

Abstract
Leptin is a hormone secreted by adipocytes that is implicated in the regulation of bone density. Serum leptin levels are decreased in rodent models of type 1 (T1-) diabetes and in diabetic patients. Whether leptin mediates diabetic bone changes is unclear. Therefore, we treated control and T1-diabetic mice with chronic (28 day) subcutaneous infusion of leptin or saline to elucidate the therapeutic potential of leptin for diabetic osteoporosis. Leptin prevented the increase of marrow adipocytes and the increased aP2 expression that we observed in vehicle-treated diabetic mice. However, leptin did not prevent T1-diabetic decreases in trabecular bone volume fraction or bone mineral density in tibia or vertebrae. Consistent with this finding, markers of bone formation (osteocalcin RNA and serum levels) in diabetic mice were not restored to normal levels with leptin treatment. Interestingly, markers of bone resorption (TRAP5 RNA and serum levels) were decreased in diabetic mice by leptin treatment. In summary, we have demonstrated a link between low leptin levels in T1-diabetes and marrow adiposity. However, leptin treatment alone was not successful in preventing bone loss.
Keywords: Leptin, Diabetes, Bone, Adipocyte, Osteoblast
INTRODUCTION
Leptin is a small protein (16 kDa) secreted by adipocytes and involved in bone mass regulation. The effects of leptin on bone are complex; it can stimulate or inhibit bone formation depending upon bone location and whether leptin is functioning directly on osteoblasts (through receptors (Cornish et al., 2002; Reseland et al., 2001)) or indirectly through the hypothalamus (Cornish et al., 2002; Ducy et al., 2000; Hamrick, 2004; Hamrick et al., 2007; Karsenty, 2001). In children and during adolescence, decreases in serum leptin levels, associated with reduced food intake and some disease conditions, are thought to contribute to reduced bone formation and growth (Chan and Mantzoros, 2005; Hamrick, 2004). Absence of leptin in mice also results in bone loss as well as increased bone marrow adiposity (Hamrick et al., 2004; Steppan et al., 2000). Similarly, increased leptin levels, as observed in obesity (Considine et al., 1996), are correlated with increased bone mass (Goulding and Taylor, 1998). Several studies have tested the potential therapeutic benefits of leptin treatment on bone loss and marrow adipocyte accumulation. In vitro studies demonstrate that leptin promotes bone marrow stromal cells to exhibit an osteoblast rather than adipocyte phenotype (Cornish et al., 2002; Reseland et al., 2001; Thomas et al., 1999). Consistent with this finding, subcutaneous infusion of leptin with osmotic mini-pumps reduces marrow adiposity and increases bone mass in ob/ob mice (Hamrick et al., 2005). Similarly, leptin treatment reduces bone loss from ovariectomy (Martin et al., 2005) and tail suspension (Burguera et al., 2001). In sum, these studies suggest the efficacy of leptin treatment to restore bone density under conditions of bone loss.
Diabetes mellitus (types 1, 2 and gestational forms) is a metabolic disease marked by hyperglycemia due to defective insulin action (lack of insulin or tissue insulin resistance), which results in impaired glucose uptake by insulin responsive cells. Type 1 (T1) diabetes is generally first diagnosed in adolescents or young adults and affects nearly one million children and adults in the United States. In T1-diabetes, insulin-secreting pancreatic β-cells are destroyed as a result of autoimmunity. This causes severe hyperglycemia that must be controlled by insulin delivery in humans. In addition to hyperglycemia, weight loss can occur in patients that do not have an adequate insulin therapy. T2-diabetes, generally diagnosed in adults, also results in hyperglycemia but insulin levels, body weight, and serum lipids are increased. Whereas effects of T2-diabetes on bone remain controversial due to increased fracture risk and increased bone mineral density in these patients (Vestergaard, 2007), it is recognized that osteoporosis is a serious complication of T1-diabetes, leaving child and adult patients at risk for bone loss, fracture and impaired fracture healing (Auwerx et al., 1988; Janghorbani et al., 2007; Kemink et al., 2000; Levin et al., 1976).
Previously we demonstrated that streptozotocin-induced T1-diabetic mice exhibit weight loss, decreased body fat mass and bone loss (Botolin et al., 2005). Bone formation by osteoblasts is decreased while resorption is unchanged or decreased in diabetic humans and rodents (Botolin et al., 2005; Bouillon et al., 1995; Goodman and Hori, 1984). Interestingly, both STZ-induced and spontaneous mouse models of T1-diabetes exhibit an increase in bone marrow adiposity despite the loss of peripheral fat depots (Botolin et al., 2005; Botolin and McCabe, 2007; Fowlkes et al., 2008). A reciprocal relationship between bone volume/density and marrow adipocyte number (consistent with altered mesenchymal stem cell lineage selection) has been implicated as a mechanism for diabetic (Botolin et al., 2005; Botolin and McCabe, 2007; Martin and McCabe, 2007; McCabe, 2007), age-related (Nuttall et al., 1998; Verma et al., 2002), and unloading-induced (Ahdjoudj et al., 2002) bone loss. However, while an increase in adiposity was observed in tibia, femur and calvaria in T1-diabetic mice, vertebrae did not exhibit an increase in adiposity (Martin and McCabe, 2007). In this respect, the T1-diabetic bone phenotype resembles that of leptin-deficient mice, which have increased marrow adiposity and bone loss in the femur but no change in marrow adiposity in the vertebrae (Hamrick et al., 2004). Because of this finding, we measured fed serum leptin and found that it was decreased in both male and female T1-diabetic mice (Martin and McCabe, 2007), consistent with reports in STZ-diabetic rats (Gulen and Dincer, 2007).
These findings led us to hypothesize that decreased serum leptin levels contribute to T1-diabetic bone loss and altered mesenchymal lineage selection. Therefore we treated control and diabetic mice with leptin and examined bone parameters. Consistent with our hypothesis, we observed reduced bone marrow adiposity in leptin-treated diabetic mice; however, bone loss was not prevented.
MATERIALS AND METHODS
Animals
Forty 9-week old BALB/c mice were obtained from Harlan Sprague Dawley (Indianapolis, IN). Mice were maintained on standard lab chow and had food and water ad libitum. At 14 weeks, mice were divided into four treatment groups: control + vehicle, control + leptin, diabetic + vehicle and diabetic + leptin. Mice were anesthetized with isofluorane and implanted subcutaneously with Alzet mini-osmotic pumps (model 2004, Durect Corporation, Cupertino, CA) that contained either 0.9% sterile saline vehicle (n=20) or 1.3 mg/mL leptin (Amylin, San Diego, CA) (n=20). Osmotic pumps had a mean pumping rate of 0.21 ± 0.01 μL/hour and therefore delivered 6.6 μg leptin per day. Wounds were closed with staples and mice were given a one-time injection of 0.15 mg carprofen (Pfizer, New York, NY). Starting on the day of implantation (day 0), mice were given five consecutive daily intraperitoneal injections of 50 mg/kg streptozotocin (n=24) or 0.1 M citrate buffer pH 4.5 vehicle (n=16). Body mass and food intake were monitored throughout the experiment. Staples were removed on day 9.
On day 28, mice were fasted 2-3 hours prior to being euthanized. Blood glucose was measured at the time of harvest with an AccuChek Compact glucometer (Roche, Nutley, NJ). Blood was collected at the time of harvest, allowed to rest at room temperature for five minutes, then centrifuged at 4000 rpm for ten minutes. Serum was removed and stored at −80°C and pellet discarded. Tibias were removed and either fixed in 10% formalin or frozen in liquid nitrogen and stored at −80°C. Femoral fat pads and tibialis anterior muscles were removed and weighed, fixed and frozen. All animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee.
Serum measurements
Serum was stored at −80°C and put through no more than one freeze/thaw cycle. Leptin was measured using the Assay Designs Mouse Leptin Titer Zyme kit (Ann Arbor, MI) according to the manufacturer protocol. Insulin was measured using a Crystal Chem Inc. Ultra Rat Insulin ELISA kit (Downers Grove, IL) according to the manufacturer protocol.
