

Abstract
Purpose
Studies have demonstrated that selenium supplementation reduces the incidence of cancer, particularly prostate cancer. Evidence from experimental studies suggests that apoptosis is a key event in cancer chemoprevention by selenium and reactive oxygen species play a role in induction of apoptosis by selenium compounds. The current study was designed to investigate the role of superoxide and mitochondria in selenite-induced apoptosis in human prostate cancer cells.
Methods
LNCaP cells were transduced with adenoviral constructs to overexpress four primary antioxidant enzymes: manganese superoxide dismutase (MnSOD), copper-zinc superoxide dismutase (CuZnSOD), catalase (CAT), or glutathione peroxidase 1 (GPx1). Cell viability, apoptosis, and superoxide production induced by sodium selenite were analyzed by the MTT assay, chemiluminescence, flow cytometry, western blot analysis, and Hoechst 33342 staining following overexpression of these antioxidant enzymes.
Results
Our study shows the following results: (1) selenite induced cancer cell death and apoptosis by producing superoxide radicals; (2) selenite-induced superoxide production, cell death, and apoptosis were inhibited by overexpression of MnSOD, but not by CuZnSOD, CAT, or GPx1; and (3) selenite treatment resulted in a decrease in mitochondrial membrane potential, release of cytochrome c into the cytosol, and activation of caspases 9 and 3, events that were suppressed by overexpression of MnSOD.
Conclusions
This study demonstrates that selenite induces cell death and apoptosis by production of superoxide in mitochondria and activation of the mitochondrial apoptotic pathway and MnSOD plays an important role in protection against prooxidant effects of superoxide from selenite. The data suggest that superoxide production in mitochondria is, at least in part, a key event in selenium-induced apoptosis in prostate cancer cells.
Keywords: antioxidant enzymes, apoptosis, mitochondria, selenium, superoxide
Introduction
Selenium (Se) is an essential trace element for human health and necessary for the antioxidant enzyme (AE) function of glutathione peroxidases (GPxs) and other selenoproteins. In addition, experimental and clinical studies have demonstrated that Se has anticancer activity [1-3]. A recent clinical trial showed that selenized yeast supplementation was effective in reduction of prostate cancer incidence by 60% compared with placebo treatment [4, 5]. Epidemiological studies also demonstrated an inverse association between levels of Se in serum or toenails and incidence of prostate cancer [6-8]. Thus, selenomethione is currently being evaluated as a chemopreventive agent for prostate cancer prevention by Se [9].
Although Se has been demonstrated to be a promising chemopreventive agent for cancer, the underlying mechanisms are still not fully understood. Data from experimental studies indicate that induction of apoptosis may account for the anticancer effect of Se [10]. A number of studies have demonstrated that several Se compounds, particularly those with a redox-cycling property, can produce reactive oxygen species (ROS) and subsequently induce prooxidant effects and cancer cell apoptosis, which can be suppressed by AEs or small molecular weight antioxidant compounds [11-15]. Based on these data, it has been proposed that a prooxidant effect may be one of the anticancer mechanisms for some Se compounds [11, 16].
Our previous study showed that selenite induced apoptosis and growth inhibition of human prostate cancer cells in association with production of ROS, reduction of intracellular reduced glutathione (GSH) and generation of oxidized glutathione (GSSG), and mitochondrial damage [17]. Selenite treatment also up-regulated AEs manganese superoxide dismutase (MnSOD), copper-zinc superoxide dismutase (CuZnSOD), and GPx. These effects of selenite were inhibited by a synthetic superoxide dismutase (SOD) mimic. We also demonstrated that overexpression of MnSOD prevented cell death from selenite treatment [18]. Additionally, our previous studies documented that treatment with selenite or combined selenomethione and methionase resulted in cancer cell apoptosis with p53 translocation to mitochondria, upregulation of the proapoptotic protein Bax, increased levels of superoxide, release of cytochrome c into the cytosol, activation of caspase 9, and induction of cell apoptosis [19, 20]. Taken together, these results suggest that mitochondria may be the target and the generation of the superoxide radical may play an important role in Se-induced cell apoptosis.
Studies have shown that Se induced apoptosis via its prooxidant activity in various types of neoplastic cells, including prostate cancer, colon cancer, liver cancer, leukemia, and lymphoma [19-24]. At least four prostate cancer cell lines have been studied [18-20, 25]. Superoxide and H2O2 are considered to be the ROS that result in the prooxidative effects of Se [11, 16, 19, 20, 26]. Previous studies used synthetic SOD mimics, N-acetyl-cysteine, or buthionine sulfoximine to assess the involvement of ROS in the prooxidant effects of Se compounds in cancer cells [13, 17, 19, 20, 26]. However, the specificity and subcellular effects of these approaches have not been clearly defined. Although it was reported that selenite-induced cell death in a prostate cancer cell line (RWPE2) was inhibited by overexpressing MnSOD by gene transfection using plasmid constructs, effects of CuZnSOD, GPx, or catalase (CAT) were not analyzed in that study [18]. In this study, we transduced LNCaP cells with recombinant adenoviral constructs to separately overexpress MnSOD, CuZnSOD, CAT, or GPx1 in human LNCaP cells and subsequently treated with selenite to determine whether superoxide was the main ROS involved in apoptosis and whether mitochondria were the target organelles. Our results show that only overexpression of the mitochondrial-localized antioxidant enzyme MnSOD decreased selenite-induced superoxide production and apoptosis in human prostate cancer cells. Our study also provides evidence that selenite induced apoptosis by damaging mitochondria through production of superoxide.
Materials and methods
Chemicals and antibodies
Sodium selenite and anti-β-actin antibody were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Lucigenin (bis-N-methylacridinium nitrate), dihydroethidium (DHE), and EnzChek Caspase-3 Assay Kit #1 were purchased from Molecular Probes (Eugene, OR, USA). Caspase-Glo™ 9 Assay Kit was purchased from Promega Co. (Madison, WI, USA). M-Per Mammalian Protein Extraction Reagent, Mitochondria Isolation Kit, and SuperSignal West Pico Stable Peroxide/Luminol Enhancer Solutions were purchased from Pierce Biotechnology Inc. (Rockford, IL, USA). Anti-cytochrome c antibody was purchased from Cell Signaling Technology (Danvers, MA, USA). Adenoviral constructs containing MnSOD, CuZnSOD, GPx1, or CAT cDNA and adenoviral empty constructs were purchased from the Gene Transfer Vector Core of the University of Iowa (Iowa City, IA, USA) [27-29].
Cell culture
LNCaP cells were obtained from the American Type Culture Collection (ATCC) and routinely maintained in 100-mm tissue culture dishes in RPMI 1640 supplemented with 5% heat-inactivated fetal bovine serum and 1% antibiotic-antimycotic (Life Technologies Inc., Rockville, MD, USA) at 37°C in a humidified atmosphere of 95% air and 5% CO2.
