ, Klebanoff SJ, Lewis DB. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin Immunol. 2000;94(3):179–91. doi: 10.1006/clim.1999.4831. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22(3):329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Gaynor R. Cellular transcription factors involved in the regulation of HIV-1 gene expression. AIDS. 1992;6(4):347–63. doi: 10.1097/00002030-199204000-00001. [DOI] [PubMed] [Google Scholar]
- Giffin MJ, Stroud JC, Bates DL, von Koenig KD, Hardin J, Chen L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR kappa B element. Nat Struct Biol. 2003;10(10):800–6. doi: 10.1038/nsb981. [DOI] [PubMed] [Google Scholar]
- Hamilton BJ, Genin A, Cron RQ, Rigby WF. Delineation of a novel pathway that regulates CD154 (CD40 ligand) expression. Mol Cell Biol. 2003;23(2):510–25. doi: 10.1128/MCB.23.2.510-525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes D, Knudsen G, Mackey-Cushman S, Su L. FoxP3 enhances HIV-1 gene expression by modulating NFkappa B occupancy at the LTR in human T cells. J Biol Chem. 2007;282(22):15973–80. doi: 10.1074/jbc.M702051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Humeau LM, Binder GK, Lu X, Slepushkin V, Merling R, Echeagaray P, Pereira M, Slepushkina T, Barnett S, Dropulic LK, Carroll R, Levine BL, June CH, Dropulic B. Efficient lentiviral vector-mediated control of HIV-1 replication in CD4 lymphocytes from diverse HIV+ infected patients grouped according to CD4 count and viral load. Mol Ther. 2004;9(6):902–13. doi: 10.1016/j.ymthe.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4(4):337–42. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, Nolan GP. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity. 1997;6(3):235–44. doi: 10.1016/s1074-7613(00)80326-x. [DOI] [PubMed] [Google Scholar]
- Kinter A, McNally J, Riggin L, Jackson R, Roby G, Fauci AS. Suppression of HIV-specific T cell activity by lymph node CD25+ regulatory T cells from HIV-infected individuals. Proc Natl Acad Sci U S A. 2007a;104(9):3390–5. doi: 10.1073/pnas.0611423104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinter AL, Horak R, Sion M, Riggin L, McNally J, Lin Y, Jackson R, O’Shea A, Roby G, Kovacs C, Connors M, Migueles SA, Fauci AS. CD25+ regulatory T cells isolated from HIV-infected individuals suppress the cytolytic and nonlytic antiviral activity of HIV-specific CD8+ T cells in vitro. AIDS Res Hum Retroviruses. 2007b;23(3):438–50. doi: 10.1089/aid.2006.0162. [DOI] [PubMed] [Google Scholar]
- Lemieux AM, Pare ME, Audet B, Legault E, Lefort S, Boucher N, Landry S, van Opijnen T, Berkhout B, Naghavi MH, Tremblay MJ, Barbeau B. T-cell activation leads to poor activation of the HIV-1 clade E long terminal repeat and weak association of nuclear factor-kappaB and NFAT with its enhancer region. J Biol Chem. 2004;279(51):52949–60. doi: 10.1074/jbc.M409896200. [DOI] [PubMed] [Google Scholar]
- Levine BL, Cotte J, Small CC, Carroll RG, Riley JL, Bernstein WB, Van Epps DE, Hardwick RA, June CH. Large-scale production of CD4+ T cells from HIV-1-infected donors after CD3/CD28 costimulation. J Hematother. 1998;7(5):437–48. doi: 10.1089/scd.1.1998.7.437. [DOI] [PubMed] [Google Scholar]
- Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, Ochs HD, Ziegler SF. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol. 2006;177(5):3133–42. doi: 10.4049/jimmunol.177.5.3133. [DOI] [PubMed] [Google Scholar]
- Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, Levine SS, Fraenkel E, von Boehmer H, Young RA. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445(7130):931–5. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murawski MR, Litherland SA, Clare-Salzler MJ, Davoodi-Semiromi A. Upregulation of Foxp3 expression in mouse and human Treg is IL-2/STAT5 dependent: implications for the NOD STAT5B mutation in diabetes pathogenesis. Ann N Y Acad Sci. 2006;1079:198–204. doi: 10.1196/annals.1375.031. [DOI] [PubMed] [Google Scholar]
- Oswald-Richter K, Grill SM, Shariat N, Leelawong M, Sundrud MS, Haas DW, Unutmaz D. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLoS Biol. 2004;2(7):E198. doi: 10.1371/journal.pbio.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport E, Casella CR, Ikle D, Mustafa F, Isaak D, Finkel TH. Mapping of HIV-1 determinants of apoptosis in infected T cells. Virology. 1998;252(2):407–17. doi: 10.1006/viro.1998.9459. [DOI] [PubMed] [Google Scholar]
- Robichaud GA, Barbeau B, Fortin JF, Rothstein DM, Tremblay MJ. Nuclear factor of activated T cells is a driving force for preferential productive HIV-1 infection of CD45RO-expressing CD4+ T cells. J Biol Chem. 2002;277(26):23733–41. doi: 10.1074/jbc.M201563200. [DOI] [PubMed] [Google Scholar]
- Romanchikova N, Ivanova V, Scheller C, Jankevics E, Jassoy C, Serfling E. NFAT transcription factors control HIV-1 expression through a binding site downstream of TAR region. Immunobiology. 2003;208(4):361–5. doi: 10.1078/0171-2985-00283. [DOI] [PubMed] [Google Scholar]
- Selliah N, Finkel TH. HIV-1 NL4-3, but not IIIB, inhibits JAK3/STAT5 activation in CD4(+) T cells. Virology. 2001;286(2):412–21. doi: 10.1006/viro.2001.0994. [DOI] [PubMed] [Google Scholar]
- Selliah N, Zhang M, Desimone D, Kim H, Brunner M, Ittenbach RF, Rui H, Cron RQ, Finkel TH. The gammac-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T cells. Virology. 2006;344(2):283–91. doi: 10.1016/j.virol.2005.09.063. [DOI] [PubMed] [Google Scholar]
- Sempere JM, Soriano V, Benito JM. T regulatory cells and HIV infection. AIDS Rev. 2007;9(1):54–60. [PubMed] [Google Scholar]
- Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, Mathis D, Benoist C, Chen L, Rao A. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375–87. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445(7130):936–40. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108(5):1571–9. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]