Bone Histology and Histomorphometry
Bones were fixed in 10% formalin and transferred to 70% EtOH after 24 hours. Fixed samples were processed on an automated Thermo Electron Excelsior tissue processor for dehydration, clearing and infiltration using a routine overnight processing schedule. Samples were then embedded in Surgipath embedding paraffin on a Sakura Tissue Tek II embedding center. Paraffin blocks were sectioned at 5 μm on a Reichert Jung 2030 rotary microtome. Slides were stained with hematoxylin and eosin. Visible adipocytes, greater than 30 μm, were counted in the tibia trabecular region ranging from the proximal growth plate to 2 mm away distally.
Micro Computed Tomography (μCT) Analyses
Fixed tibias were scanned using a GE Explore Locus μCT system at a voxel resolution of 20 μm obtained from 720 views. Beam angle of increment was 0.5 and beam strength was set at 80 peak kV and 450 μA. Each run included control and diabetic, saline and leptin-treated bones and a calibration phantom to standardize grayscale values and maintain consistency. Based on autothreshold and isosurface analyses of multiple bone samples, a fixed threshold (1400) was used to separate bone from bone marrow. Cortical bone analyses were made in a defined 2 × 2 × 2 mm cube in the mid-diaphysis immediately proximal to the distal tibial-fibular junction, with the exception of cortical bone mineral content (BMC) and bone mineral density (BMD), which were made in a 0.1 × 0.1 × 0.1 mm cube. Trabecular bone analyses were done in a region of trabecular bone defined at 0.17 mm (∼1% of the total length) distal to the growth plate of the proximal tibia extending 2 mm toward the diaphysis, and excluding the outer cortical shell. Cortical BMC, BMD, moment of inertia (MOI), thickness, perimeter and area, and trabecular BMC, BMD, volume fraction (BVF), and thickness (TbTh) values were computed by a GE Healthcare MicroView software application for visualization and analysis of volumetric image data. Cortical isosurface images were taken from a section immediately proximal to the tibial-fibular junction measuring 0.3 mm thick. Trabecular isosurface images were taken from a cylindrical region in the tibia immediately distal to the proximal growth plate measuring 0.8 mm in length and 0.8 mm in diameter.
RNA Analyses
Immediately after euthanasia, tibias were cleaned of muscle and connective tissue, snap frozen in liquid nitrogen and stored at −80 °C. Frozen tibias were crushed under liquid nitrogen conditions with a Bessman Tissue Pulverizer (Spectrum Laboratories, Inc., Rancho Dominguez, CA). RNA was isolated with Tri Reagent (Molecular Research Center, Inc., Cincinnati, OH) and integrity was assessed by formaldehyde-agarose gel electrophoresis. cDNA was synthesized by reverse transcription with Superscript II Reverse Transcriptase Kit and oligo dT(12-18) primers (Invitrogen, Carlsbad, CA) and amplified by real time PCR with iQ SYBR Green Supermix (Biorad, Hercules, CA) and gene-specific primers synthesized by Integrated DNA Technologies (Coralville, IA). HPRT mRNA levels do not fluctuate in diabetes or with increased serum leptin and were used as an internal control. HPRT was amplified using 5′-AAG CCT AAG ATG AGC GCA AG-3′ and 5′-TTA CTA GGC AGA TGG CCA CA-3′ (Vengellur and LaPres, 2004). aP2 was amplified using 5′-GCG TGG AAT TCG ATG AAA TCA- 3′ and 5′-CCC GCC ATC TAG GGT TAT GA-3′ (Li et al., 2003). Osteocalcin was amplified using 5′-ACG GTA TCA CTA TTT AGG ACC TGT G-3′ and 5′-ACT TTA TTT TGG AGC TGC TGT GAC-3′ (Ontiveros and McCabe, 2003). TRAP5 was amplified using 5′-AAT GCC TCG ACC TGG GA-3′ and 5′-CGT AGT CCT CCT TGG CTG CT-3′ (Wiren et al., 2004).
Statistical Analyses
All measurements are presented as the mean ± standard error of the mean (SEM). Statistical significance was determined with a student's t-test (assuming equal variance) using Microsoft Excel (Microsoft Corporation).
RESULTS
Serum leptin, glucose and insulin
To determine if correction of T1-diabetic serum leptin levels could prevent diabetic bone loss and marrow adiposity, osmotic pumps containing either leptin (delivering 6.6 μg leptin per day) or saline (vehicle) were implanted subcutaneously in BALB/c mice and T1-diabetes was induced by 5 daily injections of streptozotocin. Leptin treatment was successful in raising fasting serum leptin levels in both control and diabetic mice (Table I). Similar to our past results, serum leptin levels in vehicle-treated diabetic mice trended to be lower than vehicle-treated controls, however in this case the difference did not reach statistical significance. This result is likely due to the variability in control mouse leptin levels observed in this study and the difference in fasted (this study) versus fed (past study) serum leptin measurements (Akirav et al., 2004). Analysis of blood glucose levels demonstrated that leptin treatment of control mice causes a decrease in blood glucose levels compared to untreated controls, as previously shown by others in rats (Hidaka et al., 2002; Sindelar et al., 1999). As expected, fasting blood glucose levels were increased in diabetic compared to control mice in both normal (501 vs 161 mg/dl, respectively) and high leptin conditions (250 vs 94 mg/dl, respectively) (Table I). Although the blood glucose level in leptin-treated diabetic mice was significantly lower (by 50%) than vehicle-treated diabetic mice, the fold increase in blood glucose levels compared to corresponding treatment controls was similar (untreated diabetic/untreated control = 3 fold; leptin-treated diabetic/leptin-treated control = 2.7 fold). Differences in blood glucose levels between leptin-treated and untreated mice may result from the reduced food intake seen in both control leptin-treated and diabetic leptin-treated mice. Examination of fasting serum insulin levels demonstrated that diabetic vehicle-treated mice have lower fasting insulin levels compared to control vehicle-treated mice. Leptin-treated diabetic mice exhibited lower serum insulin levels compared to untreated diabetic mice; however, insulin levels were not lower than leptin-treated controls. This is likely due to leptin treatment effectively reducing insulin levels in non-diabetic mice (Table I), and enhancing insulin sensitivity, thereby reducing glucose-stimulated insulin release from the pancreas, as previously shown (Barzilai et al., 1997; Sivitz et al., 1997; Wang et al., 1999).
Table I.
Serum and tissue mass measurements.
| CONTROL |
DIABETIC |
|||
|---|---|---|---|---|
| Vehicle (n=6) |
Leptin (n=7) |
Vechicle (n=9) |
Leptin (n=9) |
|
| Leptin (pg/mL) | 65 ± 18 | 317 ± 105a | 45 ± 7b | 344 ± 75a,c |
| Glucose (mg/dL) | 161 ± 10 | 94 ± 10a | 501 ± 22a,b | 250 ± 29a,b,c |
| Insulin (ng/mL) | 487 ± 46 | 68 ± 14a | 186 ± 32a,b | 129 ± 21a,b |
| Fat Pads (mg) | 218 ± 15 | 79 ± 7a | 114 ± 13a | 92 ± 8a |
| Tibialis Anterior (mg) | 44 ± 3 | 39 ± 3 | 36 ± 3a | 43 ± 3 |
p < 0.05 compared to C + Vehicle
p < 0.05 compared to C + Leptin
p < 0.05 compared to D + Vehicle.
Food consumption and body mass
Food intake and body mass were monitored throughout the experiment. As expected, leptin treatment decreased food intake in control mice and suppressed T1-diabetic hyperphagia compared to untreated diabetic mice (Figure 1). The decrease in food intake corresponded to a decrease in body mass (Figure 1) in control but not diabetic mice. Differences were apparent throughout the time course beginning at diabetes confirmation (not shown).
Figure 1. Leptin treatment prevented diabetic hyperphagia compared to vehicle-treated mice, but did not prevent weight loss.
