

Abstract
To investigate the effects of Trypanosoma cruzi on the mechanical properties of infected host cells, cytoskeletal stiffness and remodeling dynamics were measured in parasite-infected fibroblasts. We find that cell stiffness decreases in a time-dependent fashion in T. cruzi-infected human foreskin fibroblasts without a significant change in the dynamics of cytoskeletal remodeling. In contrast, cells exposed to T. cruzi secreted/released components become significantly stiffer within two hours of exposure and exhibit increased remodeling dynamics. These findings represent the first direct mechanical data to suggest a physical picture in which an intact, stiff, and rapidly remodeling cytoskeleton facilitates early stages of T. cruzi invasion and parasite retention, followed by subsequent softening and disassembly of the cytoskeleton to accommodate intracellular replication of parasites. We further suggest that these changes occur through protein kinase A and inhibition of the Rho/Rho kinase signaling pathway. In the context of tissue infection, changes in host cell mechanics could adversely affect the function of the infected organs, and may play an important role on the pathophysiology of Chagas' disease.
Keywords: cytoskeleton, stiffness, mechanics, infection
Introduction
Trypanosoma cruzi, the causative agent of human Chagas' disease, is an obligate intracellular parasite that replicates in the cytoplasm of infected mammalian cells. T. cruzi invades and replicates within most nucleated cell types in vitro and its presence has been demonstrated in many host tissues in the acute stages of infection (Kirchhoff, 1996). During the chronic stage of infection, parasites persist mainly in cardiac and smooth muscle, causing chronic inflammation, hypertrophy and fibrosis that can eventually lead to heart failure and the serious gastrointestinal disorders that are commonly associated with chronic Chagas' disease.
Host cell invasion by T. cruzi involves multiple interactions with, and effects upon, the host cell that facilitate the steps that are necessary for successful establishment of infection (reviewed in Andrade and Andrews, 2005; Burleigh, 2005; Yoshida, 2006). The trypomastigote form of T. cruzi is a highly motile, non-replicating stage of the parasite that is remarkably adapted for penetration of non-professional phagocytes. Prior to, and during entry, trypomastigotes activate a number of host cell signaling pathways implicated in invasion including those that involve intracellular calcium transients (Scharfstein et al., 2000; Tardieux et al., 1994; Yoshida, 2006), phosphatidylinositol-3 kinases (Chuenkova et al., 2001; Wilkowsky et al., 2001; Woolsey et al., 2003) and cyclic AMP (Rodriguez et al., 1999). The T. cruzi trypomastigote surface is covered with a dense layer of glycosylphosphatidylinositol-(GPI)-anchored proteins, belonging to three major gene families, that can be secreted/released by the parasite into the culture medium (Affranchino et al., 1989) in soluble form or associated with small membrane vesicles or ‘exosomes’ (Goncalves et al., 1991). Shed trypomastigote components present in ‘parasite-conditioned medium’ (PCM) have been implicated in triggering signaling cascades in host cells to facilitate invasion (Yoshida, 2006), stimulation of pro-inflammatory responses (Almeida et al., 2000), activation of anti-apoptotic responses (Chuenkova and PereiraPerrin, 2005) and repression of extracellular matrix gene expression (Unnikrishnan and Burliegh, 2004). Once inside the host cell, trypomastigotes are contained within a tight membrane-delimited vacuole that arises from, or rapidly fuses with, host cell lysosomes (Tardieux et al., 1992; Woolsey et al., 2003). Within this parasitophorous vacuole, a process of parasite differentiation is initiated and parasites begin to egress from the vacuole at ∼8 hours, taking up residence in the host cell cytoplasm (Ley et al., 1990). By 24 hours post-invasion, fully differentiated amastigotes begin to divide in the cytosol once every 12 hours for 3-5 days before the cell is disrupted.
While previous studies using fluorescence imaging have shown a loss of certain cytoskeletal elements at late time points in T. cruzi-infected cells (Melo et al., 2004; Melo et al., 2006; Pereira et al., 1993), the role of the host cell actin cytoskeleton in T. cruzi invasion, and the impact of replicating parasites in the cytoplasm on host cell mechanics is not well understood. Disruption of the cytoskeleton with cytochalasin D was shown to dramatically increase internalization of T. cruzi trypomastigotes into fibroblasts and epithelial cells (Tardieux et al., 1992). Coupled with the observation that transient increases in the host cell cytosolic free calcium concentration, [Ca2+]i triggered by live T. cruzi trypomastigotes or parasite lysates promotes transient rearrangements in actin microfilaments and facilitates entry (Rodriguez et al., 1995; Tardieux et al., 1994), it was proposed that actin depolymerization facilitates lysosome recruitment and fusion during T. cruzi entry. More recently, we have demonstrated that in fact, disruption of the actin cytoskeleton using cytochalasin D abolishes lysosome-mediated entry of T. cruzi and results in failure to retain intracellular parasites (Woolsey and Burleigh, 2004). Upon removal of cytochalasin D, both parasite retention and lysosome fusion with the parasitophorous vacuole are recovered, suggesting that actin polymerization or dynamic actin remodeling are required for successful invasion by this pathogen (Woolsey and Burleigh, 2004). Although these observations make a compelling case that T. cruzi infection profoundly impacts the host cell cytoskeleton at different stages of the intracellular life cycle, modification of the cytoskeleton mechanical properties and its remodeling dynamics have never been measured and therefore remain open questions.