MTT assay
Cells were seeded at 1 × 105 cells/well in 24-well plates overnight before treatment with different agents and then allowed to grow for an additional 5 days. MTT solution (10 μl; 5 mg/ml in PBS) was added to each well of the plate and incubated for 3 h at 37°C. MTT lysis buffer (100 μl of 10% SDS, 45% dimethyl formamide, adjusted to pH 4.5 by glacial acetic acid) was then added to dissolve the formazan. The optical density was measured at 570 nm using a Beckman Coulter DU-640 Spectrophotometer (Beckman Coulter Inc., Fullerton, CA, USA). The percentage of viable cells was calculated as the relative ratio of optical density to the control.
Adenovirus transduction
LNCaP cells (2 × 106) were plated in 10 ml complete medium in a 100 mm dish and allowed to attach for 48 h. Adenoviral constructs (AdMnSOD, AdCuZnSOD, AdGPx1, AdCAT, or AdEmpty) suspended in 3% sucrose were added to cell cultures in 6 ml serum- and antibiotic-free medium, respectively, at multiplicities of infectivity (MOI) as indicated. Cells were incubated for 18 h and then 6 ml of medium with 10% FBS were added to the cultures. Cells were allowed to grow for 24 h and then treated with 0, 0.5, 1.5, or 2.5 μM selenite for 5 days for cell viability assay or 2.5 μM selenite for 18 h before harvesting for other analyses.
Western blot analysis
Cell pellets were lysed with M-PER mammalian protein extraction reagent and protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). Cell lysates (20−50 μg) were electrophoresed in 12.5 % SDS polyacrylamide gels and then transferred onto nitrocellulose membranes. After blotting in 5% nonfat dry milk in Tween 20 Tris-buffered saline (TTBS), the membranes were incubated with primary antibodies at 1:1,000−2,000 dilutions in TTBS overnight at 4°C, and then secondary antibodies conjugated with horseradish peroxidase at 1:10,000 dilution in TTBS for 1 hr at room temperature. Protein bands were visualized on X-ray film using an enhanced chemiluminescence system (Pierce Biotechnology, Rockford, IL, USA).
Activity gel assays for MnSOD, CuZnSOD, CAT, or GPx1
Cells transduced with AdMnSOD, AdCuZnSOD, AdCAT, AdGPx1, or AdEmpty were washed with PBS and scraped off tissue culture dishes in cold PBS. Protein concentrations were determined by the Bradford method. Activity gel assays were preformed as previously described [30]. Two hundred μg of total protein were separated in a native polyacrylamide gel (5% for stacking and 8% for separating). The gel was run in the pre-electrophoresis buffer at 40 mA for 1 h, and then left at 4°C overnight. The electrophoresis process was performed in two steps: the gel with samples was run in the pre-electrophoresis buffer at 4°C for 3 h and then in the electrophoresis buffer at 4°C for another 4 h. After electrophoresis, the gel was incubated in NBT (nitroblue tetrazolium) solution for 20 min and then in riboflavin-TEMED for 15 min at room temperature in the dark. The gel was then washed with distilled water and illuminated under a bright fluorescent light for achromatic bands indicating the presence of SOD. CuZnSOD and MnSOD were differentiated by adding sodium cyanide to inhibit CuZnSOD. Activity gels for GPx1 and catalase were performed using cumene hydroperoxide as the substrate. After electrophoresis, the gel was rinsed with 1 mM GSH three times for 7 min each and then incubated in 75 ml of distilled H2O containing 0.008% cumene hydroperoxide plus 1 mM GSH for 10 min with gentle shaking. After briefly rinsing twice with distilled H2O, the gel was stained with a freshly made 1% ferric chloride and potassium ferricyanide solution and mixed immediately from equal volumes of a 2% stock solution of each before use. The achromatic bands corresponding to GPx and catalase were illuminated under a fluorescent light and photographed.
Superoxide measurements
Lucigenin-dependent chemiluminescence (CL) in cells was measured by a modified method as described previously [19, 31]. The stock solution of lucigenin (10 mM) was prepared in PBS and stored at −20°C in the dark. Lucigenin (100 μM) was added to 1 × 105 cells in 100 μl PBS after transduction with AdMnSOD, AdCuZnSOD, AdCAT, AdGPx1, or AdEmpty for 48 h. The reaction was initiated by the addition of lucigenin and selenite to the cells and the CL level was monitored as relative light units (RLU) using a luminometer (Lumat LB9501, Berthold, Oak Ridge, TN, USA) for a total period of 10 min at 30 s intervals. To measure superoxide using DHE, 5 × 105 LNCaP cells were suspended in 1 ml of serum-free medium and incubated with 2.5 μM selenite and 5 μM DHE for 5 min at room temperature. Fluorescence was immediately measured using a flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA) [32].
Flow cytometric analysis of apoptosis
Cell samples were prepared and analyzed as described previously [17]. Cells were washed with PBS/EDTA/BSA buffer (PBS, 1 mM EDTA, and 0.1% BSA) and fixed in 100 μl of PBS/EDTA/BSA buffer plus 900 μl of 70% ethanol for 30 min at −20°C. After washing with phosphate-citric acid buffer (0.192 M Na2HPO4 and 4 mM citric acid, pH 7.8), the cells were stained in 500 μl of propidium iodide staining solution (33 μg/ml propidium iodide, 200 μg/ml DNase-free RNase A, and 0.2% Triton X-100) overnight at 4°C. Both cell cycle distribution and sub-G1 cells were simultaneously measured using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Detection of apoptosis by Hoechst 33342 staining
For morphological analysis of apoptosis, cells were spread onto glass slides by centrifugation at 700 rpm for 5 min using a Shandon Cytospin 3 centrifuge (Shandon Cytospin, Sewickey, PA, USA). The slides were incubated in a Hoechst 33342 staining solution (3 μl of 10 mg/ml Hoechst 33342 in 10 ml PBS) for 20 min after fixing in 95% alcohol for 15 min. The stained cells were examined under a fluorescence microscope. Cells with nuclear fragmentation were designated as apoptotic, and the apoptotic index was determined by counting a total of 300 cells on each slide from three slides.
Measurement of mitochondrial membrane potential
For mitochondrial membrane potential measurement, cells were suspended in 1 ml of serum-free medium containing 2.5 mmol/l JC-1 dye (Molecular Probes, Eugene, OR, USA) and incubated at 37°C for 20 min at room temperature. After washing twice with PBS, fluorescence in cells was immediately measured using a flow cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA). Mitochondrial depolarization was determined by the decrease in the ratio of the red signal at 590 nm emission to the green signal at 530 nm emission.
Cytosol fractionation
Cells were seeded at 6 × 105 in 100-mm tissue culture dishes and allowed to grow to 60% confluence. Cells were treated with or without 2.5 μM selenite for 18 h and then mitochondria and cytosol fractions were separated using Mitochondria Isolation Kit according to the manufacturer's instructions (Pierce Biotechnology, Rockford, IL, USA).
Caspase activity assays
Caspase 9 activity was determined using Caspase-Glo™ 9 Kit per manufacturer's instructions. Cells were seeded at 3 × 104 cells/well in a 96-well plate with 100 μl medium. Approximately 16 h later, cells were treated with 2.5 μM selenite for 18 h and then Caspase-Glo™ 9 Reagent (100 μl) was directly added into each well to a final volume of 200 μl/well. Chemiluminescence was measured using a Tropix TR717 Microplate Luminometer (Applied Biosystems, Bedford, MA, USA). Caspase-3 activity was measured using EnzChek Caspase-3 Kit #1 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's protocol. Briefly, LNCaP cells were treated with 2.5 μM selenite for 18 h after adenoviral transduction for 48 h. The cells were harvested and suspended in 50 μl of 1x cell lysis buffer followed by three cycles of freeze-thaw. Samples equivalent to 200 μg protein were incubated with 50 μl of 2x substrate solution at room temperature for 30 min. Fluorescence produced by Caspase-3-mediated cleavage of the substrate was determined using a fluorescence microplate reader (PerkinElmer, MA, USA) with settings of excitation wavelength at 380 nm and emission wavelength at 480 nm. Fluorescence intensity (arbitrary units) was normalized by the protein concentration.
Statistical analysis
All data are presented as means ± SD with N = 3 or more and independent experiments were repeated at least 3 times. Student's t test was used to determine the significance of statistical differences between data at the level of P <0.05.
Results
Effect of selenite and superoxide on cell viability and growth
To assess the effect of selenite in LNCaP cells, cell viability was determined using the MTT assay after 5 days treatment with different doses of selenite, while growth inhibition was determined by counting cell number using a Coulter counter following treatment with different doses of selenite for different times. Cells were treated with 0, 0.5, 1.0, 1.5, 2.0, or 2.5 μM selenite for 5 days (Fig. 1a) or with 0, 0.5, 1.5, or 2.5 μM selenite for 3, 5, 7, and 9 days (Fig. 1b). As shown in Fig. 1a, selenite treatment induced a dose-dependent cell death. Significant cell killing occurred in cells treated with 1.5 μM and higher doses of selenite and 50% of cell death (LD50) occurred at the 2.0 μM concentration. There was no significant decrease in cell viability in cells treated with 0.5 or 1.0 μM selenite. Cell growth inhibition by selenite was also time-dependent (Fig. 1b). Significant cell growth inhibition was observed on day 7 in cells treated with 1.5 μM selenite and on day 5 in cells treated with 2.5 μM selenite. There was no significant effect in cells treated with 0.5 μM selenite compared to the control (no selenite). These results demonstrate that cellular effects of selenite in LNCaP cells are dose- and time-dependent.
Fig. 1.
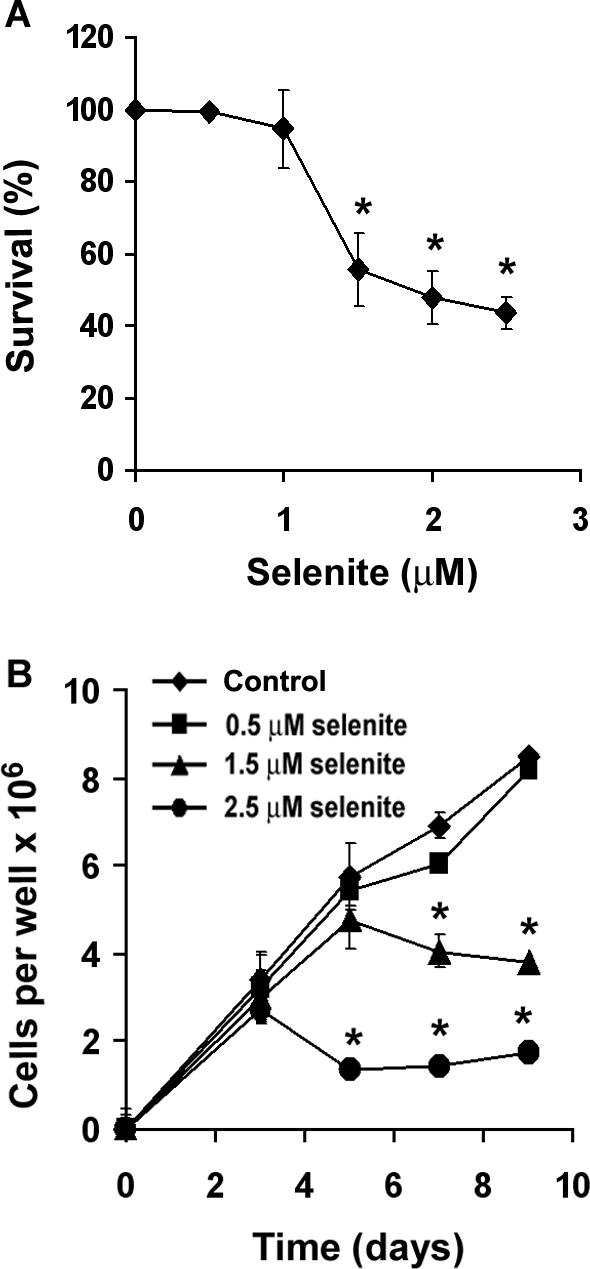
Effect of selenite on viability and growth in LNCaP cells: a effect of selenite on cell viability. Percentage of survival is expressed as cell viability relative to control without selenite treatment. Cells were treated with selenite for 5 days and viability was measured by the MTT assay. Data represent mean ± SD, n = 3. * P < 0.05 compared with control without selenite. b effect of selenite on cell growth in LNCaP cells. Cell numbers were counted using a Coulter counter. Data represent mean ± SD, n = 3. * P < 0.05 compared with control at the corresponding time points.
Effect of overexpression of AEs on superoxide production and cell viability by selenite
In order to determine involvement and localization of ROS in cellular effects of selenite, MnSOD, CuZnSOD, GPx1, or CAT were overexpressed in LNCaP cells by recombinant adenoviral construct transduction. Levels of protein and activity of these AEs were determined using western blotting analysis and native gel assays. As expected, there were dose-dependent increases in both immunoreactive proteins and enzymatic activities of MnSOD, CuZnSOD, GPx1, or CAT in LNCaP cells following specific adenoviral transduction for 48 h (Fig. 2a, b). Densitometry showed that levels of immunoreactive protein increased sixfold in cells transduced with 50 MOI AdMnSOD or AdCuZnSOD and 15-fold in cells transduced with 100 MOI AdMnSOD compared to control cells (no Ad transduction) or AdEmpty transduced cells (Fig. 2a). Levels of CAT and GPx1 dramatically increased in cells transduced with AdCAT, or GPx1. There were no significant changes in these four AEs in cells transduced with AdEmpty. Enzyme activity is the most important parameter for determining the function of AEs, and therefore, activities of these antioxidant enzymes were determined by activity gel assays. As shown in Fig. 2b, activity gel electrophoresis demonstrated that cells transduced with adenoviral constructs containing individual AE cDNA had significant increases in activities of all four AEs in a dose-dependent manner at 48 h. The activity gel results agree with those observed by western blotting analysis. Compared to 100 MOI viral transduction, 50 MOI already achieved significant increases in AEs, particularly enzymatic activity, and showed lower cytotoxicity. Thus, 50 MOI was chosen as the test-dose in the remainder of the experiments.
Fig. 2.

Adenovirus-mediated overexpression of MnSOD, CuZnSOD, CAT or GPx1 in LNCaP cells. Cells were transduced with adenoviral constructs for 48 h and cell lysates were harvested for western and activity gel analyses. 1 Control (no adenoviral transduction); 2 50 MOI AdEmpty; 3 100 MOI AdEmpty; 4 50 MOI AdMnSOD, AdCuZnSOD, AdCAT, or AdGPx1; 5 100 MOI AdMnSOD, AdCuZnSOD, AdCAT, or AdGPx1. a Western blot analysis of expression of MnSOD, CuZnSOD, CAT and GPx1. Twenty micrograms of total cellular protein were loaded per lane. b Native gel analysis of MnSOD, CuZnSOD, CAT or GPx1 enzymatic activities. A total amount of 200 μg of protein was loaded per lane. Data presented are one representative experiment of three independent experiments that showed similar results.
To assess possible involvement of superoxide in selenite-induced cell death, cellular levels of superoxide were measured by the lucigenin-dependent chemiluminescence assay. Fig. 3a shows that intracellular chemiluminescence was elevated in AdEmpty-transduced cells following treatment with 2.5 μM selenite for 6 min. Elevated chemiluminescence was suppressed by overexpression of MnSOD only, but not by overexpression of CuZnSOD, CAT, or GPx1. Elevation of intracellular superoxide was also confirmed by flow cytometric analysis of DHE fluorescence. As shown in Fig. 3b, DHE fluorescence was elevated in AdEmpty-transduced cells for 48 h following selenite treatment for 5 min, but not in AdMnSOD-transduced cells. Cell viability assay showed that AdMnSOD protected against cell death from selenite treatment, while transduction of AdCuZnSOD, AdCAT, or AdGPx1 had no protection against cell death induced by selenite treatment (Fig. 3c). Cells transduced with AdEmpty, AdCuZnSOD, AdGPx1, or AdCAT showed 50% cell death at 2.5 μM selenite treatment, while those transduced with AdMnSOD showed only 30% cell death. Dose-dependent protection against selenite-induced cell death by overexpression of MnSOD was observed in LNCaP cells and protection of overexpression of MnSOD against selenite-induced cell death was also observed in PC3 human prostate cancer cells (data not shown). On the other hand, only AdCAT transduction, but not AdMnSOD, protected against cell death from H2O2 treatment (Fig. 3d). These data not only demonstrate that selenite treatment produces superoxide in cells, but also indicate that superoxide production following selenite treatment is mainly in mitochondria.
Fig. 3.
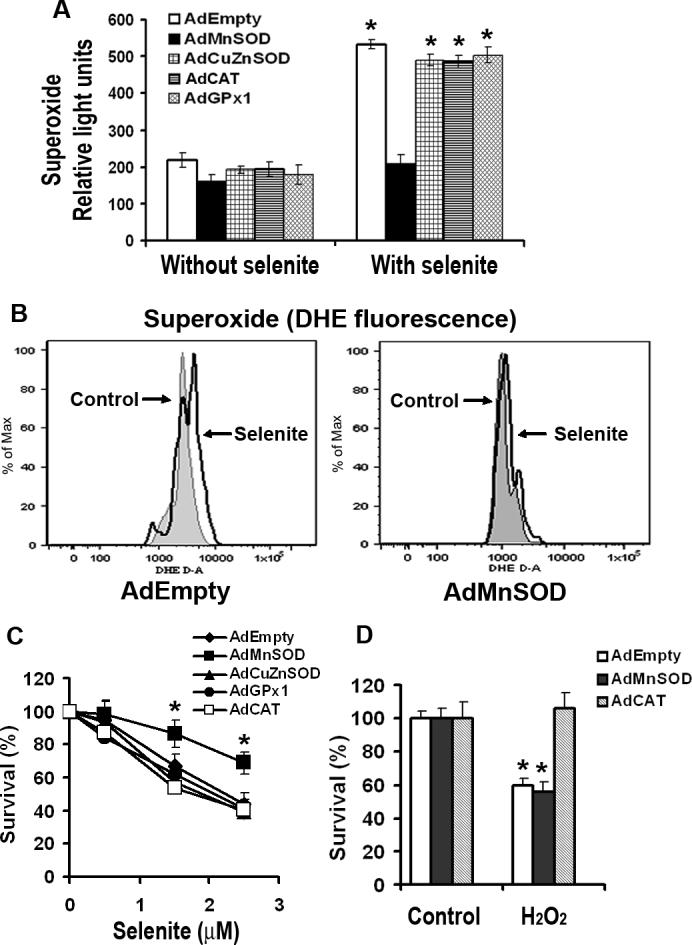
Effect of overexpression of AEs on selenite-induced production of superoxide and viability in LNCaP cells. a Levels of selenite-induced superoxide production in LNCaP cells with overexpression of MnSOD, CuZnSOD, CAT, or GPx1. Cells were pretreated with 2.5 μM selenite for 6 min and then harvested for chemiluminescence assay using a luminometer after transduction with 50 MOI AdEmpty, AdMnSOD, AdCuZnSOD, AdCAT, or AdGPx1 for 48 h. The data were obtained from three independent experiments and shown as means ± SD. *P < 0.05 compared with control cells without selenite treatment. b Levels of superoxide in cells transduced with AdEmpty or AdMnSOD. After transduction with 50 MOI AdEmpty or AdMnSOD for 48 h, cells were harvested in suspension and then incubated with 2.5 μM selenite and 5 μM DHE for 5 min. DHE fluorescence was detected using a flow cytometer. c MTT assay of cell viability. Cells were plated in 96-well plates overnight, transduced with 50 MOI AdEmpty, AdMnSOD, AdCuZnSOD, AdCAT, or AdGPx1 for 48 h, and then treated with different concentrations of selenite for an additional 5 days. Data are presented as means ± SD of three independent experiments. * P < 0.05 compared with AdEmpty at the corresponding concentrations of selenite. d MTT assay of cell viability. Cells were grown in 96-well plates overnight, transduced with 50 MOI AdEmpty, AdMnSOD, or AdCAT for 48 h, and then treated with 50 μM H2O2 for 24 h. Data are presented as means ± SD of three independent experiments. * P < 0.05 compared with control cells without H2O2 treatment.
Effect of overexpression of MnSOD on apoptosis induced by selenite
To determine whether superoxide was responsible for apoptosis induced by selenite, apoptosis was analyzed by Hoechst 33342 staining to detect nuclear fragmentation and by flow cytometry to measure sub-G1 cells following AE overexpression and selenite treatment. Cells were transduced with 50 MOI AdEmpty or AdMnSOD for 48 h prior to selenite treatment for an additional 18 h. As shown in Fig. 4a, selenite treatment significantly increased the level of apoptosis (9.4%) in control cells compared to cells without selenite treatment (3.5%), while the level of apoptosis was only slightly increased in AdMnSOD-transduced cells with selenite treatment (5.7%) in comparison to AdMnSOD-transduced cells without selenite treatment (4.2%). In contrast, apoptosis was increased from 4.5% to 11.5% in AdEmpty-transduced cells following selenite treatment, which was even slightly higher than that observed in control cells treated with selenite. This was most likely due to stress induced by adenoviral infection because apoptosis was also slightly increased in AdEmpty- or AdMnSOD-transduced cells without selenite treatment. Flow cytometric analysis also showed that selenite treatment resulted in an increase in sub-G1 cells from 5% to 17% in AdEmpty-transduced cells (Fig. 4b). In contrast, there were only 8% sub-G1 cells in AdMnSOD-transduced cells following selenite treatment, which was twofold lower than that in AdEmpty-transduced cells (Fig. 4b). The results indicate that superoxide is involved in selenite-induced apoptosis and also suggest that mitochondria may be the target organelles.
Fig. 4.
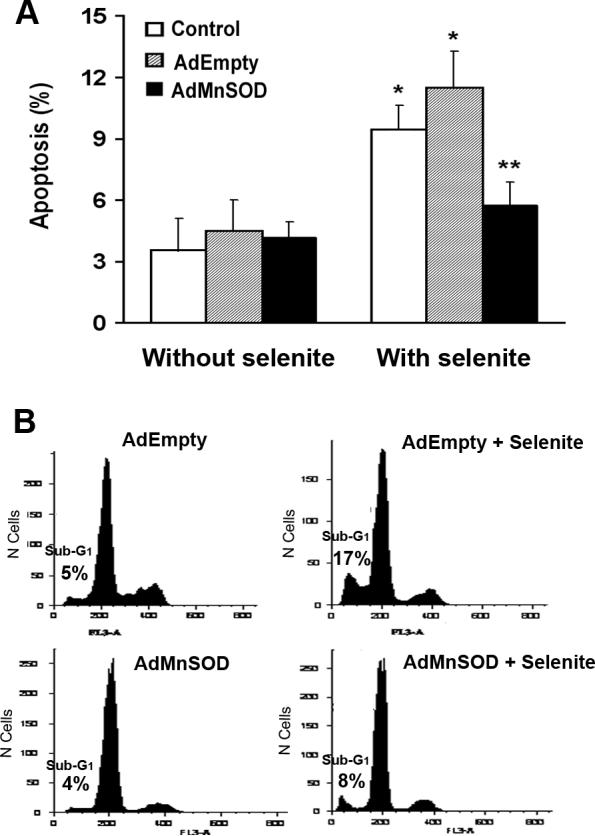
Inhibition of selenite-induced apoptosis by overexpression of MnSOD. a Apoptosis detected by Hoechst 33342 staining. Cells were transduced with 50 MOI AdEmpty or AdMnSOD for 48 h and then treated with 2.5 μM selenite for 18 h. Cells were stained with Hoechst dye and apoptosis was analyzed using a fluorescence microscope. At least 300 nuclei were counted per sample in triplicate. Data are presented as means ± SD of three independent experiments. * P < 0.05 compared with cells without selenite treatment. ** P < 0.05 compared with control and AdEmpty-transduced cells with selenite treatment. b Sub-G1 cell population measurement by flow cytometry for apoptosis. After transduction with 50 MOI AdEmpty or AdMnSOD for 48 h, cells were treated with 2.5 μM selenite for 24 h and harvested in suspension. After fixation, cell suspensions were stained with propidium iodide and sub-G1 cell populations were measured using a flow cytometer.
Mitochondria as targets in apoptosis induced by selenite
To confirm mitochondria as targets in selenite-induced apoptosis, we next analyzed the mitochondrial membrane potential, cytochrome c release into the cytosol, and activation of caspases 9 and 3 in LNCaP cells following 2.5 μM selenite treatment. Figure 5 shows a time-dependent effect on the mitochondrial membrane potential and activation of caspases 9 and 3. A significant decrease in the mitochondrial membrane potential (Fig. 5a) and significant increases in caspase 9 (Fig. 5b) and caspase 3 (Fig. 5c) were observed at 3 h and reached peaks between 12 and 18 h following selenite treatment. These events occurred almost simultaneously, but much later than the event of superoxide production (Fig. 3). As shown in Fig. 6a, selenite treatment significantly decreased the mitochondrial membrane potential in AdEmpty-transduced cells, indicating disruption of polarization of mitochondrial membranes, while AdMnSOD transduction inhibited alteration of mitochondrial membrane potential by selenite. Western blot analysis showed that the protein level of cytochrome c in the cytosol was significantly elevated in AdEmpty-transduced cells following selenite treatment, while selenite treatment only slightly increased the level of cytochrome c in AdMnSOD-transduced cells (Fig. 6b). Chemiluminescence analysis showed that AdMnSOD transduction significantly suppressed activation of caspase 9 in cells following selenite treatment compared to control and AdEmpty (Fig. 7a). Meanwhile, AdMnSOD transduction also slightly decreased caspase 9 activity in cells without selenite treatment compared to control and AdEmpty. AdMnSOD transduction significantly decreased caspase 3 activity in cells without selenite treatment and suppressed caspase 3 activation induced by selenite treatment compared to AdEmpty-transduced cells (Fig. 7b). These results demonstrate that selenite induces apoptosis by damaging mitochondria via superoxide-mediated damage with subsequent mitochondrial membrane depolarization, cytochrome c release into the cytosol, and activation of caspases 9 and 3.
Fig. 5.
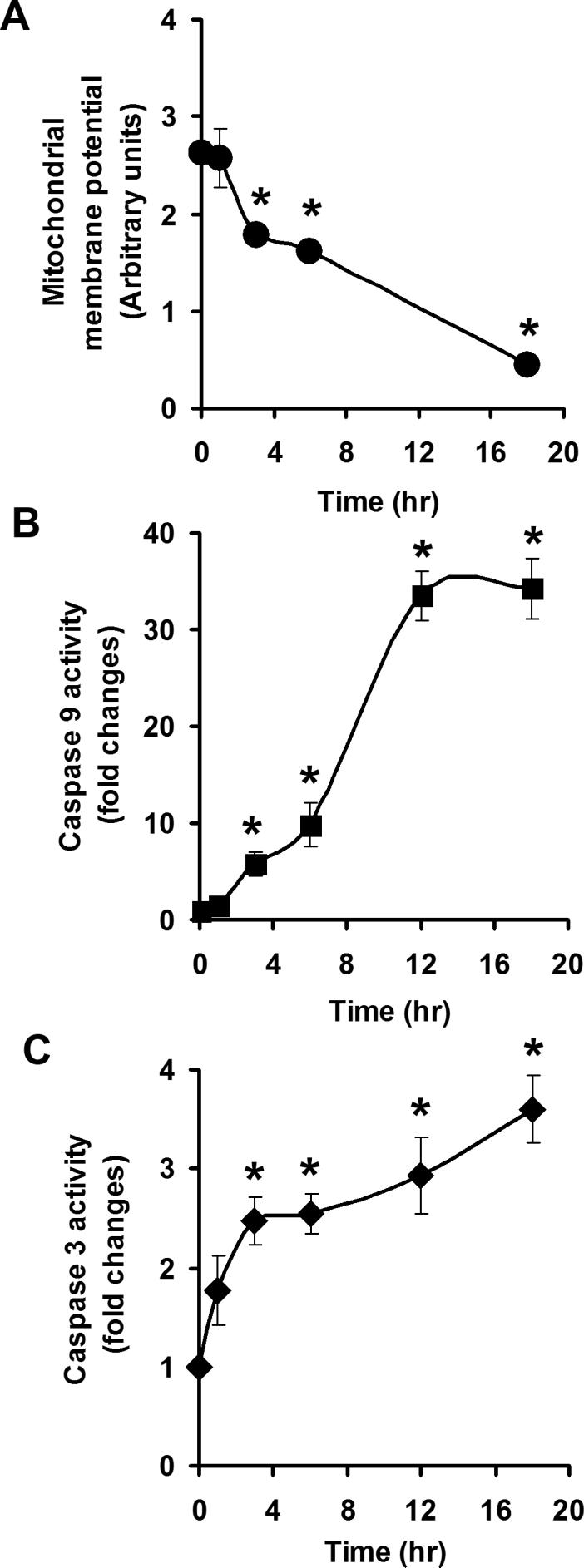
Effects of selenite on the mitochondrial membrane potential and caspases 9 and 3. a Fluorescence analysis of mitochondrial membrane potential. b Chemiluminescence analysis of caspase 9 activity. c Fluorescence analysis of caspase 3 activity. LNCaP cells were treated with 2.5 μM selenite for the times indicated. The mitochondrial membrane potential was measured using a flow cytometer. Casapases 9 and 3 were measured using a luminometer or a fluorescence microplate reader, respectively. Data are presented as means ± SD (n = 3). * P < 0.05 compared to cells without selenite treatment (time zero).
Fig. 6.

Protection of mitochondrial damage from selenite by overexpression of MnSOD in LNCaP cells. a Fluorescence analysis of the mitochondrial membrane potential by flow cytometry. Cells were transduced with 50 MOI AdEmpty or AdMnSOD for 48 hr and then treated with 2.5 μM selenite for 18 h. * P < 0.05 compared with AdEmpty control and both AdMnSOD control and selenite. b Western blot analysis of cytochrome c release into the cytosol. Cells were transduced with 50 MOI AdEmpty or AdMnSOD for 48 h and then treated with 2.5 μM selenite for 18 h.
Fig. 7.

Inhibition of activation of caspases 3 and 9 by selenite by overexpression of MnSOD in LNCaP cells. a Chemiluminescence analysis of caspase 9 activity. Cells were transduced with 50 MOI AdEmpty or AdMnSOD for 48 h and then treated with 2.5 μM selenite for 18 h. Twenty micrograms of total protein from LNCaP cells with or without AdEmpty or AdMnSOD transduction were mixed with the caspase-9 analysis reagent in 96-well plates. Chemiluminescence was measured using a luminometer. Each bar represents the average of three wells. Data are presented as means ± SD of three independent experiments. * P < 0.05 compared with cells without selenite treatment and AdMnSOD with selenite treatment. b Fluorescence analysis of caspase 3 activity. Cells were transduced with 50 MOI AdEmpty or AdMnSOD for 48 h and then treated with 2.5 μM selenite for 18 h. Fluorescence was measured using a fluorescence microplate reader. Data are presented as means ± SD of three independent experiments. * P < 0.05 compared with AdEmpty without selenite. ** P < 0.05 compared with AdEmpty without selenite and AdMnSOD with selenite.
Discussion
In this study, we document that selenite treatment produced superoxide and induced apoptosis in human prostate cancer cells, which were suppressed by overexpression of MnSOD, but not by overexpression of CuZnSOD, CAT, or GPx1. Selenite treatment resulted in a decrease in mitochondrial membrane potential, cytochrome c release into the cytosol, activation of caspases 9 and 3, in which all measurements were inhibited by the overexpression of only MnSOD.
Studies suggest that induction of apoptosis may be the key event for cancer prevention by Se and prooxidant effects of ROS produced by some Se compounds may be responsible for induction of apoptosis [11, 16]. Studies have demonstrated that different chemical forms of Se compounds have different anticancer efficacy, and Se compounds with superoxide production capability usually have a better anticancer activity [11, 12], suggesting that the subtoxic yet prooxidative effects of these Se compounds are, at least in part, responsible for anticancer activity of Se. It is known that some Se compounds catalytically produce ROS, particularly superoxide, in the presence of glutathione [11, 33]. Certain metabolites of Se compounds, such as the selenide anion (RSe−) and hydrogen selenide (H2Se), can be oxidized by O2 to produce superoxide through a redox cycling process [11, 34]. Importantly, this redox cycling process produces significant amounts of superoxide from low to moderate concentrations of Se over time.
It has been reported that selenite-induced cell death or apoptosis is inhibited by SOD mimics or by stable overexpression of MnSOD in several types of cancer cells [13, 14, 17-20, 26]. It has been shown that cancer cells usually have lower levels of MnSOD than their normal counterparts [35]. Recent studies demonstrated that normal prostatic epithelial cells had higher levels of MnSOD and lower sensitivity to selenite compared to prostate cancer cells [36, 37]. These observations suggest that high levels of MnSOD may protect normal cells against superoxide generated apoptosis by Se. Studies also showed that selenite enhanced cancer cell killing by radiation, suggesting a synergism of prooxidant effects of Se and radiation in cancer cells [38, 39]. These data support the notion that prooxidant effects of superoxide and other ROS are responsible for apoptosis induced by certain Se compounds. Se may selectively target cancer cells in vivo because they usually have lower levels of MnSOD than normal cells.
To protect against the damaging effects of ROS, cells possess several AEs including SODs, GPxs, and CAT. MnSOD and CuZnSOD are the two major intracellular AEs reacting with superoxide. A major function of MnSOD is to protect cells against oxidative stress by converting superoxide into hydrogen peroxide in mitochondria, while CuZnSOD mainly protects against superoxide damage in the cytosol and lysosomes, although smaller amounts of CuZnSOD are also present in the mitochondrial intermembrane space and the nucleus. GPx and CAT are AEs protecting against H2O2 cytotoxicity by converting H2O2 into O2 and H2O. Our current study demonstrated that only overexpression of MnSOD protected against cell death induced by selenite, indicating that superoxide generated from selenite damages mitochondria and subsequently triggers apoptosis of prostate cancer cells. Overexpression of CuZnSOD had no effect on superoxide production and cell death by selenite treatment, suggesting that selenite may produce superoxide mainly in or on the surface of mitochondria. Overexpression of GPx1 or CAT had no effect on selenite-induced cell death, indicating that H2O2 is not the primary ROS responsible for selenite-induced apoptosis in prostate cancer cells. Mitochondria are the major site of intracellular ROS production and also play an important role in apoptosis in cells [40, 41]. An increase in superoxide production during apoptosis has been shown to be a direct consequence of increased mitochondrial membrane permeability and release of cytochrome c into the cytosol. After release from mitochondria into the cytosol, cytochrome c together with apaf-1 activates caspase 9 and subsequently caspase 3 to trigger the apoptotic cascade. Our study demonstrates that selenite treatment induced cancer cell apoptosis with sequential events of elevation of superoxide, a decrease in the mitochondrial membrane potential, release of cytochrome c, and activation of caspases 9 and 3. Our results are in agreement with those observed by others [42]. These results indicate selenite-induced apoptosis is superoxide-mediated via the mitochondrial apoptotic pathway.
Previous studies have used synthetic SOD mimics, N-acetyl-cysteine, or buthionine sulfoximine to assess the involvement of superoxide or other ROS in the prooxidant effects of Se compounds in cancer cells [13, 17, 19, 20, 26]. However, the specificity and subcellular effects of these approaches are difficult to determine. In a previous study, we demonstrated that stable overexpression of MnSOD protected against cell death induced by selenite in the RWPE-2 human prostate cancer cell line [18]. However, this experimental approach using stable overexpression of MnSOD in cells has potential pitfalls arising from clonal selection and adaptation of cells. Also, there are no previous studies documenting whether overexpression of other AEs, such as CuZnSOD, GPx, and CAT, would protect against Se cytotoxicity. In the current study, we used adenoviral constructs to overexpress MnSOD in LNCaP cells to verify the results of our previous study in which plasmid vectors were used to stably overexpress MnSOD. In addition, we also overexpressed CuZnSOD, GPx1, and CAT using adenoviral constructs to assess whether these other AEs would have any impact on the cytotoxicity of selenite. Our study demonstrates that only overexpression of MnSOD suppressed selenite-induced superoxide production, apoptosis, and other apoptotic events. Our results indicate that superoxide production by selenite treatment is mainly in mitochondria and mitochondria are the targets that produce apoptosis. The results are consistent with observations from previous studies [15, 17-20, 25, 26] and more precisely define that superoxide is the ROS targeting mitochondria to trigger apoptosis by redox Se compounds.
Studies have also shown that Se can induce cell death by mitochondrial-independent apoptotic pathways, endoplasmic reticulum stress, autophagy, or necrosis [11, 25, 43, 44]. Our results show that the cell death could be induced by higher levels of selenite treatment than just apoptotic levels of treatment, suggesting that other mechanisms may also be involved in selenite-induced cell death. Our results clearly demonstrate that apoptosis induced by selenite is, at least in part, superoxide-mediated via the mitochondrial pathway, which may contribute to chemoprevention of prostate cancer.
In conclusion, we have demonstrated that selenite induced cell death in LNCaP prostate cancer cells in association with superoxide production and apoptosis. Superoxide production was mainly in or adjacent to mitochondria and triggered the mitochondrial pathway of apoptosis. Only overexpression of MnSOD suppressed selenite-induced superoxide production and apoptosis. Our study suggests that levels of MnSOD in prostate and other cancer cells may influence the efficacy of Se in supplementation in cancer chemoprevention. Since cancer cells usually have lower levels of MnSOD, they should be more sensitive to Se than their normal cell counterparts. Therefore, in cancer prevention, Se may be found to selectively induce apoptosis of cancer cells without causing significant damage to normal cells.
Acknowledgments
We thank Dr. Jeanne Bourdeau-Heller for her technical assistance. This work was funded, in part, by grants from the National Cancer Institute (CA114281) and the Office of Research and Development, Biomedical Laboratory Research and Development Service, Department of Veterans Affairs.
References
- 1.Combs GF, Jr, Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. 1998;79:179–192. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 2.Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20:1657–1666. doi: 10.1093/carcin/20.9.1657. [DOI] [PubMed] [Google Scholar]
- 3.Ip C. Lessons from basic research in selenium and cancer prevention. J Nutr. 1998;128:1845–1854. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- 4.Clark LC, Combs GF, Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL, Jr, Park HK, Sanders BB, Jr, Smith CL, Taylor JR. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. J Am Med Assoc. 1996;276:1957–1963. [PubMed] [Google Scholar]
- 5.Combs GF. Status of selenium in prostate cancer prevention. Br J Cancer. 2004;91:195–199. doi: 10.1038/sj.bjc.6601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshizawa K, Willett WC, Morris SJ, Stampfer MJ, Spiegelman D, Rimm EB, Giovannucci E. Study of prediagnostic selenium level in toenails and the risk of advanced prostate cancer. J Natl Cancer Inst. 1998;90:1219–1224. doi: 10.1093/jnci/90.16.1219. [DOI] [PubMed] [Google Scholar]
- 7.Li HJ, Stampfer MJ, Giovannucci EL, Morris JS, Willett WC, Gaziano JM, Ma J. A prospective study of plasma selenium levels and prostate cancer risk. J Natl Cancer Inst. 2004;96:696–703. doi: 10.1093/jnci/djh125. [DOI] [PubMed] [Google Scholar]
- 8.Brooks JD, Metter EJ, Chan DW, Sokoll LJ, Landis P, Nelson WG, Muller D, Andres R, Carter HB. Plasma selenium level before diagnosis and the risk of prostate cancer development. J Urol. 2001;166:2034–2038. [PubMed] [Google Scholar]
- 9.Klein EA, Thompson IM, Lippman SM, Goodman PJ, Albanes D, Taylor PR, Coltman C. SELECT: the next prostate cancer prevention trial. J Urol. 2001;166:1311–1315. doi: 10.1016/s0022-5347(05)65759-x. [DOI] [PubMed] [Google Scholar]
- 10.Sinha R, El-Bayoumy K. Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr Cancer Drug Targets. 2004;4:13–28. doi: 10.2174/1568009043481614. [DOI] [PubMed] [Google Scholar]
- 11.Spallholz JE. On the nature of selenium toxicity and carcinostatic activity. Free Radic Biol Med. 1994;17:45–64. doi: 10.1016/0891-5849(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 12.Stewart MS, Spallholz JE, Neldner KH, Pence BC. Selenium compounds have disparate abilities to impose oxidative stress and induce apoptosis. Free Radic Biol Med. 1999;26:42–48. doi: 10.1016/s0891-5849(98)00147-6. [DOI] [PubMed] [Google Scholar]
- 13.Shen HM, Yang CF, Ong CN. Sodium selenite-induced oxidative stress and apoptosis in human hepatoma HepG2 cells. Int J Cancer. 1999;81:820–828. doi: 10.1002/(sici)1097-0215(19990531)81:5<820::aid-ijc25>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 14.Jung U, Zheng X, Yoon SO, Chung AS. Se-methylselenocysteine induces apoptosis mediated by reactive oxygen species in HL-60 cells. Free Radic Biol Med. 2001;31:479–489. doi: 10.1016/s0891-5849(01)00604-9. [DOI] [PubMed] [Google Scholar]
- 15.Kim TS, Yun BY, Kim IY. Induction of the mitochondrial permeability transition by selenium compounds mediated by oxidation of the protein thiol groups and generation of the superoxide. Biochem Pharmacol. 2003;66:2301–2311. doi: 10.1016/j.bcp.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 16.Drake EN. Cancer chemoprevention: selenium as a prooxidant, not an antioxidant. Med Hypotheses. 2006;67:318–322. doi: 10.1016/j.mehy.2006.01.058. [DOI] [PubMed] [Google Scholar]
- 17.Zhong W, Oberley TD. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. 2001;61:7071–7078. [PubMed] [Google Scholar]
- 18.Zhong W, Yan T, Webber MM, Oberley TD. Alteration of cellular phenotype and responses to oxidative stress by manganese superoxide dismutase and a superoxide dismutase mimic in RWPE-2 human prostate adenocarcinoma cells. Antioxid. Redox Signal. 2004;6:513–522. doi: 10.1089/152308604773934279. [DOI] [PubMed] [Google Scholar]
- 19.Zhao R, Xiang N, Domann FE, Zhong W. Expression of p53 enhances selenite-induced superoxide radical production and apoptosis in human prostate cancer cells. Cancer Res. 2006;66:2296–2304. doi: 10.1158/0008-5472.CAN-05-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao R, Domann FE, Zhong W. Apoptosis induced by selenomethionine and methioninase is superoxide mediated and p53 dependent in human prostate cancer cells. Mol Cancer Ther. 2006;5:3275–3284. doi: 10.1158/1535-7163.MCT-06-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson MA, Goulet AC, Jacobs ET, Lance P. Studies into the anticancer effects of selenomethionine against human colon cancer. Ann N Y Acad Sci. 2005;1059:26–32. doi: 10.1196/annals.1339.016. [DOI] [PubMed] [Google Scholar]
- 22.Wei Y, Cao X, Qu Y, Lu J, Xing C, Zheng R. SeO(2) induces apoptosis with down-regulation of Bcl-2 and up-regulation of p53 expression in both immortal human hepatic cell line and hepatoma cell line. Mutat Res. 2001;490:113–121. doi: 10.1016/s1383-5718(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Zuo L, Shen T, Xu CM, Zhang ZN. Induction of apoptosis by sodium selenite in human acute promyelocytic leukemia NB4 cells: involvement of oxidative stress and mitochondria. J Trace Elem Med Biol. 2003;17:19–26. doi: 10.1016/S0946-672X(03)80041-X. [DOI] [PubMed] [Google Scholar]
- 24.Last K, Maharaj L, Perry J, Strauss S, Fitzgibbon J, Lister TA, Joel S. The activity of methylated and non-methylated selenium species in lymphoma cell lines and primary tumours. Ann Oncol. 2006;17:773–779. doi: 10.1093/annonc/mdl004. [DOI] [PubMed] [Google Scholar]
- 25.Hu H, Jiang C, Schuster T, Li GX, Daniel PT, Lü J. Inorganic selenium sensitizes prostate cancer cells to TRAIL-induced apoptosis through superoxide/p53/Bax-mediated activation of mitochondrial pathway. Mol Cancer Ther. 2006;5:1873–1882. doi: 10.1158/1535-7163.MCT-06-0063. [DOI] [PubMed] [Google Scholar]
- 26.Li GX, Hu H, Jiang C, Schuster T, Lu J. Differential involvement of reactive oxygen species in apoptosis induced by two classes of selenium compounds in human prostate cancer cells. Int J Cancer. 2007;120:2034–2043. doi: 10.1002/ijc.22480. [DOI] [PubMed] [Google Scholar]
- 27.Zwacka RM, Dudus L, Epperly MW, Greenberger JS, Engelhardt JF. Redox gene therapy protects human IB-3 lung epithelial cells against ionizing radiation-induced apoptosis. Hum Gene Ther. 1998;9:1381–1386. doi: 10.1089/hum.1998.9.9-1381. [DOI] [PubMed] [Google Scholar]
- 28.Brown MR, Miller FJ, Jr, Li WG, Ellingson AN, Mozena JD, Chatterjee P, Engelhardt JF, Zwacka RM, Oberley LW, Fang X, Spector AA, Weintraub NL. Overexpression of human catalase inhibits proliferation and promotes apoptosis in vascular smooth muscle cells. Circ Res. 1999;85:524–533. doi: 10.1161/01.res.85.6.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q, Sanlioglu S, Li S, Ritchie T, Oberley LW, Engelhardt JF. GPx-1 gene delivery modulates NFkappaB activation following diverse environmental injuries through a specific subunit of the IKK complex. Antioxid Redox Signal. 2001;3:415–432. doi: 10.1089/15230860152409068. [DOI] [PubMed] [Google Scholar]
- 30.Yan T, Li S, Oberley LW, Jiang X. Altered levels of primary antioxidant enzymes in progeria skin fibroblasts. Biochem Biophys Res Commun. 1999;257:163–167. doi: 10.1006/bbrc.1999.0423. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Zhu H, Kuppusamy P, Roubaud V, Zweier JL, Trush MA. Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J Biol Chem. 1998;273:2015–2023. doi: 10.1074/jbc.273.4.2015. [DOI] [PubMed] [Google Scholar]
- 32.Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]
- 33.Spallholz JE, Palace VP, Reid TW. Methioninase and selenomethionine but not Se-methylselenocysteine generate methylselenol and superoxide in an in vitro chemiluminescent assay: Implications for the nutritional carcinostatic activity of selenoamino acids. Biochem Pharmacol. 2004;67:547–554. doi: 10.1016/j.bcp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Chaudiere J, Courtin O, Leclaire J. Glutathione oxidase activity of selenocystamine: a mechanistic study. Arch Biochem Biophys. 1992;296:328–336. doi: 10.1016/0003-9861(92)90580-p. [DOI] [PubMed] [Google Scholar]
- 35.Oberley LW, Beuttner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 36.Husbeck B, Nonn L, Peehl DM, Knox SJ. Tumor-selective killing by selenite in patient-matched pairs of normal and malignant prostate cells. Prostate. 2006;66:218–225. doi: 10.1002/pros.20337. [DOI] [PubMed] [Google Scholar]
- 37.Menter DG, Sabichi AL, Lippman SM. Selenium effects on prostate cell growth. Cancer Epidemiol Biomarker Prev. 2000;9:1171–1182. [PubMed] [Google Scholar]
- 38.Husbeck B, Peehl DM, Knox SJ. Redox modulation of human prostate carcinoma cells by selenite increases radiation-induced cell killing. Free Radic Biol Med. 2005;38:50–57. doi: 10.1016/j.freeradbiomed.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 39.Shin SH, Yoon MJ, Kim M, Kim JI, Lee SJ, Lee YS, Bae S. Enhanced lung cancer cell killing by the combination of selenium and ionizing radiation. Oncol Rep. 2007;17:209–216. [PubMed] [Google Scholar]
- 40.Cai J, Jones DP. Superoxide in apoptosis. Mitochondrial generation triggered cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 41.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 42.Shen HM, Yang CF, Ding WX, Liu J, Ong CN. Superoxide radical-induced apoptotic signaling pathway in selenite-treated HEPG2 cells: mitochondria serve as the main target. Free Radic Biol Med. 2001;30:9–21. doi: 10.1016/s0891-5849(00)00421-4. [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Zhang H, Dong Y, Park YM, Ip C. Endoplasmic reticulum stress signal mediators are targets of selenium action. Cancer Res. 2005;65:9073–9079. doi: 10.1158/0008-5472.CAN-05-2016. [DOI] [PubMed] [Google Scholar]
- 44.Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67:6314–6324. doi: 10.1158/0008-5472.CAN-06-4217. [DOI] [PubMed] [Google Scholar]