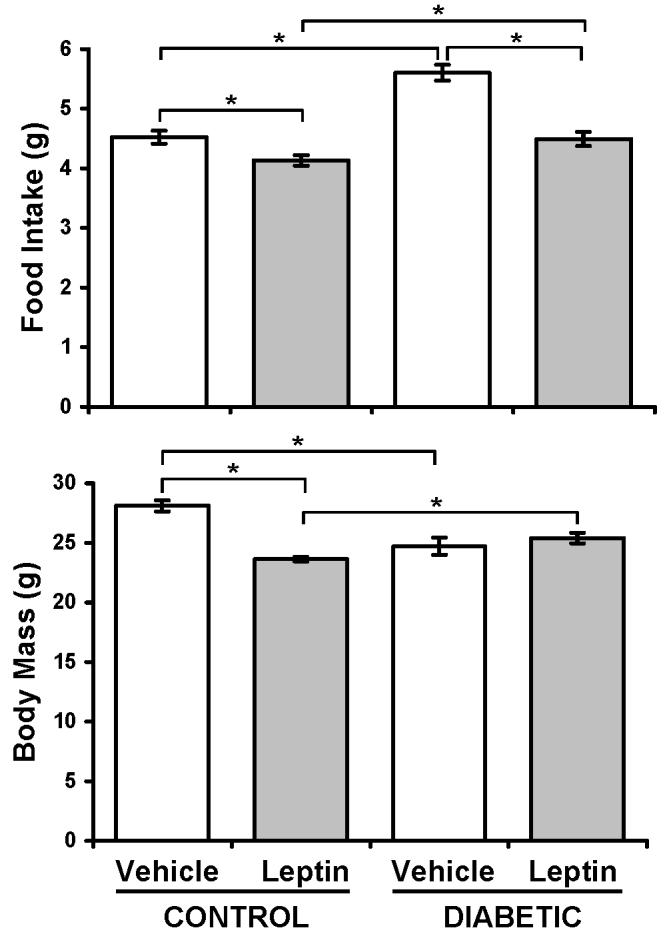
Food intake and body mass from vehicle-treated (white bars) and leptin-treated (gray bars), control and diabetic mice were monitored throughout the experiment. Shown are average values obtained 28 days after diabetes induction. Values are representative of all time points after diabetes was induced. Bars represent mean ± standard error. n≥6 per condition. *p<0.05 by Student's t-test.
To determine if the weight loss was the result of fat and/or muscle mass loss, these parameters were also examined. As expected and consistent with our past studies, diabetic vehicle-treated mice lost 50% of femoral fat pad mass compared to vehicle-treated controls (Table I). Even more fat pad mass was lost in control leptin versus vehicle-treated mice (64%), consistent with previous reports (Hamrick et al., 2005; Martin et al., 2007). No differences were seen between leptin-treated diabetic, vehicle-treated diabetic or leptin-treated control mouse fat pad mass. However, after normalizing for body mass changes, diabetic leptin-treated mice had significantly lower fat pad mass than diabetic vehicle-treated mice (3.6 ± 0.3 mg/g versus 4.5 ± 0.4 mg/g, respectively). Normalizing fat pad mass for body mass did not alter the statistical comparisons in any other case. Similar to our previous findings, diabetic mice had decreased tibialis anterior muscle mass (Table I). Leptin treatment did not affect muscle weight in control mice, but did prevent diabetic muscle mass loss compared to leptin-treated controls. However, when normalized for body weight, no statistical differences were apparent between groups, indicating that all muscle mass changes were directly proportional to body weight changes.
Diabetic marrow adiposity was prevented by leptin treatment
T1-diabetic bone loss is associated with increased marrow adiposity, implicating altered lineage selection of bone marrow stromal cells as a potential mechanism. Because leptin-treatment has been shown to prevent marrow adipocyte accumulation in ob/ob mice (Hamrick et al., 2005), we hypothesized that it should prevent marrow adiposity in diabetic mice. As expected based on previous reports (Botolin et al., 2005; Botolin and McCabe, 2007), untreated diabetic tibias exhibited an increase in marrow adipocyte number and aP2 mRNA levels (a marker of mature adipocytes; Figure 2). Consistent with our hypothesis, leptin treatment completely prevented adipocyte accumulation and aP2 mRNA induction in diabetic bone marrow (Figure 2).
Figure 2. Leptin treatment prevented T1-diabetic marrow adiposity.
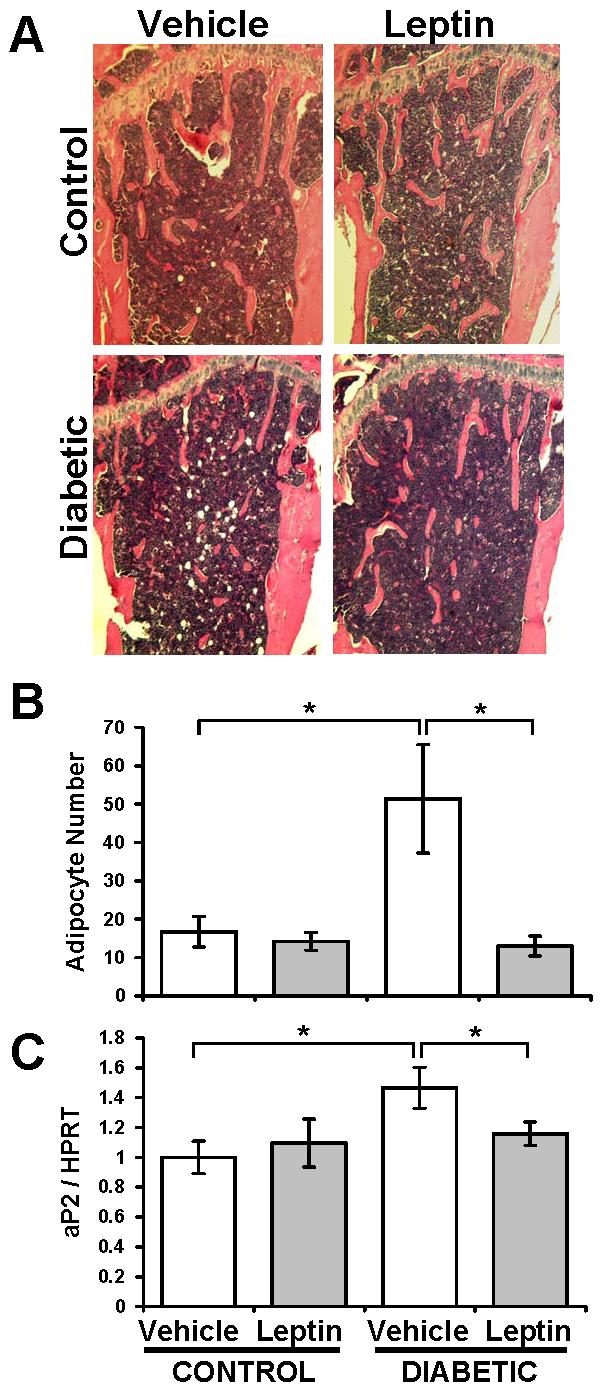
(A) Representative hematoxylin and eosin stained sections of decalcificed tibias from vehicle- (white bars) or leptin-treated (gray bars), control and diabetic mice. (B) Marrow adipocyte counts in the proximal tibia, immediately distal to the growth plate. (C) aP2 gene expression was measured by RT-PCR in whole tibia cDNA synthesized from mRNA. aP2 levels were expressed relative to HPRT, an unmodulated housekeeping gene control. Bars represent mean ± standard error. n≥6 per condition. *p<0.05 by Student's t-test.
Leptin did not prevent diabetic bone loss
Given the inverse relationship between bone density and adiposity in diabetes, we examined leptin-treated diabetic bone to see if preventing adiposity would also prevent bone loss. Because leptin can affect long bone and vertebrae differently, we examined both the proximal tibia and L2 vertebrae trabecular bone (Table II). In control mice, leptin treatment tended to reduce all bone parameters examined in both tibia and vertebrae. Significant reductions (compared to vehicle treatment) were seen in bone volume fraction (BVF) and trabecular thickness in tibia but not vertebrae, consistent with location-dependent leptin effects (Ducy et al., 2000; Hamrick et al., 2004). However, leptin-treated control vertebrae exhibited significant decreases in bone mineral content and density (BMC, BMD). As expected, untreated diabetic mice exhibited a decrease in trabecular bone mineral content and density (BMC, BMD), bone volume fraction (BVF), and trabecular thickness (TbTh) compared to untreated controls at both sites (tibia and vertebrae). These decreases were more prominent than the changes seen in untreated versus leptin-treated controls. In contrast to our original hypothesis, leptin treatment did not prevent the reduction in diabetic trabecular BMC, BMD or BVF in tibia. In vertebrae, leptin treatment did not prevent the diabetes-induced decrease in BMC or BVF. Leptin treatment did significantly increase diabetic vertebral BMD compared to vehicle-treated diabetic mice to the point of not being different from leptin-treated controls. Because diabetic- and leptin-treated mice lose body weight, we examined changes in BVF relative to total body mass. When corrected for body mass, the leptin-treated control tibia bone volume fraction (BVF/g) was similar to vehicle-treated controls (Figure 3), suggesting the reduction in bone volume is consistent with reduced body size and load. However, bone loss (BVF/g) was still evident in both untreated and leptin-treated diabetic groups (Figure 3).
Table II.
Trabecular μCT measurements.
| CONTROL |
DIABETIC |
|||
|---|---|---|---|---|
| Vehicle (n=6) | Leptin (n=7) | Vehicle (n=9) | Leptin (n=9) | |
| Tibia | ||||
| BMC (mg) | 0.82 ± 0.03 | 0.76 ± 0.02 | 0.65 ± 0.07a | 0.66 ± 0.02b |
| BMD (mg/cm2) | 271 ± 10 | 252 ± 9 | 213 ± 20a | 218 ± 5b |
| BVF (%) | 16.9 ± 1.1 | 13.8 ± 1.0a | 9.6 ± 1.9a | 10.2 ± 0.6b |
| Tb Th (mm) | 0.042 ± 0.001 | 0.038 ± 0.001a | 0.035 ± 0.002a | 0.037 ± 0.001 |
| Vertebrae | ||||
| BMC (mg) | 0.67 ± 0.03 | 0.55 ± 0.03a | 0.48 ± 0.03a | 0.51 ± 0.02 |
| BMD (mg/cm2) | 273 ± 8 | 232 ± 12a | 204 ± 9a | 220 ± 5c |
| BVF (%) | 24.1 ± 2.0 | 21.0 ± 2.8 | 11.3 ± 1.6a | 11.5 ±1.2b |
| Tb Th (mm) | 0.044 ± 0.002 | 0.040 ± 0.003 | 0.034 ± 0.002a | 0.032 ± 0.002b |
p < 0.05 compared to C + Vehicle
p < 0.05 compared to C + Leptin
p < 0.05 compared to D + Vehicle.
Abbreviations: BMC, bone mineral content. BMD, bone mineral density. BVF, bone volume fraction. Tb, trabecular. Th, thickness.
Figure 3. Leptin did not prevent T1-diabetic bone loss.
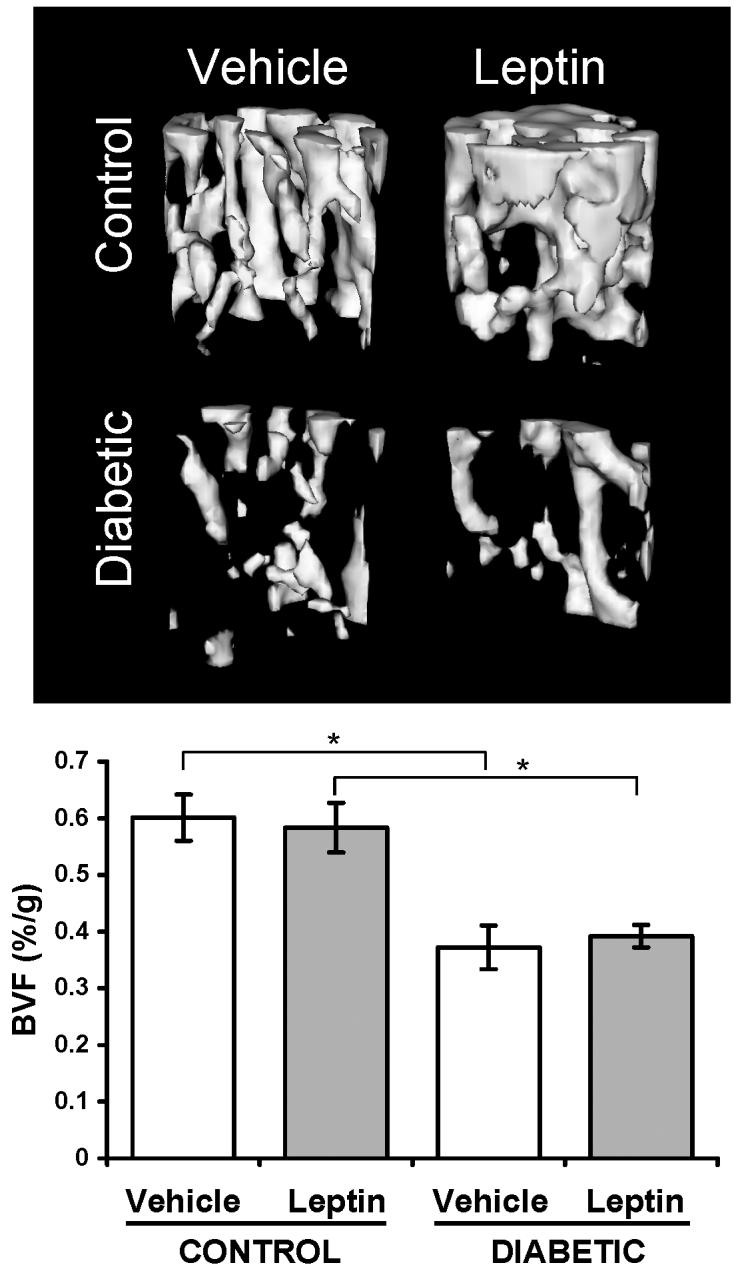
Fixed tibias were scanned by μCT and the trabecular area immediately distal to the proximal growth plate was analyzed for bone volume fraction. Top: Representative isosurface images of trabecular bone in vehicle- and leptin-treated mice, control and diabetic mice. Bottom: Bone volume fraction (BVF) was corrected for body mass. Bars represent mean ± standard error. n≥6 per condition. *p<0.05 by Student's t-test.
We further examined cortical bone parameters in the tibia metaphysis (Table III). Diabetic mice had significantly decreased cortical thickness, which can be attributed to greater inner cortical bone perimeter and increased marrow area. Leptin-treatment (in both control and diabetic mice) also reduced cortical thickness in a similar manner, suggesting the decreased cortical bone could be due to weight changes. When corrected for differences in body mass, cortical thickness was not statistically significant different between groups (Figure 4).
Table III.
Tibia cortical μCT measurements.
| CONTROL |
DIABETIC |
|||
|---|---|---|---|---|
| Vehicle (n=6) | Leptin (n=7) | Vechicle (n=9) | Leptin (n=9) | |
| Th (mm) | 0.310 ± 0.008 | 0.285 ± 0.011 | 0.285 ± 0.007a | 0.281 ± 0.005 |
| MOI (mm4) | 0.080 ± 0.006 | 0.070 ± 0.007 | 0.082 ± 0.009 | 0.084 ± 0.005 |
| Inner P (mm) | 1.48 ± 0.04 | 1.56 ± 0.05 | 1.60 ± 0.03a | 1.64 ± 0.03 |
| Outer P (mm) | 3.50 ± 0.03 | 3.46 ± 0.07 | 3.48 ± 0.09 | 3.55 ± 0.06 |
| Marrow A (mm2) | 0.145 ± 0.009 | 0.162 ± 0.012 | 0.172 ± 0.006a | 0.182 ± 0.007 |
| Cortical A (mm2) | 0.718 ± 0.019 | 0.665 ± 0.029 | 0.678 ± 0.029 | 0.681 ± 0.019 |
| BMD (mg/cm2) | 1144 ± 13 | 1097 ± 26 | 1070 ± 35 | 1073 ± 26 |
p < 0.05 compared to C + Vehicle
p < 0.05 compared to D + Vehicle
p < 0.05 compared to C + Leptin.
Abbreviations: Th, thickness. MOI, moment of inertia. P, perimeter. A, area. BMD, bone mineral density.
Figure 4. Cortical bone thickness did not differ when corrected for body mass changes.
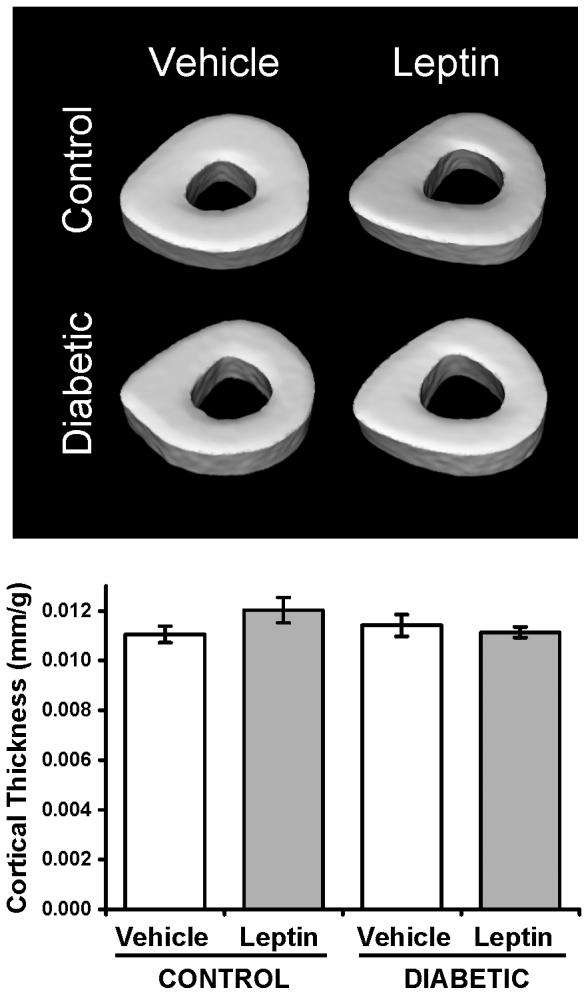
Top: representative isosurface images of cortical bone transverse sections immediately proximal to the tibial-fibular junction. Bottom: Cortical thickness expressed per gram body mass. Bars represent mean ± standard error. n≥6 per condition. *p<0.05 by Student's t-test.
Leptin treatment suppressed bone resorption in diabetes
To understand how the bone changes observed in diabetes and leptin-treatment occurred, we examined whole tibia gene expression and serum for markers of bone formation and resorption (Figure 5). Osteocalcin mRNA, a marker of bone formation, was significantly decreased in vehicle-treated diabetic bone compared to vehicle-treated control. Leptin-treatment had no effect on osteocalcin gene expression in control mice. Interestingly, osteocalcin trended to decrease in leptin-treated diabetic compared to leptin-treated control mice, although this changed did not reach statistical significance. Additionally, the level of osteocalcin mRNA was not different between vehicle-treated diabetic mice and leptin-treated diabetic mice. Both leptin groups tended to have higher serum osteocalcin levels than vehicle-treated controls, although this difference was not statistically significant.
Figure 5. Leptin treatment suppressed bone resorption diabetes.

(A) Osteocalcin and TRAP5 gene expression was measured by RT-PCR in whole tibia cDNA synthesized from mRNA and were expressed relative to HPRT, an unmodulated housekeeping gene control. (B) Osteocalcin and active TRAP5b were measured in serum collected at 28 days with commercially available ELISA kits. Bars represent mean ± standard error. n≥6 per condition. *p<0.05 by Student's t-test.
Reports indicated that T1-diabetes generally decreases or does not affect bone resorption. In this study, vehicle-treated diabetic mice had similar levels of bone resorption as vehicle-treated control mice (marked by TRAP5 gene expression and serum TRAP5b levels, Figure 5). We examined serum TRAP5 (versus urine measurements) because diabetic mice develop polyluria and can exhibit nephropathy, both of which can confound urine measurements. Similarly, TRAP5 mRNA and serum TRAP5b were not different in leptin-treated compared to vehicle-treated controls. Diabetes did however decrease serum TRAP5b in leptin-treated mice compared to leptin-treated controls. Interestingly, diabetic leptin-treated mice had reduced TRAP5 expression and serum TRAP5b compared to vehicle-treated diabetic mice, suggesting that leptin is capable of suppressing resorption in diabetic mice.
DISCUSSION
We (and others) found serum leptin levels decreased in diabetic rodent models (Akirav et al., 2004; Gulen and Dincer, 2007; Hidaka et al., 2002; Martin and McCabe, 2007). Additionally, we demonstrated that T1-diabetic mice have a location-dependent marrow adiposity phenotype similar to that of leptin-deficient ob/ob mice: increased marrow adiposity in long bones with diabetes, but no marrow adipocyte accumulation in the vertebrae (Hamrick et al., 2004; Martin and McCabe, 2007). Altered mesenchymal stem cell lineage selection is thought to be one mechanism for T1-diabetic bone loss and increased marrow adiposity. We therefore hypothesized that treating diabetic mice with leptin (through an osmotic pump) could prevent bone loss and marrow adipocyte accumulation. We found that although leptin administration completely prevented T1-diabetic marrow adiposity, it did not prevent bone loss in tibia or vertebrae.
As noted previously, serum leptin levels have been demonstrated to be reduced in diabetic rodent models (Akirav et al., 2004; Gulen and Dincer, 2007; Hidaka et al., 2002; Martin and McCabe, 2007) and in diabetic patients (Hanaki et al., 1999; Kiess et al., 1998). In this experiment, we observed a trend toward decreased serum leptin levels in diabetic compared to control mice, but it did not reach significance as we had observed previously (Martin and McCabe, 2007) (Table I). In part, we observed increased variability in our control population. In addition, the current study obtained leptin levels from fasted mice, whereas our previous measurements were obtained from fed mice. Akirav, et al. (Akirav et al., 2004) also demonstrate significant decreases in fed serum leptin levels in streptozotocin-induced diabetic compared to control rats, but found no difference between fasting diabetic and control rats. The latter was the result of reduced leptin levels (to that of diabetic rat levels) in fasted control rats. Although leptin levels are often decreased in T1-diabetic patients (Hanaki et al., 1999; Kiess et al., 1998), fasting serum leptin levels were reported to be unchanged in T1-diabetic children and adolescents aged 6-16 years (Karaguzel et al., 2006). We expect that if we had isolated serum from fed mice, leptin levels would have been decreased.
To our knowledge, this is the first report demonstrating a role leptin in the accumulation of marrow adipocytes in T1-diabetes. We found that by replacing the leptin that is normally lost by peripheral fat lipolysis in T1-diabetes, we can prevent the increase in adiposity in the marrow that is associated with T1-diabetes. It is possible that subcutaneous leptin administration prevented the increased lipid accumulation and marrow adipocyte differentiation that occurs in diabetes (Thomas et al., 1999) or alternatively acted directly or via sympathetic nervous system (SNS) stimulation on marrow adipocytes to induce apoptosis (Kim et al., 2003; Page et al., 2004). The latter is supported by a report indicating that central leptin administration to the rat ventromedial hypothalamus causes adipocyte apoptosis in fat pads and in bone marrow (Hamrick et al., 2007).
Several reports (Botolin et al., 2005; Jilka et al., 1996; Kajkenova et al., 1997; Moerman et al., 2004; Verma et al., 2002) demonstrate increased marrow adiposity with bone loss suggesting that (1) marrow fat accumulation could have negative effects on bone formation, (2) mesenchymal stem cells preferentially choose the adipocyte lineage instead of the osteoblast lineage in disease conditions, and/or (3) trabecular bone volume decreases, therefore fat accumulates, filling in the empty space it has left behind (Duque, 2008; Gimble et al., 2006; Nuttall et al., 1998; Rosen and Bouxsein, 2006). However, in this experiment inhibiting marrow adiposity with leptin treatment did not prevent diabetes-induced decreases in BMC, BMD and BVF in tibia trabecular bone (Table II, Figure 3). Consistent with this finding, diabetes induced changes in serum bone remodeling markers were not prevented by leptin treatment in rats (Gad, 2007).
Our results also indicated that leptin treatment reduced tibia trabecular bone mass and several vertebral bone parameters (BMC and BMD) in control mice. Other parameters showed trends toward decreases but were not significant. The suppression in bone parameters in control mice was somewhat unexpected since previous reports of chronic leptin treatment in wild type mice suggest that under normal conditions leptin does not affect bone mass (Steppan et al., 2000) and/or may increase bone strength (Cornish et al., 2002). To our knowledge we are the first to examine the influence of leptin treatment on BALB/c mouse bone density, which may account for some of the differences between studies. Leptin treatment has been predominantly examined in the C57BL/6 strain since it is the background for leptin deficient (ob/ob) and dysfunctional leptin receptor (db/db) genetic mouse models. Although leptin treatment of wild type C57BL/6 mice does not affect most bone parameters, labeled bone forming surface have been reported to be significantly decreased at 2.5 and 10 μg leptin/day doses (Hamrick et al., 2005). In that study, the mice were treated for 14 days, so it may be that longer treatments would have resulted in bone loss in control mice as we have seen with a 28 day treatment. It is also important to note that the majority of studies using leptin as bone loss therapeutic (ie: for ovariectomy and disuse (Burguera et al., 2001; Martin et al., 2005)) have used rats not mice, suggesting that species differences must also be considered. Regarding gender, key studies demonstrating leptin's bone anabolic potential have used female rats (unloading and ovariectomy (Burguera et al., 2001; Martin et al., 2005)) while both male and female ob/ob mouse bones positively respond to leptin treatment (Hamrick et al., 2005; Iwaniec et al., 2007). Thus, gender is less likely to play a role in our findings.
We hypothesized that leptin treatment of diabetic mice would prevent bone loss based on the finding that subcutaneous infusion of leptin with osmotic mini-pumps reduces marrow adiposity and restores bone mass in leptin deficient ob/ob mice (Hamrick et al., 2005). Similarly, hypothalamic injection of leptin can also correct skeletal abnormalities in ob/ob mice (Iwaniec et al., 2007). Leptin injection into control (leptin-replete) mice did not affect bone mass (Hamrick et al., 2005). This suggests that leptin deficiency could be critical for determining leptin effectiveness. Our diabetic mice exhibit a significant suppression in leptin levels although they are not completely deficient as seen in ob/ob mice. It is possible that the low levels of endogenous leptin affect adiposity but not bone loss. An alternative concept is based on the comparative phenotypes between leptin deficient and T1-diabetic mice. While leptin deficient mice lose long bone mass, they have increased vertebral trabecular bone mass (Ducy et al., 2000; Hamrick et al., 2004; Lorentzon et al., 1986; Mathey et al., 2002; Steppan et al., 2000; Takeda et al., 2002); this is in contrast to T1-diabetic mice, which lose bone at all sites examined to date. Thus, leptin may not be playing a role in regulating bone mass in this disease model. However, the adiposity phenotype, long bone but not vertebral adiposity, is identical to what we observe in T1-diabetic mice and is restored with leptin treatment.
We did observe that leptin treatment increased serum osteocalcin levels (although not statistically significantly) in both control and diabetic mice suggesting a potential positive effect, however at the RNA level, the suppression in osteocalcin levels by diabetes was not prevented with leptin treatment. Our studies also demonstrate that leptin treatment significantly suppresses osteoclast activity in diabetic mice as indicated by reduced active serum TRAP5b and bone TRAP5 RNA levels. Previous reports suggest that leptin treatment does not influence osteoclast activity (Hamrick et al., 2005) or can suppress activity possibly through inducing OPG expression and suppressing RANK ligand (Burguera et al., 2001).
Why was leptin unable to prevent bone loss in diabetes? We do not believe there were any technical problems with leptin administration because of its potent effect on food intake (Figure 1) and marrow adiposity (Figure 2) and because leptin treatment was successful at raising serum leptin levels (Table I). Concentration could play a role based on recent studies indicating the efficacy of leptin in restoring bone density is concentration dependent. Specifically, Martin et al. (Martin et al., 2007) demonstrated in the tail-suspended disuse rat model that only low dose leptin treatment (50 μg/kg/day) was effective in preventing femur trabecular bone loss. High dose leptin treatment (500 μg/kg/day) reduced femur bone mass in control rats and did not prevent bone loss in suspended groups. Either treatment dose prevented marrow adiposity. Previous studies in rats have used doses between ∼100-350 μg/kg/day with therapeutic success (Burguera et al., 2001; Martin et al., 2005). In ob/ob mice, successful therapeutic doses have spanned 2.5 - 10 μg/day or ∼100-400 μg/kg/day for control mice and ∼66-263 μg/kg/day for leptin-deficient mice (Hamrick et al., 2005). Our dose was intermediate (6.6 μg/day; ∼ 240 μg/kg/day): lower than the high dose in the Martin et al. study, but within the range to successfully treat bone loss in ob/ob mice (Hamrick et al., 2005). While our results exhibited similarities to the high dose leptin treatment response seen in tail-suspended rats (bone loss in all conditions and adiposity prevention) (Martin et al., 2007), the high dose (but not the low dose) in that study decreased serum osteocalcin, whereas we observed no significant change in serum osteocalcin levels (Figure 5).
Interestingly, blood glucose levels were significantly reduced by leptin treatment in both control and diabetic mice. This is consistent with a previous report in which STZ-induced, diabetic mice overexpressing leptin became hyperglycemic but were more sensitive to insulin and required lower doses (compared to mice not overexpressing leptin) to maintain euglycemia (Miyanaga et al., 2003). We do not believe the lower blood glucose in the leptin-treated mice confounded our results because we demonstrated bone loss to the same degree in leptin-treated and vehicle-treated diabetic mice. We also found leptin treatment affected other mouse parameters. For example, leptin-treated control mice exhibited lower body weights than diabetic (vehicle- or leptin-treated) mice. Still, the diabetic mice lost more bone (and more bone/gm body weight), supporting the notion that diabetic weight loss cannot fully account for the bone loss. While leptin treatment caused fat pad loss in control and diabetic mice, it prevented diabetic muscle loss, suggesting that the leptin-treated diabetic mice are leaner compared to the vehicle-treated diabetic mice. Similarly, the leptin-treated control mice were leaner than vehicle-treated control mice. A trend toward reduced fat pad and body mass was seen in wild type mice treated with 10 μg leptin/day although it did not reach significance (Hamrick et al., 2005).
Although the reciprocal relationship of fat to bone is often observed in diabetes, it is possible that they are not related to each other and occur through two completely different mechanisms. It is also possible that leptin did inhibit adipocyte differentiation (or only inhibited lipid deposition in adipocytes) and other factors may be necessary to induce osteoblast differentiation. The latter is likely, due to the complex pathology of T1-diabetes, which includes hyperglycemia, hypoinsulinemia, low serum leptin, hyperlipidemia, and inflammation. In a previous study, we demonstrated that inhibition of PPARγ2 with BADGE has a similar effect: prevention of marrow adiposity but not bone loss in diabetes (Botolin and McCabe, 2006). In this case, BADGE prevented hyperlipidemia, suggesting that excess lipids are not requisite for the process of bone loss in T1-diabetics. We have also demonstrated that loss of insulin receptor signaling in bone does not alter bone density, which suggests hypoinsulinemia cannot alone account for bone loss in T1-diabetes (Irwin et al., 2006). It is possible that other factors that have not yet been addressed or a combination of T1-diabetic complications are important for bone loss to occur.
In summary, we have demonstrated a link between low leptin levels in T1-diabetes and marrow adiposity. Although leptin treatment prevented diabetes-induced increased marrow adipocyte number and gene expression, leptin had no effect on bone loss. We conclude, therefore, that it is unlikely that leptin deficiency alone is responsible for diabetic bone loss and that other factors are necessary for the pathogenesis of T1-diabetic osteoporosis.
ACKNOWLEGEMENTS
The authors thank Amylin for providing the leptin, Regina Irwin for technical assistance and critically reviewing the manuscript and Lindsay Martin, Laura Harris, Dennean Lippner and Erin Nekritz for critically reviewing the manuscript.
Contract grant sponsor: Funded by a grant from NIH (DK061184) and the American Diabetes Association (7-07-RA-105) to LRM.
REFERENCES
- Ahdjoudj S, Lasmoles F, Holy X, Zerath E, Marie PJ. Transforming growth factor beta2 inhibits adipocyte differentiation induced by skeletal unloading in rat bone marrow stroma. J Bone Miner Res. 2002;17(4):668–677. doi: 10.1359/jbmr.2002.17.4.668. [DOI] [PubMed] [Google Scholar]
- Akirav EM, Chan O, Inouye K, Riddell MC, Matthews SG, Vranic M. Partial leptin restoration increases hypothalamic-pituitary-adrenal activity while diminishing weight loss and hyperphagia in streptozotocin diabetic rats. Metabolism. 2004;53(12):1558–1564. doi: 10.1016/j.metabol.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Auwerx J, Dequeker J, Bouillon R, Geusens P, Nijs J. Mineral metabolism and bone mass at peripheral and axial skeleton in diabetes mellitus. Diabetes. 1988;37(1):8–12. doi: 10.2337/diab.37.1.8. [DOI] [PubMed] [Google Scholar]
- Barzilai N, Wang J, Massilon D, Vuguin P, Hawkins M, Rossetti L. Leptin selectively decreases visceral adiposity and enhances insulin action. J Clin Invest. 1997;100(12):3105–3110. doi: 10.1172/JCI119865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botolin S, Faugere MC, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146(8):3622–3631. doi: 10.1210/en.2004-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botolin S, McCabe LR. Inhibition of PPARgamma prevents type I diabetic bone marrow adiposity but not bone loss. J Cell Physiol. 2006;209(3):967–976. doi: 10.1002/jcp.20804. [DOI] [PubMed] [Google Scholar]
- Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148(1):198–205. doi: 10.1210/en.2006-1006. [DOI] [PubMed] [Google Scholar]
- Bouillon R, Bex M, Van Herck E, Laureys J, Dooms L, Lesaffre E, Ravussin E. Influence of age, sex, and insulin on osteoblast function: osteoblast dysfunction in diabetes mellitus. J Clin Endocrinol Metab. 1995;80(4):1194–1202. doi: 10.1210/jcem.80.4.7714089. [DOI] [PubMed] [Google Scholar]
- Burguera B, Hofbauer LC, Thomas T, Gori F, Evans GL, Khosla S, Riggs BL, Turner RT. Leptin reduces ovariectomy-induced bone loss in rats. Endocrinology. 2001;142(8):3546–3553. doi: 10.1210/endo.142.8.8346. [DOI] [PubMed] [Google Scholar]
- Chan JL, Mantzoros CS. Role of leptin in energy-deprivation states: normal human physiology and clinical implications for hypothalamic amenorrhoea and anorexia nervosa. Lancet. 2005;366(9479):74–85. doi: 10.1016/S0140-6736(05)66830-4. [DOI] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334(5):292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- Cornish J, Callon KE, Bava U, Lin C, Naot D, Hill BL, Grey AB, Broom N, Myers DE, Nicholson GC, Reid IR. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol. 2002;175(2):405–415. doi: 10.1677/joe.0.1750405. [DOI] [PubMed] [Google Scholar]
- Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100(2):197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- Duque G. Bone and fat connection in aging bone. Curr Opin Rheumatol. 2008;20(4):429–434. doi: 10.1097/BOR.0b013e3283025e9c. [DOI] [PubMed] [Google Scholar]
- Fowlkes JL, Bunn RC, Liu L, Wahl EC, Coleman HN, Cockrell GE, Perrien DS, Lumpkin CK, Jr., Thrailkill KM. Runt-related transcription factor 2 (RUNX2) and RUNX2-related osteogenic genes are down-regulated throughout osteogenesis in type 1 diabetes mellitus. Endocrinology. 2008;149(4):1697–1704. doi: 10.1210/en.2007-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad HI. The potential osteogenic effects of systemic leptin and insulin administration in streptozotocin-induced diabetic female rats. Saudi Med J. 2007;28(8):1185–1190. [PubMed] [Google Scholar]
- Gimble JM, Zvonic S, Floyd ZE, Kassem M, Nuttall ME. Playing with bone and fat. J Cell Biochem. 2006;98(2):251–266. doi: 10.1002/jcb.20777. [DOI] [PubMed] [Google Scholar]
- Goodman WG, Hori MT. Diminished bone formation in experimental diabetes. Relationship to osteoid maturation and mineralization. Diabetes. 1984;33(9):825–831. doi: 10.2337/diab.33.9.825. [DOI] [PubMed] [Google Scholar]
- Goulding A, Taylor RW. Plasma leptin values in relation to bone mass and density and to dynamic biochemical markers of bone resorption and formation in postmenopausal women. Calcif Tissue Int. 1998;63(6):456–458. doi: 10.1007/s002239900557. [DOI] [PubMed] [Google Scholar]
- Gulen S, Dincer S. Effects of leptin on oxidative stress in healthy and Streptozotocin-induced diabetic rats. Mol Cell Biochem. 2007;302(1-2):59–65. doi: 10.1007/s11010-007-9426-5. [DOI] [PubMed] [Google Scholar]
- Hamrick MW. Leptin, bone mass, and the thrifty phenotype. J Bone Miner Res. 2004;19(10):1607–1611. doi: 10.1359/JBMR.040712. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Della Fera MA, Choi YH, Hartzell D, Pennington C, Baile CA. Injections of leptin into rat ventromedial hypothalamus increase adipocyte apoptosis in peripheral fat and in bone marrow. Cell Tissue Res. 2007;327(1):133–141. doi: 10.1007/s00441-006-0312-3. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Della-Fera MA, Choi YH, Pennington C, Hartzell D, Baile CA. Leptin treatment induces loss of bone marrow adipocytes and increases bone formation in leptin-deficient ob/ob mice. J Bone Miner Res. 2005;20(6):994–1001. doi: 10.1359/JBMR.050103. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, Pennington C, Newton D, Xie D, Isales C. Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone. 2004;34(3):376–383. doi: 10.1016/j.bone.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Hanaki K, Becker DJ, Arslanian SA. Leptin before and after insulin therapy in children with new-onset type 1 diabetes. J Clin Endocrinol Metab. 1999;84(5):1524–1526. doi: 10.1210/jcem.84.5.5653. [DOI] [PubMed] [Google Scholar]
- Hidaka S, Yoshimatsu H, Kondou S, Tsuruta Y, Oka K, Noguchi H, Okamoto K, Sakino H, Teshima Y, Okeda T, Sakata T. Chronic central leptin infusion restores hyperglycemia independent of food intake and insulin level in streptozotocin-induced diabetic rats. FASEB J. 2002;16(6):509–518. doi: 10.1096/fj.01-0164com. [DOI] [PubMed] [Google Scholar]
- Irwin R, Lin HV, Motyl KJ, McCabe LR. Normal bone density obtained in the absence of insulin receptor expression in bone. Endocrinology. 2006;147(12):5760–5767. doi: 10.1210/en.2006-0700. [DOI] [PubMed] [Google Scholar]
- Iwaniec UT, Boghossian S, Lapke PD, Turner RT, Kalra SP. Central leptin gene therapy corrects skeletal abnormalities in leptin-deficient ob/ob mice. Peptides. 2007;28(5):1012–1019. doi: 10.1016/j.peptides.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. 2007;166(5):495–505. doi: 10.1093/aje/kwm106. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Weinstein RS, Takahashi K, Parfitt AM, Manolagas SC. Linkage of decreased bone mass with impaired osteoblastogenesis in a murine model of accelerated senescence. J Clin Invest. 1996;97(7):1732–1740. doi: 10.1172/JCI118600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajkenova O, Lecka-Czernik B, Gubrij I, Hauser SP, Takahashi K, Parfitt AM, Jilka RL, Manolagas SC, Lipschitz DA. Increased adipogenesis and myelopoiesis in the bone marrow of SAMP6, a murine model of defective osteoblastogenesis and low turnover osteopenia. J Bone Miner Res. 1997;12(11):1772–1779. doi: 10.1359/jbmr.1997.12.11.1772. [DOI] [PubMed] [Google Scholar]
- Karaguzel G, Ozdem S, Boz A, Bircan I, Akcurin S. Leptin levels and body composition in children and adolescents with type 1 diabetes. Clin Biochem. 2006;39(8):788–793. doi: 10.1016/j.clinbiochem.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Karsenty G. Leptin controls bone formation through a hypothalamic relay. Recent Prog Horm Res. 2001;56:401–415. doi: 10.1210/rp.56.1.401. [DOI] [PubMed] [Google Scholar]
- Kemink SA, Hermus AR, Swinkels LM, Lutterman JA, Smals AG. Osteopenia in insulin-dependent diabetes mellitus; prevalence and aspects of pathophysiology. J Endocrinol Invest. 2000;23(5):295–303. doi: 10.1007/BF03343726. [DOI] [PubMed] [Google Scholar]
- Kiess W, Anil M, Blum WF, Englaro P, Juul A, Attanasio A, Dotsch J, Rascher W. Serum leptin levels in children and adolescents with insulin-dependent diabetes mellitus in relation to metabolic control and body mass index. Eur J Endocrinol. 1998;138(5):501–509. doi: 10.1530/eje.0.1380501. [DOI] [PubMed] [Google Scholar]
- Kim GS, Hong JS, Kim SW, Koh JM, An CS, Choi JY, Cheng SL. Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J Biol Chem. 2003;278(24):21920–21929. doi: 10.1074/jbc.M204598200. [DOI] [PubMed] [Google Scholar]
- Levin ME, Boisseau VC, Avioli LV. Effects of diabetes mellitus on bone mass in juvenile and adult-onset diabetes. N Engl J Med. 1976;294(5):241–245. doi: 10.1056/NEJM197601292940502. [DOI] [PubMed] [Google Scholar]
- Li J, Takaishi K, Cook W, McCorkle SK, Unger RH. Insig-1 “brakes” lipogenesis in adipocytes and inhibits differentiation of preadipocytes. Proc Natl Acad Sci U S A. 2003;100(16):9476–9481. doi: 10.1073/pnas.1133426100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentzon R, Alehagen U, Boquist L. Osteopenia in mice with genetic diabetes. Diabetes Res Clin Pract. 1986;2(3):157–163. doi: 10.1016/s0168-8227(86)80017-1. [DOI] [PubMed] [Google Scholar]
- Martin A, David V, Malaval L, Lafage-Proust MH, Vico L, Thomas T. Opposite effects of leptin on bone metabolism: a dose-dependent balance related to energy intake and insulin-like growth factor-I pathway. Endocrinology. 2007;148(7):3419–3425. doi: 10.1210/en.2006-1541. [DOI] [PubMed] [Google Scholar]
- Martin A, de Vittoris R, David V, Moraes R, Begeot M, Lafage-Proust MH, Alexandre C, Vico L, Thomas T. Leptin modulates both resorption and formation while preventing disuse-induced bone loss in tail-suspended female rats. Endocrinology. 2005;146(8):3652–3659. doi: 10.1210/en.2004-1509. [DOI] [PubMed] [Google Scholar]
- Martin LM, McCabe LR. Type I diabetic bone phenotype is location but not gender dependent. Histochem Cell Biol. 2007;128(2):125–133. doi: 10.1007/s00418-007-0308-4. [DOI] [PubMed] [Google Scholar]
- Mathey J, Horcajada-Molteni MN, Chanteranne B, Picherit C, Puel C, Lebecque P, Cubizoles C, Davicco MJ, Coxam V, Barlet JP. Bone mass in obese diabetic Zucker rats: influence of treadmill running. Calcif Tissue Int. 2002;70(4):305–311. doi: 10.1007/s00223-001-2077-8. [DOI] [PubMed] [Google Scholar]
- McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem. 2007;102(6):1343–1357. doi: 10.1002/jcb.21573. [DOI] [PubMed] [Google Scholar]
- Miyanaga F, Ogawa Y, Ebihara K, Hidaka S, Tanaka T, Hayashi S, Masuzaki H, Nakao K. Leptin as an adjunct of insulin therapy in insulin-deficient diabetes. Diabetologia. 2003;46(10):1329–1337. doi: 10.1007/s00125-003-1193-6. [DOI] [PubMed] [Google Scholar]
- Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. 2004;3(6):379–389. doi: 10.1111/j.1474-9728.2004.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuttall ME, Patton AJ, Olivera DL, Nadeau DP, Gowen M. Human trabecular bone cells are able to express both osteoblastic and adipocytic phenotype: implications for osteopenic disorders. J Bone Miner Res. 1998;13(3):371–382. doi: 10.1359/jbmr.1998.13.3.371. [DOI] [PubMed] [Google Scholar]
- Ontiveros C, McCabe LR. Simulated microgravity suppresses osteoblast phenotype, Runx2 levels and AP-1 transactivation. J Cell Biochem. 2003;88(3):427–437. doi: 10.1002/jcb.10410. [DOI] [PubMed] [Google Scholar]
- Page KA, Hartzell DL, Li C, Westby AL, Della-Fera MA, Azain MJ, Pringle TD, Baile CA. beta-Adrenergic receptor agonists increase apoptosis of adipose tissue in mice. Domest Anim Endocrinol. 2004;26(1):23–31. doi: 10.1016/j.domaniend.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Reseland JE, Syversen U, Bakke I, Qvigstad G, Eide LG, Hjertner O, Gordeladze JO, Drevon CA. Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization. J Bone Miner Res. 2001;16(8):1426–1433. doi: 10.1359/jbmr.2001.16.8.1426. [DOI] [PubMed] [Google Scholar]
- Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006;2(1):35–43. doi: 10.1038/ncprheum0070. [DOI] [PubMed] [Google Scholar]
- Sindelar DK, Havel PJ, Seeley RJ, Wilkinson CW, Woods SC, Schwartz MW. Low plasma leptin levels contribute to diabetic hyperphagia in rats. Diabetes. 1999;48(6):1275–1280. doi: 10.2337/diabetes.48.6.1275. [DOI] [PubMed] [Google Scholar]
- Sivitz WI, Walsh SA, Morgan DA, Thomas MJ, Haynes WG. Effects of leptin on insulin sensitivity in normal rats. Endocrinology. 1997;138(8):3395–3401. doi: 10.1210/endo.138.8.5327. [DOI] [PubMed] [Google Scholar]
- Steppan CM, Crawford DT, Chidsey-Frink KL, Ke H, Swick AG. Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept. 2000;92(1-3):73–78. doi: 10.1016/s0167-0115(00)00152-x. [DOI] [PubMed] [Google Scholar]
- Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305–317. doi: 10.1016/s0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999;140(4):1630–1638. doi: 10.1210/endo.140.4.6637. [DOI] [PubMed] [Google Scholar]
- Vengellur A, LaPres JJ. The role of hypoxia inducible factor 1alpha in cobalt chloride induced cell death in mouse embryonic fibroblasts. Toxicol Sci. 2004;82(2):638–646. doi: 10.1093/toxsci/kfh278. [DOI] [PubMed] [Google Scholar]
- Verma S, Rajaratnam JH, Denton J, Hoyland JA, Byers RJ. Adipocytic proportion of bone marrow is inversely related to bone formation in osteoporosis. J Clin Pathol. 2002;55(9):693–698. doi: 10.1136/jcp.55.9.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes--a meta-analysis. Osteoporos Int. 2007;18(4):427–444. doi: 10.1007/s00198-006-0253-4. [DOI] [PubMed] [Google Scholar]
- Wang JL, Chinookoswong N, Scully S, Qi M, Shi ZQ. Differential effects of leptin in regulation of tissue glucose utilization in vivo. Endocrinology. 1999;140(5):2117–2124. doi: 10.1210/endo.140.5.6681. [DOI] [PubMed] [Google Scholar]
- Wiren KM, Zhang XW, Toombs AR, Kasparcova V, Gentile MA, Harada S, Jepsen KJ. Targeted overexpression of androgen receptor in osteoblasts: unexpected complex bone phenotype in growing animals. Endocrinology. 2004;145(7):3507–3522. doi: 10.1210/en.2003-1016. [DOI] [PubMed] [Google Scholar]