In this study, we have quantified for the first time, the effect of T. cruzi infection on the mechanical properties and the remodeling dynamics of the host cell cytoskeleton. To facilitate interpretation of our findings, infections were carried out in a human dermal fibroblast cell line for which we have knowledge of the effect of T. cruzi on early signaling and gene expression in these cells. Our results demonstrate that initial signaling triggered in fibroblasts by components released by invasive trypomastigotes causes cells to become stiffer with an increase in cytoskeletal remodeling dynamics, correlating with an increase in filamentous actin and supporting previous observations that an intact cytoskeleton is known to be required for the initial steps of parasite invasion (Woolsey and Burleigh, 2004). As the parasite establishes residence in the cytoplasm of its host cell and begins to divide, cytoskeletal disassembly is indicated by the softening of the cell a time-dependent manner, correlating with a decrease in filamentous actin observed at 72 hours post-infection. Furthermore our data suggests that Rho/Rho kinase signaling is repressed in infected cells, possibly through the action of PKA. However, there is no obvious role for myosin light chain suggesting an alternate role for protein kinase A and inhibition of the Rho/Rho kinase signaling pathway.
Materials and Methods
Cell Culture
Human Foreskin Fibroblast (HFF) BJ cells were acquired from the American Tissue Culture Collection and maintained in DMEM + 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine at 37°C in 5% CO2. HFF were plated in glass-bottom 35 mm dishes at a density of 5 × 104 per ml 48 hr prior to the start of the experiment to achieve 95% confluence.
Parasite Infection
Tissue culture-derived T. cruzi trypomastigotes (Y strain) were generated by weekly passage in confluent monolayers of LLcMK2 cells in DMEM containing 2% FBS (DMEM-2) as previously described (Tardieux et al., 1992). Trypomastigotes harvested from cell culture supernatants were washed 3 times in cold DMEM-2, resuspended in warm DMEM-2 at 108 parasites per ml. Confluent monolayers of HFF were infected with 2 ml of parasite suspension for 1 hr and then washed 3 times in DMEM-2. Infection was allowed to proceed for 2, 24, 48 or 72 hr. Parasite-conditioned medium (PCM) was produced by incubation of freshly harvested and washed trypomastigotes in DMEM-2 at 37°C in 5% CO2 for 14-18 hr. Following removal of parasites by centrifugation, the resulting PCM was passed through a 0.2 μm membrane and adjusted to neutral pH.
Fluorescence staining for filamentous actin (F-actin)
Mock and T. cruzi-infected HFF were fixed in 2% paraformaldehyde (PFA) in PBS, washed three times in PBS and incubated for 10 min in 50 mM NH4Cl in PBS. F-actin was stained with Alexafluor 488-Phalloidin (Molecular Probes) in TBS/BSA (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% BSA) containing 0.1% saponin for 30 min followed by extensive washes over 25 min. Coverslips were washed with PBS and mounted in 10% Mowiol containing 2.5% DABCO (1,4-diazobicyclo-2,2,2-octane). Samples were then analyzed using confocal microscopy.
In vivo globular actin (G-actin)/F-actin ratio assay
G-actin/F-actin in vivo assays (Cytoskeleton Inc.) were carried out on HFF cell monolayers prepared and infected as described above and as instructed by the manufacturer. In brief, cells were harvested, disrupted, and filamentous actin was pelleted by centrifugation at 100,000 × g for 30 min at 37°C. The supernatants containing G-actin were removed, and both pellets and supernatants were prepared for western blot analysis. Densitometry was used to determine the relative amount of F-actin in cells following various treatments.
Optical Magnetic Twisting Cytometry
Mechanical properties of HFF were measured using optical magnetic twisting cytometry (OMTC), as described in Fabry et al. (Fabry et al., 2003). OMTC is a micro-rheometer where the living cell is sheared between the culture dish upon which the cell is adhering and a magnetic microbead partially embedded into the cell surface. Briefly, ferrimagnetic microbeads (∼4.5 μm in diameter) were produced in our laboratory, and then coated with a peptide containing the sequence RGD (150 μg ligand per mg of microbeads) by overnight incubation at 4°C in carbonate buffer (pH 9.4). Prior to each measurement, microbeads were added to an individual well and incubated for 20 min, leading to an average of one microbead per living cell. Such RGD-coated microbeads attached to HFF via integrin receptors, mainly through β1 subunits (Hynes, 2002; Puig-de-Morales et al., 2004), and became strongly anchored to the actin cytoskeleton (Fabry et al., 2003; Lenormand et al., 2007a; Puig-de-Morales et al., 2004). The well was then rinsed twice with DMEM + 2% FBS medium at room temperature to remove unbound microbeads, and placed on the stage on an inverted microscope (Leica, DM-IRB). The stage was mounted with a pair of magnetizing coils and a pair of twisting coils. Microbeads were first magnetized horizontally by two consecutive magnetic pulses (∼0.1 T for ∼0.1 ms) using the magnetizing coils. Then a vertical sinusoidal magnetic field, , applied by the twisting coils, induced a mechanical torque on each microbead, and caused both a rotation and a lateral displacement of the microbeads (Mijailovich et al., 2002). The individual displacements of approximately a hundred microbeads were recorded simultaneously using a CCD camera with pixel-clock synchronization (JAI CV-M10, Glostrup, Denmark) attached to the camera side-port of the microscope.
Complex elastic modulus
For each microbead, the complex elastic modulus, g*, was measured from the applied specific torque and the resulting bead displacement,
where f is the frequency of the applied torque, g'(f) is the storage modulus, g"(f) is the loss modulus, T̃ is the Fourier transform of the mechanical torque per microbead volume, d̃ is the Fourier transform of the resulting microbead displacement, and i2 = −1. This complex elastic modulus has dimensions of Pa/nm and can be transformed to the conventional complex elastic modulus, G*(f), (with units of Pa) by a geometric factor, α, through the relationship G*(f) = α g*(f). Assuming 10% of the microbead diameter embedded in a cell 5 μm high sets α to 6.8 μm (Mijailovich et al., 2002). For each single microbead, g* was measured over 4 decades in frequency, from 0.1 Hz to 1 kHz, and we report a median value of g*(f) calculated over a large number of microbeads. This number, n, is usually greater than 200 and is given in figure legends. As shown previously (Fabry et al., 2003), g* was well described by:
| (1) |
with g0 being a scale factor for stiffness, f0 being a scale factor for frequency (f0 is arbitrarily fixed at 1), Γ(.) being the gamma function, and μ being an additive Newtonian viscosity (with dimensions of Pa.s.nm−1). The first term on the right hand side of Eq. (1), known as the structural damping law, describes a relationship between the exponent of the power law, α, and the transition from a Hookean solid-like (α = 0) to a Newtonian liquid-like (α = 1) behavior.
Mean Square Displacement
Cytoskeleton remodeling was inferred from spontaneous displacement of RGD-coated microbeads. Such beads can move spontaneously only if the cytoskeletal structures to which they are attached rearrange; therefore, spontaneous bead motions report ongoing CSK remodeling in space and time (An et al., 2004; Bursac et al 2005; Bursac et al., 2007; Lenormand et al., 2007b). Spontaneous bead motions are measured by identifying the position of each bead (approximately 100 beads per field-of-view) at the rate of 12 frames per second (0.082 seconds per frame) for approximately 5 min. Bead positions are corrected for microscope stage drift; the stage drift is estimated from changes in the mean position of all beads within a field of view. We define the mean square displacement, MSD, as <r2(Δt)> = <(r(t+Δt) − r(t))2>, where r(t) is the bead position at time t, Δt is the time lag, and brackets indicate an average over t and over all beads (Bursac et al 2005; Bursac et al., 2007). In previous studies (An et al., 2004; Bursac et al 2005; Bursac et al., 2007), MSDs were found to vary with Δt as a power law, <r2(Δt)> = D*(Δt/t0)β with β typically greater than 1 for Δt longer than 1 s and t0 arbitrarily fixed at 1 s. We measured the MSD for each single microbead, and we report a median value calculated over a large number of microbeads. This number, n, is usually greater than 200 and is given in figure legends.
Cytoskeleton modulations
The properties of the cytoskeleton were modulated using Jasplakinolide (Jas) and cytochalasin-D (cytD); Jas stabilizes actin filaments whereas CytD disrupts actin filaments. Both drugs were added after bead incubation, and measurements begin 15 min after addition of 1 μM Jas or 2 μM CytD. Jas and CytD were prepared in sterile dimethylsulfoxide (DMSO); on the day of experiments, all drugs were diluted to final concentrations in serum-free media, yielding less than 0.1% DMSO in final volume.
Reagents
Tissue culture reagents and drugs used in this study were obtained from Sigma (St. Louis, MO, USA), with the following exceptions: Trypsin-ETDA solution, which was purchased from Gibco (Grand Island, NY), RGD peptide (Peptide 2000) from Telios Pharmaceuticals (San Diego, CA), and jasplakinolide from CalBiochem (La Jolla, CA). Antibodies to phospho-MLC19-20, MLC, phospho-MYPT1-Thr853, MYPT1 and phospho-PKA substrate were purchased from Cell Signaling Technologies. Antibodies to phospho-MYPT1-Ser695 was obtained from Santa Cruz Biotechnology.
Results
F-actin levels decrease in T. cruzi-infected fibroblasts
To investigate the effect of parasite infection on the host cell actin network, HFF were infected with T. cruzi for 72 hr prior to visualization of F-actin with Alexafluor 488-Phalloidin by confocal microscopy. A striking reduction in the amount of phalloidin stained filments was observed in T. cruzi-infected cells as compared to nearby uninfected cells (Fig. 1A). Applying a more quantitative approach, we assessed the relative levels of globular (G)-actin and filamentous (F)-actin present in fibroblasts infected with T. cruzi or treated with parasite-conditioned medium (PCM). Whereas exposure of cells to PCM caused a significant increase in F-actin content, infection of HFF with T. cruzi resulted in a marked reduction of F-actin (Fig. 1B, C). Jasplakinolide (Jas), a drug that stabilizes F-actin, and cytochalasin D (CytD), which disrupts F-actin, were used as controls and produced the expected increase (Jas) or decrease (CytD) in relative F-actin levels (Fig. 1B). Densitometric analysis of western blots from four independent experiments demonstrates statistically significant changes in F-actin in response to each treatment (Fig. 1C).
Figure 1. Reduction in the F-actin content in host fibroblasts.
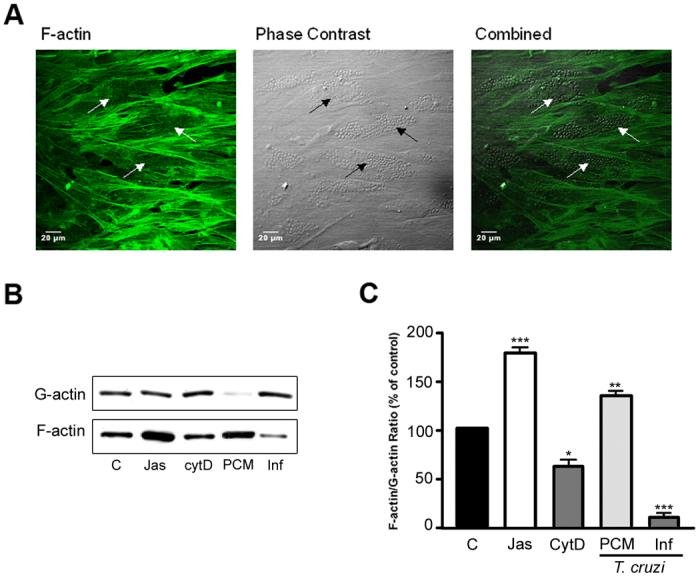
A. Alexafluor 488-phalloidin staining of T. cruzi-infected HFF at 72 hours post-infection (left panel). Parasitized cells, as shown in the phase contrast image (center panel), lacked the actin filaments present in uninfected cells. The arrows highlight three infected cells. B. Biochemical quantification of F-actin to G-actin ratio in HFF treated with Jasplakinolide (Jas), cytochalasin D (CytD) or following T. cruzi infection for 72 hours. C. Quantification of relative F-actin content (% of total actin) following infection. Results from 4 independent experiments were quantified using densitometry of western blots revealing the reduction in F-actin in T. cruzi-infected cells (*p <0.05, **p<0.01, ***p<0.005 Student's t test).
T. cruzi causes a time-dependent softening of the host cell cytoskeleton
The reduction of F-actin in T. cruzi infected cells, as well as the increase F-actin content in PCM treated cells, predicts that these conditions would have a measurable impact on the mechanical properties of the host cell. To test this prediction, we measured the storage modulus, g′(f), and the loss modulus, g″(f), of the host cell cytoskeleton at different time points after infection (2, 24, 48, and 72 hr), and after 2 hr incubation with the secreted/released parasite fraction (parasite-conditioned medium; PCM). In each case, g′ and g″ were well described using the structural damping equation, Eq. (1) (Fig. 2). For each measurement, g′ increased weakly with frequency and followed a power law, g′ ∼ fα, with an exponent, α, comprised between 0.1 and 0.3. Below 30 Hz, g″ increased with frequency with the same power law exponent. However, g″ showed a stronger frequency dependence at higher frequencies approaching a frequency dependence of 1, represented by the additive Newtonian viscosity, μ, in Eq. (1). When HFF were infected by T. cruzi, the mechanical properties of the cytoskeleton decreased over the duration of infection. The scaling stiffness g0 regularly decreased, and was divided by a factor of two within 72 hr of infection (Fig. 3A). Furthermore, the change in stiffness with infection time was well fitted by a linear relation. With the infection time, the power law exponent α decreased as well from 0.17 to 0.14, but the decrease did not follow a simple linear relationship (Fig. 3B). The Newtonian viscosity μ regularly decreased with the infection time, and also decreased by a factor of two within 72 hr of infection; furthermore μ varied linearly with the infection time (Fig. 3C). Interestingly, when HFF were incubated with PCM for two hours, the stiffness g0 and the Newtonian viscosity μ increased by ∼50%, and the power law exponent α decreased slightly (Fig. 3). A similar effect was observed when HFF were treated with PCM for 16 hours (data not shown).
Figure 2. Storage modulus g′(f) and loss modulus g″(f) under baseline condition vs. frequency.
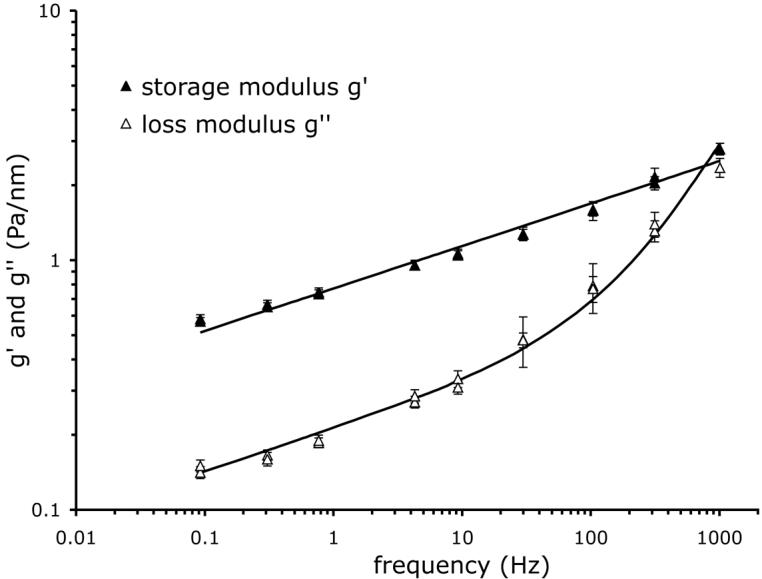
Solid line: best fit by Eq. (1). Median values over n = 1381 measurements.
Figure 3. Changes in mechanical properties in fibroblasts infected with T. cruzi or treated with PCM.
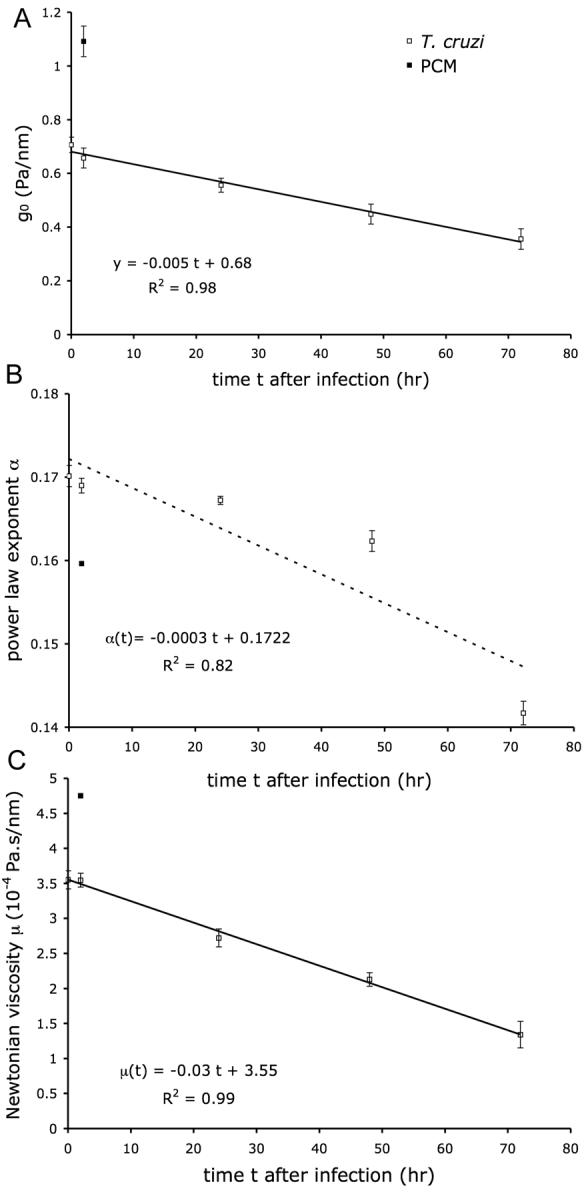
T. cruzi-infected HFF at 2, 24, 48 and 72 hr post-infection (open symbols) or PCM-treated cells after 2 hr (closed symbols) were subjected to OMTC and analyzed as follows: A. g0 vs. time. Solid line: best fit by linear function of infection data. B. Power law exponent α vs. time. Dashed line: best fit by linear function of infection data. C. Newtonian viscosity μ vs. time. Solid line: best fit by linear function of infection data. All three parameters are obtained from fit of g′ and g″ with Eq. (1). Numbers of measurements are: for PCM data 582, and for infection data 1381, 681, 1126, 616, and 572 for 0, 2, 24, 48, and 72 hr respectively.
T. cruzi infection inhibits jasplakinolide-mediated cell stiffening
To further investigate the dynamic state of the actin cytoskeleton in T. cruzi-infected cells, fibroblasts were treated with jasplakinolide (Jas) or cytochalasin-D (cytD) and changes in stiffness g0, the power law exponent α, and the Newtonian viscosity μ were measured. In uninfected controls cells, treatment with Jas caused the stiffness g0 to increase by 35%, where the power law exponent was slightly decreased from 0.159 to 0.155, and the Newtonian viscosity was increased by 37% (Fig. 4). As compared to mock-infected controls (media treatment alone), T. cruzi infection caused a 50% decrease in stiffness g0, α, and μ, which could not be overcome by the addition of Jas (Fig. 4A, C). In contrast, treatment of infected cells with CytD resulted in a further reduction of stiffness g0 (Fig. 4A) and infection did not inhibit CytD dependent increases in the power law exponent α (Fig. 4B). Collectively, these findings indicate that similar to CytD, intracellular infection with T. cruzi causes a decrease in actin polymerization and decreased cell stiffness. Given that this can be further reduced by the addition of CytD suggests that actin-dependent structures that contribute to stiffness are not completely disrupted in infected cells. In addition, the inability to overcome the parasite-imposed reduction in cell stiffness by treatment with the actin stabilizing drug jasplakinolide suggests that the cells are no longer able to form new or crosslinked actin filaments.
Figure 4. Changes in mechanical properties after challenge with jasplakinolide and cytochalasinD.
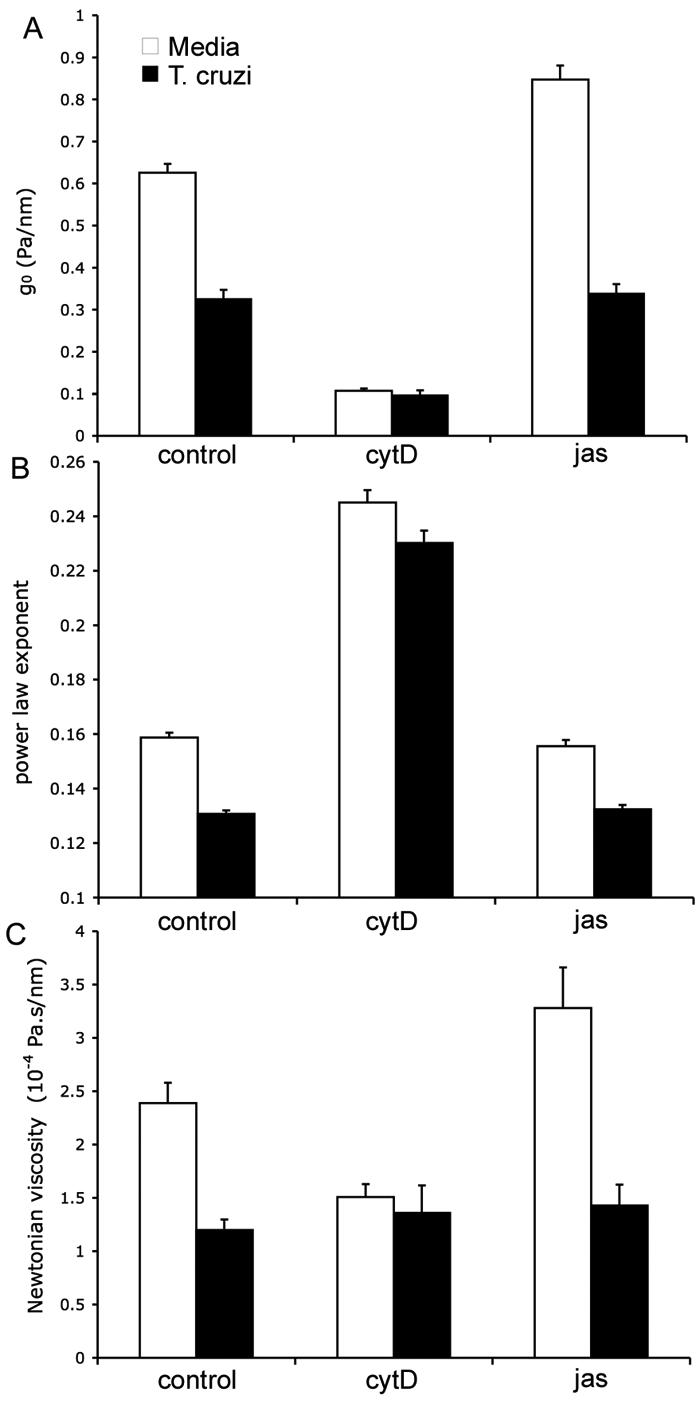
Mock- and T. cruzi-infected HFF (72 hr) were treated with Jasplakinolide (Jas) or cytochalasin-D (CytD) and analyzed for (A) stiffness, g0, (B) power law exponent, α, and (C) Newtonian viscosity, μ, are obtained from fit with Eq. (1). Error bars are standard errors. Numbers of measurements taken are between 200 and 800.
T. cruzi-infected cells showed no measurable change in cytoskeletal remodeling dynamics
We then assessed the changes in remodeling dynamics of the cytoskeleton of the host cells induced by infection with T. cruzi. This was evaluated by measuring the range of spontaneous motion of a microbead firmly attached to the host cell cytoskeleton as described in the methods section; such a microbead can move spontaneously only if the cytoskeletal structures to which it is attached rearrange. We measured the MSD under baseline condition, after 2 hr incubation with PCM, and at 2, 24, 48, and 72 hr after infection with T. cruzi (Fig. 5). In each case, for time lags, Δt, larger than 10 s, the MSD followed a power law, <r2(Δt)> = D0 Δtβ, with β typically comprised between 1.4 and 1.6. After incubation with PCM, D0 increased by a factor of two, and the exponent β slightly increased from 1.56 to 1.62. After 2 hr of infection with T. cruzi, the MSD was unchanged compared to baseline. Only after 24 hr did the MSD begin to decrease, and then went back to the baseline value after 72 hr (Fig. 5 inset), however these changes were not statistically significant.
Figure 5. Changes in mean square displacements, MSD, vs. time lag, Δt, in T. cruzi infected cells.

HFF, at different time points after infection (0, 2, 24, 48, and 72 hr) and after 2 hr incubation with PCM were analyzed for changes in MSD. Inset: MSD taken at Δt = 100 s vs. time after infection; error bars are standard errors. Baseline, n = 1623; PCM 2h, n = 876; TC 2h, n = 727; TC 24h, n = 1131; TC 48h, n = 607; TC 72h, n = 622.
T. cruzi infection fails to alter myosin light chain phosphorylation
The phosphorylation state of myosin light chain (MLC) is often used as a surrogate for cell stiffness, where The level of MLC phosphorylation in cells is regulated by opposing kinase and phosphatase activities (Martens and Radmacher, 2008; Ramachandran et al. 2008). Activation of myosin light chain kinase (MLCK) or the RhoA/Rho kinase pathway increase MLC phosphorylation and increase cell stiffness. The effects of Rho kinase are amplified by the ability of this kinase to phosphorylate the myosin light chain phosphatase regulatory subunit, MYPT1, on Thr696 and Thr853 which inhibits myosin phosphatase activity. Counteracting the Rho kinase-dependent phosphorylation and inhibition of MYPT1, phosphorylation of this protein at Ser695 by cAMP-dependent protein kinase (PKA) has been shown to block Rho kinase dependent phosphorylation of Thr696 and Thr853, resulting in activation of myosin phosphatase activity and cell softening (Murthy, 2006). To investigate the potential role of MLC in T. cruzi-induced cell softening, antibodies to phospho-MLC19-20, MYPT1-Thr853, and MYPT1-Ser695 were used to probe western blots of T. cruzi-infected cell lysates (Fig. 6A, B). Here we found that T. cruzi infection had little effect on the phosphorylation state of MLC, despite a slight reduction observed at 72 hours post-infection. In contrast, we observed a dramatic and time-dependent reduction in MYPT1-Thr853 phosphorylation (the Rho kinase phosphorylation site) and a concomitant increase in the level of MYPT1-Ser695 (PKA phosphorylation site) at 72 hours post-infection (Fig. 6 A, B). These findings suggest that Rho kinase activity is reduced in T. cruzi-infected cells and/or activation of cAMP/PKA signaling, which opposes RhoA-dependent increases in cell stiffness, is activated. Probing T. cruzi-infected cell lysates with a phospho-PKA substrate antibody revealed differential phosphorylation of a high molecular weight band (>150kDa) in T. cruzi infected cells (Fig. 6C), which is absent in both uninfected controls and in parasite lysate alone (Fig. 6C, T.c.). A similar band is observed at early time points of infection (Fig. 6D) but not in lysates of PCM-treated cells (Fig. 6D, PCM). Collectively, these data suggest that T. cruzi infection results in decreased Rho kinase-dependent phosphorylation of a myosin phosphatase inhibition site on MYPT1, which may be a result of increased PKA-dependent signaling as observed in other systems (Murthy, 2006). However, the expected outcome of these signaling events, i.e. decreased in MLC phosphorylation, was not observed. It is unlikely that parasite-driven changes in myosin activity in the cell accounts for the dramatic decrease in stiffness measured in T. cruzi-infected cells.
Figure 6. T. cruzi elicits changes in Rho kinase and PKA-dependent substrate phosphorylation.

Lysates prepared from mock- or T. cruzi-infected HFF (C or Inf) were blotted and probed with antibodies to phospho-MLC(19-20), phospho-MYPT1-Thr853, phospho-MYPT1-Ser695, stripped and reprobed with MLC or MYPT1 as appropriate (A) and densitometric analysis of the bands was carried out to determine the average relative phosphorylation levels from 2 independent experiments (B). Mock- and T. cruzi-infected (C; Inf) or PCM-treated (PCM) HFF lysates were subjected to western blotting and probed with antiphospho-PKA substrate antibody (C,D). A mixture of isolated parasites 2 × 106trypomastigote/amastigote mix was included as a control for phosphorylated parasite proteins (Tc). Arrow indicates differentially phosphorylated protein band that is not present in isolated parasites.
Discussion
The internal stiffness of a cell often correlates with the stiffness of the underlying substrate as well as extent of cell-to-cell contacts (Doornaert et al., 2003; Solon et al., 2007; Yeung et al., 2005). These interactions are largely governed by integrin-mediated contacts between the cortical actin cytoskeleton and extracellular matrix (Brakebusch and Fassler, 2003; Coppolino and Dedhar, 2000; Juliano, 2002). Changes in cell stiffness, which can be triggered directly by changes in mechanical force as well as through the action of cytokines (Goldberg et al., 2007; Leung et al., 2007; Qi et al., 2006), profoundly influence morphology, proliferation and other phenotypic properties of a cell (Atance et al., 2004; Karamichos et al., 2007; Khatiwala et al., 2006). Here, we have investigated the effect of pathogen infection on host cell stiffness and on the dynamics of actin remodeling at different stages of the infection process. We have found that intracellular infection of human dermal fibroblasts with T. cruzi leads to a progressive loss of cell stiffness, with no measurable change in cytoskeletal remodeling dynamics. In contrast, brief exposure of fibroblasts to secreted/shed parasite components (PCM) results in increased cytoskeletal remodeling with an overall increase in cell stiffness.
Cell entry by T. cruzi trypomastigotes is facilitated by activation of host cell signaling pathways involving increases in host cell cytosolic free [Ca2+] (Tardieux et al., 1994), cAMP levels (Caler et al., 2000) and stimulation of class I phosphatidyl-3-kinases (Woolsey et al., 2003). The [Ca2+]i-transients triggered locally by invading trypomastigotes promote transient rearrangements in the cortical actin cytoskeleton (Rodriguez et al., 1995) and targeted lysosome exocytosis at the parasite attachment site for formation of the parasitophorous vacuole (Tardieux et al., 1994). Because cytochalasin D pretreatment of cells significantly enhances T. cruzi invasion, it was originally assumed that actin depolymerization is required for efficient entry via the lysosome-dependent pathway (Tardieux et al., 1992). However, depolymerization of actin prevents fusion of lysosomes with parasite-containing plasma membrane invaginations (Woolsey and Burleigh, 2004). Furthermore, it is clear that a dynamic actin cytoskeleton is required for parasite retention in infected cells (Woolsey and Burleigh, 2004). Considering that the parasites shed soluble antigen and small vesicles prior to and during the invasion event, it is reasonable to speculate that the increase in actin remodeling that we observed following treatment with PCM reflects early events in the invasion process that facilitate retention (Woolsey and Burleigh, 2004). While a similar increase in actin remodeling and cell stiffness is not observed following a 2 hr exposure to live T. cruzi trypomastigotes, this is likely due to the decrease in the effective concentration of shed material, localized signaling events at the invasion site (Caler et al 2000), and the asynchronous nature of the invasion process. Over the course of the experimental infection we did observe a temporary and minor reduction in the rate of actin remodeling, which is fully recovered by 72 hours of infection, howver, none of these changes reaches the level of statistical significance. In contrast, the effect of live intracellular parasites on stiffness parameters is much more pronounced. Infection of the host cells with T. cruzi results in a linear decrease in stiffness over the course of infection. In contrast, treatment of host cells with PCM results in a sharp increase in cell stiffness, which along with the increase in the rate of actin remodeling, may play a role in the retention of motile parasites within the host cell. The observed stiffening is likely due to a direct signaling effect of a PCM component, rather than a secondary effect due to host cell cytokine secretion, given that the effect is observed immediately after PCM addition (data not shown).
Presently, the mechanistic basis for the parasite-dependent reduction in F-actin content and cell stiffness is not known. Given the link between the actin-myosin system and cell stiffness (Martens and Radmacher, 2008) we investigated the possibility that the presence of intracellular parasites affects the phosphorylation status of myosin light chain (MLC). No detectable change in MLC phosphorylation was observed in T. cruzi-infected cells at 24 and 48 hours post-infection and at 72 hours post-infection, a marginal reduction (∼15%) in MLC phosphorylation was observed. Based on these findings, it is unlikely that changes in myosin activity are the driving force behind T. cruzi-dependent changes in cell stiffness. Intriguingly, however, we observed a pronounced and time-dependent reduction in the phosphorylation of Thr853 on MYPT1, indicative of Rho kinase inhibition. Together with Thr696, these residues are phosphorylated by Rho kinase which serves as an inhibitory signal for myosin phosphatase activity. This finding suggests that T. cruzi interferes with the Rho/Rho-kinase signaling pathway in infected cells. One possible mechanism is through the activation of the cAMP/PKA signaling pathway, which has been shown to inhibit RhoA activity directly by phosphorylation on Ser188 (Lang et al. 1996) and through competitive phosphorylation of MYPT1 at Ser695. Increased phosphorylation of Ser695, a PKA phosphorylation site in MYPT1, at 72 hours post-infection is indicative of an increased activation of cAMP-dependent signaling pathways in T. cruzi-infected cells, consistent with previous findings in NRK cells (Rodriguez et al. 1999). This is supported by the observation of a differentially phosphorylated PKA substrate in lysates of T. cruzi-infected cells as early as 30 minutes post-invasion. However, given the number of T. cruzi proteins that are recognized by the phospho-PKA substrate antibody, we cannot rule out the possibility that the differentially phosphorylated band in lysates of T. cruzi –infected cells represents an amastigote protein that is differentially phosphorylated in the context of intracellular infection and is therefore absent in the parasite control lane. Regardless, while the inhibitory effects of infection on Rho kinase-dependent phosphorylation of MYPT1 are substantial, these changes do not appear to impact MLC phosphorylation. Therefore, we can only conclude that inhibition of host cell myosin is not the primary cause of cell softening that occurs in response to T. cruzi infection. More likely, a combination of parasite-dependent effects on the expression of key actin regulatory proteins and signaling status in cells, caused by the physical impact of parasite replication within the host cell cytoplasm and/or cytokine-mediated effects, operate to promote decreases in cell stiffness. In support of this notion, results from recent DNA microarray experiments which have examined changes in host cell gene expression in T. cruzi infected HFF at 24 hours post-infection, reveal decreases in transcript abundance for a number of cytoskeletal genes including many involved in the Rho pathway and FERM-domain containing proteins that bind Arp3 and promote actin polymerization downstream of integrins (Costales, Daily, Reddy and Burleigh, manuscript in preparation). We also observe that infected cells become refractory to jasplakinolide treatment after 72 h of parasite infection, which suggests that actin nucleation in these cells is impaired. A reduction in the expression of cytoskeletal genes, including those involved in actin polymerization would explain these observations and further argue that the mechanism of cell softening is multifactorial.
While the potential benefit of these cellular events for the parasite, if any, is unknown, breakdown of the constraining cytoskeleton and softening of the cell could provide a more permissive environment for parasite replication or may facilitate the eventual egress of parasites from the cell. In the context of tissue infection, changes in the mechanical properties of infected host cell is likely to adversely affect the function of infected organs such as the heart and could contribute to the pathophysiology of Chagas' disease. Clearly, a better understanding of the mechanisms involved in these processes is of great interest.
Acknowledgements
The authors are grateful to Colby Wells for maintenance of T. cruzi cultures and thank Emil Millet for producing the magnetic beads and for his help in data processing.
Contract Grant Sponsor: National Institutes of Health; Contract Grant #: R01 HL073227 (BAB); HL065960 and HL084224 (JJF).
Contract Grant Sponsor: Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Diseases (BAB).
Literature Cited
- Affranchino JL, Ibanez CF, Luquetti AO, Rassi A, Reyes MB, Macina RA, Aslund L, Pettersson U, Frasch AC. Identification of a Trypanosoma cruzi antigen that is shed during the acute phase of Chagas' disease. Mol Biochem Parasitol. 1989;34(3):221–228. doi: 10.1016/0166-6851(89)90050-9. [DOI] [PubMed] [Google Scholar]
- Almeida IC, Camargo MM, Procopio DO, Silva LS, Mehlert A, Travassos LR, Gazzinelli RT, Ferguson MAJ. Highly Purified Glycosylphosphatidylinositols from Trypanosoma cruzi are Potent Proinflammatory Agents. EMBO Journal. 2000;19(7):1476–1485. doi: 10.1093/emboj/19.7.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SS, Fabry B, Mellema M, Bursac P, Gerthoffer WT, Kayyali US, Gaestel M, Shore SA, Fredberg JJ. Role of heat shock protein 27 in cytoskeletal remodeling of the airway smooth muscle cell. J Appl Physiol. 2004;96(5):1701–1713. doi: 10.1152/japplphysiol.01129.2003. [DOI] [PubMed] [Google Scholar]
- Andrade LO, Andrews NW. The Trypanosoma cruzi-host-cell interplay: location, invasion, retention. Nat Rev Microbiol. 2005;3(10):819–823. doi: 10.1038/nrmicro1249. [DOI] [PubMed] [Google Scholar]
- Atance J, Yost MJ, Carver W. Influence of the extracellular matrix on the regulation of cardiac fibroblast behavior by mechanical stretch. J Cell Physiol. 2004;200(3):377–386. doi: 10.1002/jcp.20034. [DOI] [PubMed] [Google Scholar]
- Brakebusch C, Fassler R. The integrin-actin connection, an eternal love affair. EMBO Journal. 2003;22:2324–2333. doi: 10.1093/emboj/cdg245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh BA. Host cell signaling and Trypanosoma cruzi invasion: do all roads lead to lysosomes? Sci STKE. 2005;2005(293):pe36. doi: 10.1126/stke.2932005pe36. [DOI] [PubMed] [Google Scholar]
- Bursac P, Fabry B, Trepat X, Lenormand G, Butler JP, Wang N, Fredberg JJ, An SS. Cytoskeleton dynamics: fluctuations within the network. Biochem Biophys Res Commun. 2007;355(2):324–330. doi: 10.1016/j.bbrc.2007.01.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursac P, Lenormand G, Fabry B, Oliver M, Weitz DA, Viasnoff V, Butler JP, Fredberg JJ. Cytoskeletal remodelling and slow dynamics in the living cell. Nat Mater. 2005;4(7):557–561. doi: 10.1038/nmat1404. [DOI] [PubMed] [Google Scholar]
- Caler EV, Morty RE, Burleigh BA, Andrews NW. Dual role of signaling pathways leading to Ca(2+) and cyclic AMP elevation in host cell invasion by Trypanosoma cruzi. Infect Immun. 2000;68(12):6602–6610. doi: 10.1128/iai.68.12.6602-6610.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenkova MV, Furnari FB, Cavenee WK, Pereira MA. Trypansoma cruzi trans-sialidase: A Potent and Specific Survival Factor for Human Schwann Cells by Means of Phosphatidylinositol 3-kinase/Akt Signaling. Proceedings of the National Academy of Sciences USA. 2001;98(17):9936–9941. doi: 10.1073/pnas.161298398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenkova MV, PereiraPerrin M. A synthetic peptide modeled on PDNF, Chagas' disease parasite neurotrophic factor, promotes survival and differentiation of neuronal cells through TrkA receptor. Biochemistry. 2005;44(48):15685–15694. doi: 10.1021/bi0512039. [DOI] [PubMed] [Google Scholar]
- Coppolino MG, Dedhar S. Bi-directional signal transduction by integrin receptors. The International Journal of Biochemistry & Cell Biology. 2000;32(2):171–188. doi: 10.1016/s1357-2725(99)00043-6. [DOI] [PubMed] [Google Scholar]
- de Avalos SV, Blader IJ, Fisher M, Boothroyd JC, Burleigh BA. