

Abstract
Peripheral neuropathy is one of the major side-effects of the anticancer drug, cisplatin. Although previous work suggests that this neuropathy correlates with formation of DNA adducts in sensory neurons, growing evidence suggests that cisplatin also increases the generation of reactive oxygen species (ROS), which could cause DNA damage. Apurinic/apyrimidinic endonuclease/redox factor-1 (Ape1/Ref-1) is a multifunctional protein involved in DNA base excision repair (BER) of oxidative DNA damage and in redox regulation of a number of transcription factors. Therefore, we asked whether altering Ape1 functions would influence cisplatin induced neurotoxicity. Sensory neurons in culture were exposed to cisplatin for 24 hrs and several endpoints of toxicity were measured including production of ROS, cell death, apoptosis, and release of the immunoreactive calcitonin gene-related peptide (iCGRP). Reducing expression of Ape1 in neuronal cultures using siRNA enhances cisplatin-induced cell killing, apoptosis, ROS generation and the cisplatin-induced reduction in iCGRP release. Overexpressing wild-type (WT)-Ape1 attenuates all the toxic effects of cisplatin in cells containing normal endogenous levels of Ape1 and in cells with reduced Ape1 levels following Ape1siRNA treatment. Overexpressing the redox deficient/repair competent C65-Ape1 provides partial rescue, while the repair deficient Ape1 (N226A+R177A) does not protect neurons from cisplatin toxicity. We also observe an increase in phosphorylation of p53 following a decrease in Ape1 levels in sensory neuronal cultures. These results strongly support the notion that Ape1 is a potential translational target such that protecting Ape1 levels and particularly its DNA repair function could reduce peripheral neuropathy in patients undergoing cisplatin treatment.
Keywords: ROS, neurotoxicity, base excision repair (BER), oxidative stress, sensory neurons, DRG, redox, peripheral neuropathy, p53, Ape1
Introduction
Cisplatin is one of the most commonly used cytotoxic agents in the treatment of a variety of solid malignant tumors including testicular, bladder, lung, esophagus, stomach, ovarian cancers as well as sarcomas and lymphomas (1). One of the major limitations of the use of cisplatin, however, is peripheral neuropathy (2, 3) which appears to be caused mainly by the action of the drug on dorsal root ganglion (DRG) neurons (4). Cisplatin therapy and cytotoxicity has been shown to involved the formation of intrastrand and interstrand adducts in DNA (5) and this is thought to be one of the major mechanisms for cisplatin-induced damage in sensory neurons (3, 6) Indeed, mice deficient in nucleotide excision repair (NER), the pathway once thought to be the major repair response to cisplatin, show an increase in symptoms of neuropathy after cisplatin treatment compared to mice with NER intact (7). However, there is growing evidence suggesting that cisplatin toxicity is closely associated with increased generation of reactive oxygen species (ROS) (8–10).
Apurinic/apyrimidinic endonuclease/redox factor (Ape1/Ref-1; which will be called Ape1) is a ubiquitous multifunctional protein involved in the DNA base excision repair (BER) pathway of oxidative DNA damage that can occur from either endogenous or exogenous agents generating ROS (11–13). In addition, Ape1 functions as a major reducing-oxidizing (redox) factor augmenting the binding activity of a number of transcription factors to DNA including, but not limited to NFkB, p53, AP-1, and CREB (13). In neurons, these transcription factors play important roles in neuronal survival, stress response and various disorders (14–16).
Previous studies by our laboratory demonstrated that Ape1 protects sensory neuronal cells from oxidative DNA damage, secondary to exposure to H2O2 (17). Other groups have demonstrated that overexpression of Ape1 protected melanoma cells from cisplatin or H2O2 induced apoptosis (18) and we have shown that Ape1 overexpression protected germ cell tumor cells from radiation and bleomycin treatment (19). Thus, there is precedent in mitotic cancer cell lines that Ape 1 plays a role in cisplatin induced cell death. The question remains, however, whether Ape 1 could be neuroprotective against cisplatin-induced toxicity in postmitotic sensory neurons and thus have the potential to reduce peripheral neuropathy. In this article, we demonstrate that cisplatin produces significant amounts of ROS in sensory neuronal cultures and that reducing Ape1 expression in these cultures results in an increase in cisplatin-induced neurotoxicity and an increase in the activity of the p53 signaling pathway. Conversely overexpressing wild type and redox deficient Ape 1 in neuronal cultures attenuates cisplatin-induced toxicity while a repair deficient Ape1 does not. These findings have potential translational applications such that modifying or protecting the function of Ape1 in neuronal cells may have chemoprotective effects for patients undergoing chemotherapy with platinating agents.
Materials and Methods
Cell culture
Adult dorsal root ganglion neuronal cell cultures were prepared as previously described (20, 21). Briefly, adult male (150–175g) Sprague-Dawley rats (Harlan, Indianapolis, IN) were euthanized by CO2 asphyxiation. Dorsal root ganglia (DRG) were collected from the spinal column, incubated in collagenase, and mechanically dissociated. For studies measuring cell viability, ROS, apoptosis and signaling approximately 60,000 cells were plated into each well of poly-D-lysine and laminin coated 6-well culture plates. For studies measuring CGRP release, approximately 15,000 cells were plated in 12 well culture plates. Cells were maintained in F-12 media (Invitrogen, Carlsbad, CA) supplemented with 10% horse serum, 2mM glutamine, 100μg/ml normocin, 50μg/ml penicillin, 50μg/ml streptomycin, 50μM 5-fluoro-2′-deoxyuridine (Invitrogen), 150μM uridine, and 30ng/ml of NGF(Harlan Bioproducts for Science, Inc. Indianapolis, IN) in 3% CO2 at 37°C.
Cell viability assays
To assay cell viability, trypan blue exclusion analysis was performed as previously described (17). Cells were resuspended using a 0.05% trypsin EDTA solution. Equal volumes of the cell suspension and 0.4% (w/v) trypan blue in phosphate-buffered saline (PBS) were mixed and the cells scored under phase contrast microscope.
ROS Measurement
Sensory neuronal cell cultures were treated with cisplatin for 24 hrs. After washing with PBS, the cells were incubated with 10μM carboxy-H2DCFDA (Invitrogen, Carlsbad, CA) in fresh PBS for 60 min (22). Excessive probe was washed off with PBS, and cells incubated in PBS for another 60 min at 37°C. The cells were harvested with trypsin and fluorescence of the labeled cells was measured by using a Coulter EPICS XL flow cytometer (Coulter). An average of 10,000 cells from each sample was counted.
Ape1siRNA transfection
To decrease Ape1 in sensory neuronal cell cultures, Ape1siRNA and SCsiApe1 (scrambled control) were used as described previously (17, 23). Briefly, on the fourth day in culture, the growth media was replaced with 0.5 ml of Optimem I medium containing 10 μl of the transfecting reagent Neuroporter in the absence or presence of the 21-mer oligonucleotide double-stranded siRNA to Ape1 (Ape1siRNA) or scrambled Ape1siRNA (SCsiRNA). Fresh medium (0.5 ml) without antibiotics was added after 24 hrs of incubation and after an additional 24 hrs, the medium was replaced with normal medium containing antibiotics and cell growth maintained.
Adenoviral infection
Adult sensory neuronal cell cultures were grown for eight days and then infected with either the wild-type Ad5 HA-Ape1 (WT-Ape1), the redox deficient/DNA repair competent Ad5 HA-C65A (C65-Ape1), DNA repair deficient/redox competent Ad5 HA-N226A+R177A (N226+R177-Ape1), or vector control Ad5 IRES2EGFP (Vector) adenovirus at 150 pfu/cell for 24 hrs by adding the virus directly to the growth medium as previously described (17). The N226A+R177A Ape1 double mutant has been shown to decrease the ability of this mutant Ape1 to bind to AP sites in DNA (24). The constructs used contained the human Ape1 sequence so that the rat Ape1-siRNA oligonucleotides could not bind to it because of sequence differences between human and rat. However, the codons are highly conserved as are their functions. The Hemagglutinin epitope (HA) tag was added to the amino terminus of Ape in order to distinguish exogenous transgene overexpression from endogenous Ape1 gene levels and has been routinely used by us (17). After 24 hrs, the medium containing adenovirus was aspirated and replaced with culture medium. Infection efficiencies of transduced cells were determined 24 hrs after infection by fluorescence microscopy.
Cisplatin treatment
Adult sensory neuronal cultures were grown in culture for 11 days, with or without siRNA transfection and with or without adenovirus infection (vector, WT-Ape1, 226+177-Ape1, C65-Ape1), the cells were washed, treated with cisplatin at various concentrations and analyzed as described in the results section. Cisplatin (Sigma-Aldrich, St. Louis, MO) was prepared fresh for each experiment by dissolving in N,N-dimethylformamide (Sigma-Aldrich, St. Louis, MO) with the final N,N-dimethylformamide concentration less than 0.005%.
Western Blot Analysis
Cells in culture were harvested and lysed in an ice-cold RIPA lysis buffer (Santa Cruz Biotechnology, Inc. Santa Cruz, CA). Protein concentration was quantified using the detergent-compatible (25). Lowry-based protein assay (Bio-Rad Laboratories, Hercules, CA). Protein (20–40μg) was electrophoresed in SDS gel-loading buffer on a 10% SDS-polyacrylamide gel. After electrophoresis, the gel was transferred to a PVDF membrane. The membrane was incubated with a primary antibody against Ape1 (17, 26), phosphor-histone H2AX (Ser139) (Upstate Cell Signaling Solutions, Charlottesville, VA), phosphor-p53 (Ser15) (Cell signaling Technology, Beverly MA), p53, Gadd45a (C-4) (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Generally antibodies were used at dilutions of 1:1000 or 1:1500. After incubated with HRP-conjugated secondary antibody (Santa Cruz Biotechnology Inc.), Antibody binding was detected using chemiluminescence (Roche Diagnostics Corp., Indianapolis, IN), and equal loading was confirmed by probing with β-actin monoclonal antibody (NeoMarkers, Inc., Fremont, CA).
Cell apoptosis Analysis
Apoptosis was quantitated using flow cytometry after cell treatment using Annexin V-APC and 7-amino-actinomycin D (7-ADD) and the Annexin V-Apoptosis Detection kit (BD Biosciences, San Diego, CA) according to the manufacturer’s instructions. Apoptotic cells were defined as those positive for Annexin V with or without 7ADD staining.
Calcitonin gene-related peptide (CGRP)
Release studies were performed on the cells as previously described (17). Briefly, the neuronal cultures were washed once with HEPES buffer consisting of (in mM) 25 HEPES, 135 NaCl, 3.5 KCl, 2.5 CaCl2, 1 MgCl2, 3.3 D-glucose, and 0.1% bovine serum albumin, pH 7.4 and maintained at 37 C, then incubated for successive 10 min intervals with 0.4 ml of the same buffer in the absence or presence of drugs. Basal or resting release was determined by exposing the cells to HEPES buffer alone, whereas stimulated release was determined by exposing the cultures to 30 nM capsaicin. Cells then were reexposed to HEPES buffer without drugs for one or two 10 min incubations to reestablish resting release. During incubations, the cells were maintained in at 37°C. After each incubation, the buffer was removed to measure the amount of CGRP using radioimmunoassays (RIAs) as previously described (17). At the end of each release experiment, the cells are scraped and sonicated in 0.4 M HCl and an aliquot taken to measure total CGRP content in the cultures using radio immuno assay (RIA). Total content is determined by adding the total amount released in all incubations to the amount remaining in the cells. The release data is calculated as a % of the total peptide content in the cells. Because CGRP was measured by RIA, results were expressed as CGRP-like immunoreactivity (iCGRP).
Statistical Analysis
Data are expressed as the mean ± standard error, for at least three independent experiments from separate harvests. Statistical analysis was performed using ANOVA. *p<0.05 vs. control group.
Results
Effects of cisplatin on sensory neuronal cultures
Cisplatin-induced cytotoxicity
In the first series of experiments, we determined the cytotoxic effect of cisplatin on sensory neuronal cultures by exposing cultures to various concentrations of cisplatin for 24 hrs then measuring cell viability using the trypan blue exclusion assay. As shown in figure 1A, exposing sensory neurons to cisplatin causes a concentration dependent decrease in cell survival. Twenty-five μM cisplatin for 24 hr had no significant effect on cell viability, whereas with 400 μM cisplatin only 4 ± 0.01 % of the cells survived. The calculated LD50 for cisplatin in these experiments was 80 μM.
Figure 1. Effect of cisplatin treatments on sensory neuronal cells.

A. Sensory neuronal cell cultures were incubated with different concentrations of cisplatin for 24 hrs. Cytotoxicity was determined using the trypan blue exclusion assay as described. Each point represents the mean ± SE for three independent harvests of cells. Asterisks indicates a statistically significant (*p<0.05) difference from control.
B. ROS generation in sensory neuronal cell cultures following cisplatin treatment. After incubation with cisplatin for 24 hrs, sensory neuronal cell cultures were loaded with 10 μM carboxy-H2DCFDA and incubated for 1 hour. Carboxy-H2DCFDA is an indicator for reactive oxygen species (ROS) and only fluoresces when hydrolyzed by esterases and oxidation occurs within the reaction system. The top portion of the figure shows the fluorescence of the oxidized form of carboxy-H2DCFDA as analyzed in a representative flow cytometry experiment. As a positive control for oxidative stress, cells were treated for 1 hr with 100 μM TBHP. The panel at the bottom represents the summary of three experiments expressed as the amount of change in fluorescent cells following cisplatin treatment. Each point represents the mean ± SE for three independent harvests of cells. Statistically different points from controls are indicated with an asterisk (*p<0.05).
C. Ape1 protein expression levels following cisplatin treatment. The panel on the top shows a representative Western blot of the expression of Ape1 after sensory neuronal cell cultures were incubated with different cisplatin concentrations for 24 hrs. The panel at the bottom represents the summary of Ape1 expression following cisplatin treatment normalized to the amount of actin detected by densitometry. Each point represents the mean ± SE for three independent harvests of cells. Statistically different points from controls are indicated with an asterisk (*p<0.05).
Cisplatin-induced ROS generation
Although it is clear that cisplatin results in DNA adducts in sensory neurons (6, 7), recent evidence suggests that the drug also can produce ROS in non-neuronal cells (27). To determine if cisplatin could produce ROS in sensory neurons, cultures were exposed to increasing concentrations of cisplatin for 24 hours and ROS measured using carboxy-H2DCFDA and FACS analysis as described in methods. In a manner analogous to cell eviability studies, 25 μM cisplatin was not effective, i.e. it did not increase ROS, whereas with concentrations of 50 μM, 100 μM and 200 μM cisplatin ROS was significantly higher then untreated controls. As a positive control in these experiments, cultures were exposed to 100 μM Tert-Butyl hydroperoxide (TBHP) for 1 hour (28), but the increase in ROS was not as high as that observed with higher concentrations of cisplatin. These data support our contention that ROS and oxidative DNA damage are important components of cisplatin’s deleterious effects on neuronal cells.
Cisplatin-induced increase in Ape1 expression
Several studies have demonstrated that ROS enhanced Ape1 expression in gastric epithelial, ovary, macrophages and liver cells (29, 30). The question of whether chemotherapeutic agents, especially those typically classified as DNA crosslinking agents alter Ape1 expression has not been addressed. Therefore, we examined whether Ape1 expression in sensory neuronal cultures would be altered by exposing cells to cisplatin. For these experiments, cultures were exposed to various concentrations of cisplatin for 24 hrs and Ape1 protein expression analyzed using immunoblotting. As shown in figure 1C, Ape1 protein levels were not significantly increased when cells were exposed to 6.25 μM or 12.5 μM cisplatin, but were significantly higher than controls at 25 μM and higher. Increased Ape1 expression levels were also observed in cells 48 hrs after treatment with cisplatin (data not shown).
Reduced expression of Ape1 enhances cisplatin-induced cytotoxicity and ROS production in sensory neuronal cultures
Because cisplatin increased ROS and Ape1 expression in sensory neurons, we examined whether reducing Ape1 expression in sensory neuronal cultures would augment cisplatin-induced cell death. Exposing neuronal cultures to Ape1siRNA significantly reduced the expression of Ape1 compared to levels in cultures exposed to SCsiRNA (see figure 2A for a representative western blot). This reduction of Ape1 resulted in a statistically significant increase in cisplatin-induced cytotoxicity compared to the SCsiRNA controls (figure 2B). The increase in cell killing was approximately 20% greater when Ape1 levels were decreased compared to cells treated with SCsiRNA.. This result demonstrates that reduction of Ape1 expression in sensory neuronal cells significantly increases cisplatin-induced cytotoxicity.
Figure 2. Effect of Ape1 siRNA knockdown on sensory neuronal cells following cisplatin treatment.
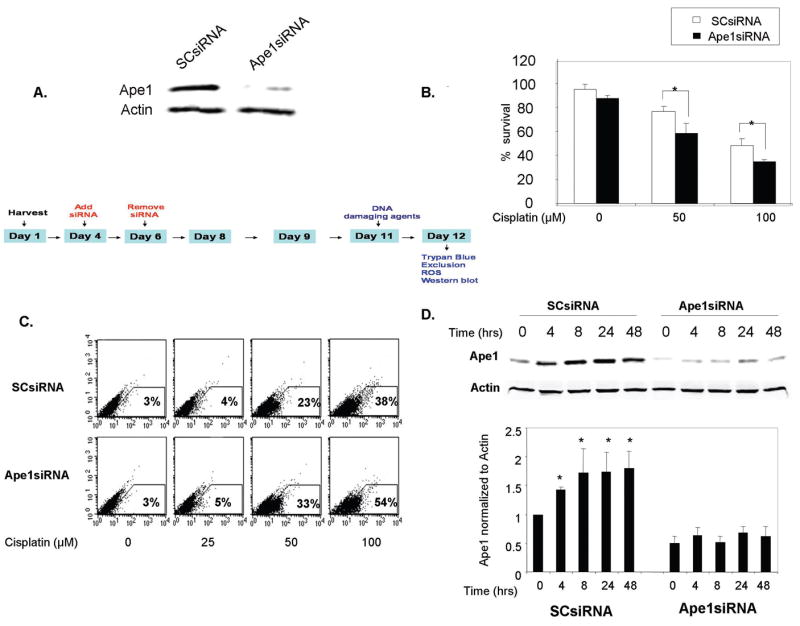
A. Sensory neuronal cells were incubated with 100 nM Ape1siRNA on day four in culture and the Ape1siRNAs were removed on day six. Ape1 expression was analyzed on day eleven by western blot. SCsiRNA (100 nM) was used as negative control.
B. Ape1siRNA knockdown and SCsiRNA transfected sensory neuronal cells were incubated with cisplatin (0, 50, 100μM) for 24 hrs. The ordinate represents the percent of cell survival measured by trypan blue exclusion. Each point represents the mean ± SE for three independent harvests of cells. Statistically different points from controls are indicated with an asterisk (*p<0.05).
C. ROS generation. The fluorescence of the oxidized form of carboxy-H2DCFDA was analyzed by flow cytometry in SCsiRNA and Ape1siRNA knockdown cells. These data are representative of four individual experiments. Statistically different points from controls are indicated with an asterisk (*p<0.05).
D. Ape1 protein expression levels in SCsiRNA and Ape1siRNA knockdown DRG cells following cisplatin 50μM treatment at indicated timepoints (left) as analyzed by Western blot. The summary of Ape1 expression was normalized to the amount of actin detected by densitometry (right). Each point represents the mean ± SE for three independent harvests of cells. Statistically different points from controls are indicated with an asterisk (*p<0.05).
We also examined ROS production in cultures exposed to SCsiRNA or Ape1SiRNA. In these experiments Ape1siRNA treatment reduced Ape1 expression by 70% compared to cultures exposed to SCsiRNA (data not shown). This decrease in Ape1 expression resulted in a significant increase in the ROS generated by a 24 hr exposure to 50 μM or 100 μM cisplatin, to 10 and 16 percent, respectively (figure 2C) compared to SCsiRNA treated cultures.
Cisplatin treatment (50 μM) enhanced the level of Ape1 expression in cultures from 4 to 48 hrs after a 24 hr exposure in cultures treated with SCsiRNA (figure 2D) in a manner a comparable to that observed in previous experiments (figure 1A), but this did not occur in cultures exposed to Ape1siRNA.
Ape1 overexpression in sensory neuronal cells in cultures protects against cisplatin-induced cytotoxicity
Because reducing Ape1 expression in sensory neuronal cultures augments cisplatin-induced toxicity, we determined whether overexpressing Ape1 could be neuroprotective. WT-Ape1, C65-Ape1, or vector control adenoviral constructs containing an IRES site and an enhanced green fluorescent protein (EGFP) construct were used to infect sensory neuronal cell cultures pretreated with either SCsiRNA or Ape1siRNA. The cells were transfected with Ape1siRNA on days 4–6 in culture, infected with WT-Ape1 or the C65-Ape1 adenovirus on day 8, treated with cisplatin (50uM or 100uM) on day 11 and cell viability assessed using trypan blue after 24 hrs of exposure to cisplatin. In all experiments we confirmed expression of the transgene proteins by performing western blots on cultures at the end of each experiment and using HA-antibody. As shown in representative Western blot in figure 3A, after transfecting with Ape1siRNA the expression of Ape1 was lower than Ape1 levels in cultures treated with SCsiRNA. After infection with viral vectors, however, the Ape1 expression was higher (figure 3A). When cells treated with SCsiRNA and the viral vector control were exposed to cisplatin there was a significant reduction in cell survival (figure 3B, left panel). Cell viability was reduced from 95% in cells not exposed to cisplatin to 65 +/− 5% and 51 +/− 3% after 50 μM and 100 μM cisplatin, respectively. With a reduction in Ape1 expression, vector treated cells showed an augmentation of cisplatin-induced neurotoxicity (figure 3B, right panel) and these results are analogous to the finding presented above. Overexpressing either WT-Ape1 or C65-Ape1 significantly rescued cells from killing by cisplatin. This occurred independent of whether the cells had reduced Ape1 levels secondary to siRNA treatment. However, the protective ability of C65-Ape1 was not equivalent to that of the WT-Ape1. For example, in cells treated with Ape1siRNA, overexpressing WTApe1 increased cells viability after 100 μM cisplatin to 50 +/− 6%, whereas the C65-Ape only increased viability to 32 +/− 3%.
Figure 3. Effect of Ape1 overexpression on sensory neuronal cell viability after cisplatin treatment.
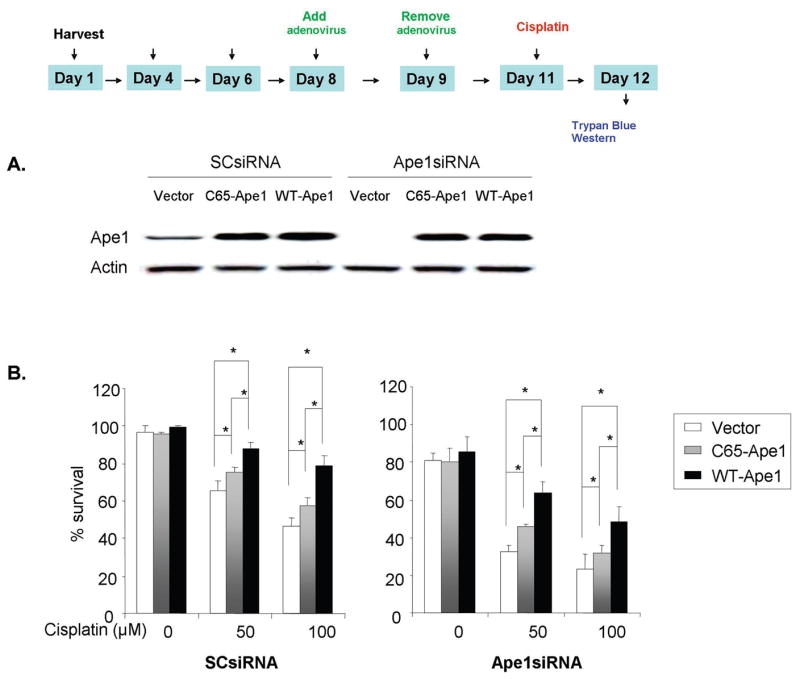
A. Sensory neuronal cells were incubated with 100 nM Ape1siRNA on day four in culture and the Ape1siRNAs were removed on day six. The cells were then infected with one of three adenoviral constructs; vector control, WT-Ape1or C65-Ape1 on day eight for 24 hrs. The level of adenovirus infection was measured on day eleven by fluorescence microscope. Ape1 expression was analyzed on day eleven by western blot.
B. Survival of sensory neuronal cells following cisplatin treatment with or without Ape1 knockdown and the addition of transgene Ape1 constructs. The ordinate represents percent of cells surviving at 24 hrs after various doses of cisplatin treatment as measured by trypan blue exclusion in DRG cells, without (left) or with (right) Ape1 knockdown. Each point represents the mean ± SE for three independent harvests of cells. Statistically different points from controls are indicated with an asterisk (*p<0.05).
Overexpression of Ape1 reduces cisplatin-induced an apoptosis in sensory neuronal cultures
Apoptosis has been observed in sensory neurons following cisplatin treatment in vitro and in vivo (31, 32). To ascertain whether Ape1 affects cisplatin-induced apoptosis in sensory neuronal cultures, we performed apoptosis assays using Annexin-V/PI flow cytometry analysis. The time line for these experiments is analogous to the previous studies but after 24 hr exposure to 100 μM cisplatin GFP positive cells were gated and cells undergoing apoptosis were detected by Annexin and 7ADD staining. As shown in the representative experiment in figure 4A and the summary data from 6 experiments in figure 4B, 22 ± 4% of cells from cultures exposed to SCsiRNA, viral vector and cisplatin were apoptotic. In cells treated with Ape1siRNA, vector and cisplatin 46 ± 6 were apoptotic. Overexpressing C65-Ape1 did not significantly rescue the cells from cisplatin-induced apoptosis, whereas over expressing WT-Ape significantly decreased the number of apoptotic cells independent of whether the cells were exposed to SCsiRNA or Ape1siRNA (figure 4). Even though we do not see a significant difference between the overexpression of C65-Ape1 compared to the vector control, there does appear to be a trend in the direction of the C65-Ape1 providing protection.
Figure 4. Effect of Ape1 levels on the apoptosis levels of sensory neuronal cells following cisplatin treatment.
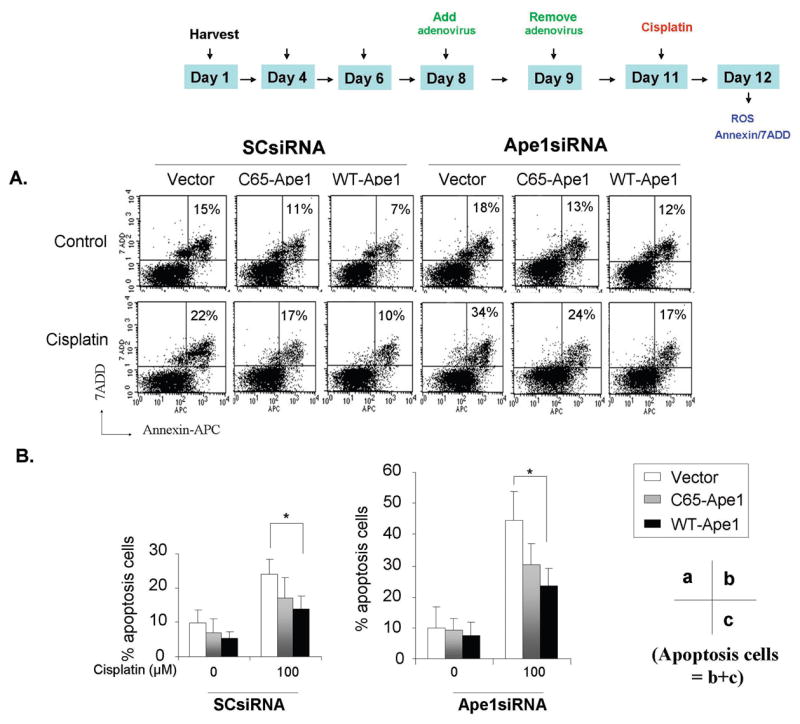
A. Sensory neuronal cell cultures were incubated with Ape1siRNA or SCsiRNA control oligos and were then infected with either WT-Ape1 or C65-Ape1. The cells were then treated with cisplatin (100uM) for 24hrs. The level of cells undergoing apoptosis were detected by Annexin-V and 7ADD staining and FACS analysis as discussed in materials and methods.
B. Quantification of the data presented in A. The percentage of cells undergoing apoptosis (c, lower right quadrant) plus late apoptosis (b, upper right quadrant) are shown. Statistically different points from controls are indicated with an asterisk (*p<0.05).
Effect of Ape1 on cisplatin- altered evoked release of iCGRP from sensory neurons
We examined the effects of cisplatin in the absence or presence of Ape1 manipulations on the release of iCGRP from sensory neurons in culture as an integrated functional output from these cells. For these studies, sensory neuronal cultures in the absence or presence of Ape1 manipulations were exposed to various concentrations of cisplatin for 24 hours, then release studies were performed as outlined in the methods. For internal consistency the release studies were always performed on cells grown in culture for 12 days. Exposing sensory neurons in culture to various concentrations of cisplatin results in a concentration dependent decrease in the capsaicin-evoked release of iCGRP (figure 5A). 3 μM cisplatin did not alter release compared to untreated cells (11.3 ± 0.3 % of total iCGRP content for controls versus 12.9 ± 0.9 % for treated cells). In contrast both 10 μM and 30 μM cisplatin reduced the capsaicin-evoked release to 6.3 ± 0.2 and 3.5 ± 0.1 % of total content, respectively. Cisplatin treatment had no significant effect on basal release of iCGRP from sensory neurons. Basal release in untreated cells was 7.7 ± 2.0 fmol/well of cells/10 min and was 7.0 ± 0.1, 7.4 ± 2.0, and 7.1 ± 0.2 fmol/well/10 min after 3, 10, or 30 μM cisplatin, respectively (data not shown). For the remaining studies we used a concentration of 10 μM cisplatin since it produces a significant decrease in iCGRP release, but does not cause cell death in the cultures (see figure 1).
Figure 5. Effect of altered Ape1 levels on cisplatin-induced iCGRP release from sensory neuronal cells.
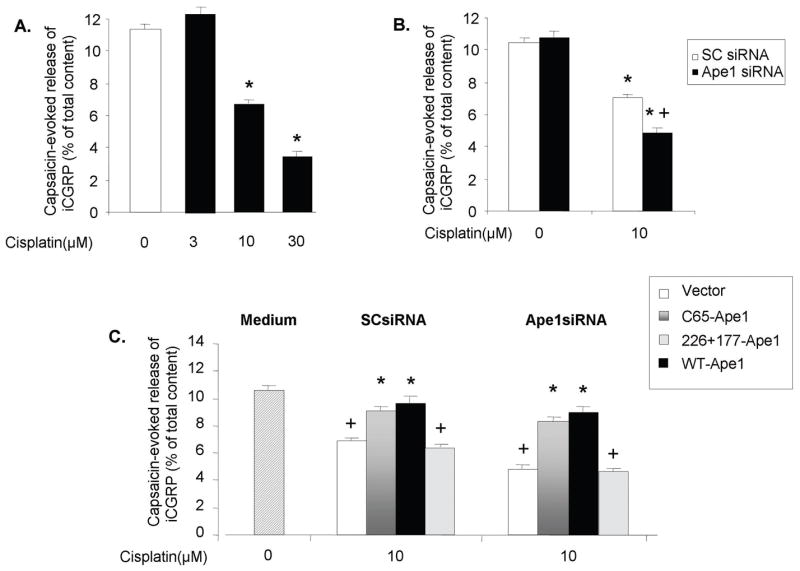
A. Sensory neuronal cells were treated for 24 hrs with increasing amounts of cisplatin. There was a decrease in the amount of iCGRP released in the capsaicin-evoked release of iCGRP. Each column represents the mean +/- SE of the percent of total content of iCGRP after exposure to 30 nM capsaicin for nine wells of neurons.
B. Sensory neuronal cells were treated with cisplatin, Ape1-siRNA or SC-siRNA and/or virus as indicated. Compared to untreated controls, SCsiRNA and Ape1siRNA treated cells had a statistically significant (*p < 0.05) decrease in release after exposed to cisplatin.
C. Sensory neuronal cells were treated with cisplatin at 10 uM and either SCsiRNA or Ape1-siRNA and then either adenoviral vector control or adenovirus with either WT-Ape1, C65-Ape1 or N226A+R177A Ape1. There was a significantly significant increase in the release in the C65A-Ape1 and the WT-Ape1 transgenes compared to the vector or the N226A+R177A-Ape1 (p < 0.05).
When neuronal cultures were exposed to SCsiRNA or Ape1siRNA in the absence of cisplatin treatment, there was no significant effect on capsaicin-evoked release compared to untreated controls (figure 5B). When cells were exposed to SCsiRNA, which did not reduce levels of Ape1, 10 μM cisplatin for 24 hrs significantly reduced cisplatin-evoked release to 7.0 ± 0.2 % of total iCGRP content, which is to the values observed in control cells treated with 10 μM cisplatin (figure 5A). Reducing Ape1 expression using siRNA, however, significantly augmented the ability of cisplatin to reduce peptide release to 4.8 ± 0.3 % of total content (Figure 5B).
As in previous studies, we next determined if overexpression of Ape1 or Ape1 mutants could reverse the effects of cisplatin on iCGRP release. As in previous experiments, cultures were first treated with siRNAs, infected with viral vectors, exposed to 10 μM cisplatin for 24 hrs, than basal and capsaicin-evoked iCGRP release measures. When cells treated with SCsiRNA or Ape1siRNA were infected with the vector control, cisplatin treatment resulted in a significant decrease in capsaicin-evoked release of iCGRP in a manner analogous to untreated cells (figure 5C). In contrast overexpression of WT-Ape1 or C65-Ape1 significantly attenuated the effects of cisplatin (figure 5C). The addition of siRNA and viral vectors in cells not exposed to cisplatin did not alter basal or capsaicin evoked release of iCGRP (data not shown). In addition no treatment regime significantly affected basal release.
These data suggest that the repair component of Ape1 is involved in reversing the effects of cisplatin on peptide release from sensory neurons. To further substantiate this notion, experiments were performed using overexpression of another mutant Ape1, N226A+R177A-Ape1 (24). This mutant has the redox component of WT-Ape1, but no repair function (24). When this mutant was overexpressed in sensory neuronal cultures it had no effect on the ability of cisplatin to reduce capsaicin-evoked release of iCGRP (figure 5C).
Effect of Ape1 on DNA damage and expression of transcription factors in sensory neuronal cultures
Although the data above indicate that Ape1 is neuroprotective against cisplatin induced neurotoxicity, the question remains what is the mechanism of this protection. Thus, in order to determine if exposing sensory neurons to cisplatin would alter the phosphorylation of histone H2A.X as an indicator of double strand breaks (26) and transcription factors that are downstream of Ape1 that are under redox control by Ape1 such as the transcription factor, p53 (33). Sensory neuronal cultures were treated with either SCsiRNA orApe1siRNA, followed by 50 uM cisplatin and the cells harvested after 8, 24 and 48 hr exposure. When cells treated with SCsiRNA were exposed to cisplatin, there was a small increase in H2AX phosphorylation and phosphorylation of p53 at the serine 15 position over time (figure 6A and 6B). Figure 6A shows a representative Western blot, whereas figure 6 B shows summary results from three experiments. These increases were not significantly different from cultures not exposed to cisplatin (time zero). There also was no significant change in Gadd45a expression as measured by western blotting. In contrast, in cells treated with Ape1siRNA phosphorylation of H2AX was significantly higher after 48 hrs of cisplatin exposure (figure 6A and 6B). Phosphorylation of p53 was also significantly higher in cultures with reduced Ape1 expression after 24 and 48 hrs of cisplatin treatment. Gadd45a protein was also greatly augmented at 24 hours in the Ape1siRNA knockdown sensory neuronal cells compared with SCsiRNA controls. This time course indicates an increase in the p53 response pathway leading to an increase in Gadd45a following p53 activation 24–48 hrs after cisplatin treatment and with the reduction of Ape1 in cells.
Figure 6. Effect of altering Ape1 levels on the p53 signaling and dsb pathway following cisplatin treatment of sensory neuronal cells.
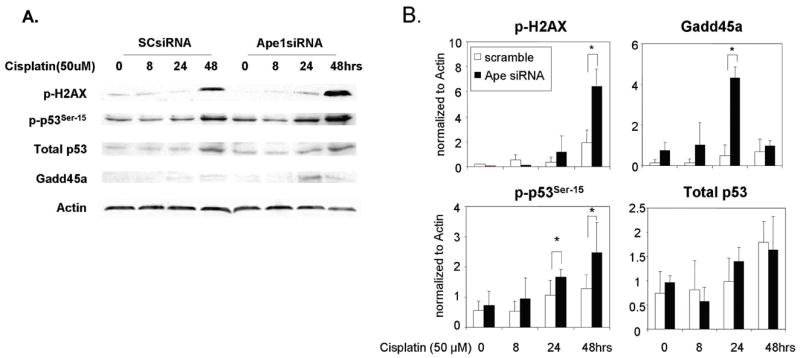
A. A representative western blot analysis of the protein levels of p53 and Gadd45a and the phosphorylation of p53ser15 and phosphorylation levels of H2AX. Sensory neuronal cell cultures were incubated with Ape1siRNA or SCsiRNA control oligos and were then treated with 50 μM cisplatin for 48 hrs.
B. The protein levels of three independent experiments following normalization to actin. Each point represents the mean ± SE for at least three independent harvests of cells. Differences were considered significant at *p<0.05.
Discussion
This report is the first data demonstrating a role of a BER protein, Ape1, in sensory neuronal survival and function following cisplatin treatment. Furthermore, we demonstrate that it is largely Ape1’s repair function that is involved in the response of sensory neuronal cells to cisplatin stress and that cisplatin causes an alteration of the p53 signaling pathway. We chose to examine the effects of cisplatin on sensory neurons since the neuropathy caused by this anticancer drug is largely sensory and often involves paresthesia and/or overt pain (34). Furthermore, platinum binding to DNA in sensory neurons is high (3, 6) and exposing isolated sensory neurons to cisplatin causes apoptosis (32). In this report, we observed a concentration-dependent increase in sensory neuronal cell killing and apoptosis after cisplatin, confirming the previous studies. Of interest, however is the fact that a 10 μM over 24 hours significantly reduced the release of iCGRP from sensory neurons. We did not observe any significant change in the content of iCGRP in the cultures after exposure to cisplatin (data not shown), thus the cisplatin-induced alterations in release were not secondary to cell death or loss of transcription. Our observation of a decrease in evoked release of iCGRP after cisplatin exposure, are similar to in situ studies demonstrating that treating rats with 1.5 mg/kg cisplatin once per day for 5 days results in a decrease in stimulated-release of the neuropeptides substance P, somatostatin and CGRP from the lower trachea and main bronchi (35). The cisplatin-induced decrease in CGRP release from sensory neurons suggests a loss of function of these neurons that could have important consequences. The release of CGRP from peripheral terminals of small diameter sensory neurons is associated with local vasodilation that contributes to neurogenic inflammation (36). Indeed, CGRP is a potent vasodilator and a decrease in its release could negatively impact local blood flow and contribute to symptoms of peripheral neuropathy. At central terminals of sensory neurons, CGRP may contribute to thermal sensation as well as nociception. Thus a decrease in its release in response to stimuli could result in reduced sensory input. The potential impact of a decrease in CGRP release on nociception still needs to be determined. Cisplatin has been shown to induce ROS generation in a selected number of cell systems (10, 37) and the increase in ROS could damage lipids, proteins, and nuclear and mitochondrial DNA, leading to cell death (38). Oxidative damage has been suggested as the main cause of cisplatin-induced renal cell death and several antioxidants and radical scavengers alleviate cisplatin-induced nephrotoxicity in vitro and in vivo (39, 40). Recently, it was demonstrated that inhibiting mitochondrial respiration results in an enhancement of cisplatin toxicity to leukemic cells (10) and an imbalancing of the BER pathway in mitochondria using the OGG1 protein leads to enhanced cisplatin toxicity (10). In line with these studies, our results demonstrate a significant increase in intracellular ROS concentration in sensory neuronal cells follow cisplatin exposure (figures 1 and 2). The basal increase in ROS production in sensory neuronal cells parallels a significant increase in Ape1 expression; the first time an induction of Ape1 has been seen following cisplatin treatment. Furthermore, reducing Ape1 expression augments cisplatin-induced ROS production. This correlates with previous studies in tumor cell lines that demonstrated cisplatin apoptosis is mediated by ROS production and Ape1 overexpression can suppress this ROS production (18, 41, 42).
The question remains as to the mechanisms by which cisplatin induced toxicity in sensory neurons. Previous work shows a correlation between the platinum binding to DNA and the toxicity in sensory neurons (3, 6). This would imply that the nucleotide excision repair pathway is important in regulating cisplatin-induced toxicity and in mice deficient in NER function; there is an increase in accumulation of unrepaired cross-links and in neurotoxicity (7). Our current findings, however, demonstrate that Ape1 plays a role in reducing cisplatin-induced neurotoxicity. We demonstrate that reducing Ape1 by siRNA increased cisplatin-induced ROS production (figure 2) and also increased cell killing and apoptosis (figures 3 and 4) and deceased iCGRP release (figure 5). Moreover, over expression of Ape1 in DRG cells or siRNA knockdown DRG cells resulted in increased protection from cisplatin. Our current results further suggest that the repair component of Ape1 plays a major role in its ability to protect against cisplatin-induced toxicity. In most studies both wild-type Ape1 and the C65-Ape1 repair competent/redox deficient protein protected the cells, although overexpression of the C-65 mutant was effective at an intermediate level. In contrast, the N226A+R177A-Ape1 which has the redox component of WT-Ape1, but no repair function (24), was not effective in blocking the ability of cisplatin to decrease iCGRP release.
In order to begin to determine the mechanistic and signaling pathways that may be involved in the response of DRG cells to cisplatin induced stress and the role of Ape1 in this process we studied the role of the p53 stress response pathway. We chose this pathway to begin our analyses given the previously demonstrated interactions between Ape1 and p53 in tumor and normal, dividing cells (33, 43) and since the p53 tumor suppressor protein plays a major role in cellular response to DNA damage and other genomic aberrations, particularly in mitotically growing cells. p53 is a nuclear phosphoprotein and phosphorylation of Ser15, a key phosphorylation target during the p53 activation process, has been shown as being critical for p53-dependent transactivation(44). DNA damage induces phosphorylation of p53 at Ser15 and leads to reduced interaction between p53 and its negative regulator, oncoprotein MDM2 (45, 46). To begin to examine this pathway, we performed studies using western blot analysis and found that altering Ape1 levels leads to alterations in the amount phosphorylated-p53 and an increase in Gadd45a protein levels (figure 6). Our results indicate an induction of p53 phosphorylation at Ser15 following the alteration of Ape1 levels and cisplatin treatment which is the first time a relationship between Ape1 protein levels and p53 phosphorylation has been observed. This change in p53 phosphorylation was correlated with an induction of Gadd45a and implicates this pathway as the primary signaling pathway involving Ape1, cisplatin and ROS in DRG cells. Gadd45a which is involved in the DNA repair, maintenance of genomic stability, cell cycle control and apoptosis and its induction is primarily p53-dependent (47). The p53-GADD45a pathway is also intimately linked to the repair of DNA damage related to cross-linking agents or those that are normally repaired by the NER pathway (33, 48). Therefore, our findings reported here are consistent with alterations in Ape1 levels and functions impinging on DRG survival and function by altering both BER and NER pathways.
While we present a number of novel findings in this report; induction of Ape1 by cisplatin, knocking down Ape1 levels leading to increased ROS in post-mitotic cells, role of Ape1 redox and repair functions in cisplatin survival and function of sensory neurons and the perturbation of the p53-Gadd45a signaling pathway by Ape1, we have not completely delineated all the mechanisms that may be acting with Ape1 perturbation in the DRG neuron. One of the areas that will require additional study involves the role of the mitochondria in these cells and in the DRG response to chemotherapeutic agents. For example, in a recent report, apoptosis induced by cisplatin was shown to require the production of ROS, but was independent of damage to nuclear DNA (49). Additionally, mtDNA has been shown to be naturally more sensitive to oxidative damage than nuclear DNA (50). Therefore, studying the effects of cisplatin on the mitochondrial genome and Ape1’s role in the repair of mtDNA damage following cisplatin treatment warrants further investigation. It may turn out that the main response of DRG neurons to cisplatin treatment is mediated through mitochondria ROS production such that altering Ape1 leads to a suboptimal response of the mitochondria and a chain of events resulting in DRG dysfunction or death.
References
- 1.Manju K, Muralikrishna B, Parnaik VK. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J Cell Sci. 2006;119:2704–14. doi: 10.1242/jcs.03009. [DOI] [PubMed] [Google Scholar]
- 2.Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249:9–17. doi: 10.1007/pl00007853. [DOI] [PubMed] [Google Scholar]
- 3.McDonald ES, Randon KR, Knight A, Windebank AJ. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: a potential mechanism for neurotoxicity. Neurobiol Dis. 2005;18:305–13. doi: 10.1016/j.nbd.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Holmes J, Stanko J, Varchenko M, et al. Comparative neurotoxicity of oxaliplatin, cisplatin, and ormaplatin in a Wistar rat model. Toxicol Sci. 1998;46:342–51. doi: 10.1006/toxs.1998.2558. [DOI] [PubMed] [Google Scholar]
- 5.Wu F, Lin X, Okuda T, Howell SB. DNA polymerase zeta regulates cisplatin cytotoxicity, mutagenicity, and the rate of development of cisplatin resistance. Cancer Res. 2004;64:8029–35. doi: 10.1158/0008-5472.CAN-03-3942. [DOI] [PubMed] [Google Scholar]
- 6.Ta LE, Espeset L, Podratz J, Windebank AJ. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. Neurotoxicology. 2006;27:992–1002. doi: 10.1016/j.neuro.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Dzagnidze A, Katsarava Z, Makhalova J, et al. Repair capacity for platinum-DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J Neurosci. 2007;27:9451–7. doi: 10.1523/JNEUROSCI.0523-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feghali JG, Liu W, Van De Water TR. L-n-acetyl-cysteine protection against cisplatin-induced auditory neuronal and hair cell toxicity. Laryngoscope. 2001;111:1147–55. doi: 10.1097/00005537-200107000-00005. [DOI] [PubMed] [Google Scholar]
- 9.van den Berg JH, Beijnen JH, Balm AJ, Schellens JH. Future opportunities in preventing cisplatin induced ototoxicity. Cancer Treat Rev. 2006;32:390–7. doi: 10.1016/j.ctrv.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Haihong Z, Takatsugu M, Jaime C-T, et al. Targeting human 8-oxoguanine DNA glycosylase (hOGG1) to mitochondria enhances cisplatin cytotoxicity in hepatoma cells. Carcinogenesis. 2007;28:1629. doi: 10.1093/carcin/bgm072. [DOI] [PubMed] [Google Scholar]
- 11.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 12.Duguid JR, Eble JN, Wilson TM, Kelley MR. Differential cellular and subcellular expression of the human multifunctional apurinic/apyrimidinic endonuclease (APE/ref-1) DNA repair enzyme. Cancer Res. 1995;55:6097–102. [PubMed] [Google Scholar]
- 13.Fritz G. Human APE/Ref-1 protein. Int J Biochem Cell Biol. 2000;32:925–9. doi: 10.1016/s1357-2725(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 14.Di Giovanni S, Knights CD, Rao M, et al. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. Embo J. 2006;25:4084–96. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–60. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 16.Reme CE, Grimm C, Hafezi F, Iseli HP, Wenzel A. Why study rod cell death in retinal degenerations and how? Doc Ophthalmol. 2003;106:25–9. doi: 10.1023/a:1022423724376. [DOI] [PubMed] [Google Scholar]
- 17.Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair (Amst) 2005;4:367–79. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Yang S, Irani K, Heffron SE, Jurnak F, Meyskens FL., Jr Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in human melanoma and identification of the therapeutic potential of resveratrol as an APE/Ref-1 inhibitor. Mol Cancer Ther. 2005;4:1923–35. doi: 10.1158/1535-7163.MCT-05-0229. [DOI] [PubMed] [Google Scholar]
- 19.Robertson KA, Bullock HA, Xu Y, et al. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001;61:2220–5. [PubMed] [Google Scholar]
- 20.Southall MD, Vasko MR. Prostaglandin E(2)-mediated sensitization of rat sensory neurons is not altered by nerve growth factor. Neurosci Lett. 2000;287:33–6. doi: 10.1016/s0304-3940(00)01158-7. [DOI] [PubMed] [Google Scholar]
- 21.Lindsay RM. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci. 1988;8:2394–405. doi: 10.1523/JNEUROSCI.08-07-02394.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pines A, Bivi N, Romanello M, et al. Cross-regulation between Egr-1 and APE/Ref-1 during early response to oxidative stress in the human osteoblastic HOBIT cell line: evidence for an autoregulatory loop. Free Radic Res. 2005;39:269–81. doi: 10.1080/10715760400028423. [DOI] [PubMed] [Google Scholar]
- 23.Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–86. [PubMed] [Google Scholar]
- 24.McNeill DR, Wilson DM., 3rd A Dominant-Negative Form of the Major Human Abasic Endonuclease Enhances Cellular Sensitivity to Laboratory and Clinical DNA-Damaging Agents. Mol Cancer Res. 2007;5:61–70. doi: 10.1158/1541-7786.MCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 25.Rinne M, Caldwell D, Kelley MR. Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol Cancer Ther. 2004;3:955–67. [PubMed] [Google Scholar]
- 26.Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin Cancer Res. 2007;13:260–7. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 27.Song JD, Lee SK, Kim KM, et al. Redox factor-1 mediates NF-κB nuclear translocation for LPS-induced iNOS expression in murine macrophage cell line RAW 264.7. Immunology. 124:58–67. doi: 10.1111/j.1365-2567.2007.02736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao K, Zhao GM, Wu D, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–90. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 29.Ding SZ, O’Hara AM, Denning TL, et al. Helicobacter pylori and H2O2 increase AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology. 2004;127:845–58. doi: 10.1053/j.gastro.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 30.Flaherty DM, Monick MM, Carter AB, Peterson MW, Hunninghake GW. Oxidant-mediated increases in redox factor-1 nuclear protein and activator protein-1 DNA binding in asbestos-treated macrophages. J Immunol. 2002;168:5675–81. doi: 10.4049/jimmunol.168.11.5675. [DOI] [PubMed] [Google Scholar]
- 31.Fischer SJ, McDonald ES, Gross L, Windebank AJ. Alterations in cell cycle regulation underlie cisplatin induced apoptosis of dorsal root ganglion neurons in vivo. Neurobiol Dis. 2001;8:1027–35. doi: 10.1006/nbdi.2001.0426. [DOI] [PubMed] [Google Scholar]
- 32.Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101:2842–50. doi: 10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seo YR, Kelley MR, Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci U S A. 2002;99:14548–53. doi: 10.1073/pnas.212319799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson SW, Davis LE, Kornfeld M, Hilgers RD, Standefer JC. Cisplatin neuropathy. Clinical, electrophysiologic, morphologic, and toxicologic studies. Cancer. 1984;54:1269–75. doi: 10.1002/1097-0142(19841001)54:7<1269::aid-cncr2820540707>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 35.Horvath P, Szilvassy J, Nemeth J, Peitl B, Szilasi M, Szilvassy Z. Decreased sensory neuropeptide release in isolated bronchi of rats with cisplatin-induced neuropathy. Eur J Pharmacol. 2005;507:247–52. doi: 10.1016/j.ejphar.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 36.Maggi CA. Tachykinins and calcitonin gene-related peptide (CGRP) as co-transmitters released from peripheral endings of sensory nerves. Prog Neurobiol. 1995;45:1–98. doi: 10.1016/0301-0082(94)e0017-b. [DOI] [PubMed] [Google Scholar]
- 37.Kruidering M, Van de Water B, de Heer E, Mulder GJ, Nagelkerke JF. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J Pharmacol Exp Ther. 1997;280:638–49. [PubMed] [Google Scholar]
- 38.Zuliani T, Denis V, Noblesse E, et al. Hydrogen peroxide-induced cell death in normal human keratinocytes is differentiation dependent. Free Radic Biol Med. 2005;38:307–16. doi: 10.1016/j.freeradbiomed.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 39.Zhang JG, Zhong LF, Zhang M, Xia YX. Protection effects of procaine on oxidative stress and toxicities of renal cortical slices from rats caused by cisplatin in vitro. Arch Toxicol. 1992;66:354–8. doi: 10.1007/BF01973631. [DOI] [PubMed] [Google Scholar]
- 40.Sadzuka Y, Shoji T, Takino Y. Mechanism of the increase in lipid peroxide induced by cisplatin in the kidneys of rats. Toxicol Lett. 1992;62:293–300. doi: 10.1016/0378-4274(92)90033-g. [DOI] [PubMed] [Google Scholar]
- 41.Huang HL, Fang LW, Lu SP, Chou CK, Luh TY, Lai MZ. DNA-damaging reagents induce apoptosis through reactive oxygen species-dependent Fas aggregation. Oncogene. 2003;22:8168–77. doi: 10.1038/sj.onc.1206979. [DOI] [PubMed] [Google Scholar]
- 42.Chiarini LB, Freitas FG, Petrs-Silva H, Linden R. Evidence that the bifunctional redox factor/AP endonuclease Ref-1 is an anti-apoptotic protein associated with differentiation in the developing retina. Cell Death Differ. 2000;7:272–81. doi: 10.1038/sj.cdd.4400639. [DOI] [PubMed] [Google Scholar]
- 43.Hanson S, Kim E, Deppert W. Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene. 2005;24:1641–7. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- 44.Bulavin DV, Saito S, Hollander MC, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. Embo J. 1999;18:6845–54. doi: 10.1093/emboj/18.23.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ko LJ, Shieh SY, Chen X, et al. p53 is phosphorylated by CDK7-cyclin H in a p36MAT1-dependent manner. Mol Cell Biol. 1997;17:7220–9. doi: 10.1128/mcb.17.12.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 47.Daino K, Ichimura S, Nenoi M. Both the basal transcriptional activity of the GADD45A gene and its enhancement after ionizing irradiation are mediated by AP-1 element. Biochim Biophys Acta. 2006;1759:458–69. doi: 10.1016/j.bbaexp.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Seo YR, Fishel ML, Amundson S, Kelley MR, Smith ML. Implication of p53 in base excision DNA repair: in vivo evidence. Oncogene. 2002;21:731–7. doi: 10.1038/sj.onc.1205129. [DOI] [PubMed] [Google Scholar]
- 49.Berndtsson M, Hagg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer. 2007;120:175–80. doi: 10.1002/ijc.22132. [DOI] [PubMed] [Google Scholar]
- 50.Sawyer DE, Roman SD, Aitken RJ. Relative susceptibilities of mitochondrial and nuclear DNA to damage induced by hydrogen peroxide in two mouse germ cell lines. Redox Rep. 2001;6:182–4. doi: 10.1179/135100001101536157. [DOI] [PubMed] [Google Scholar]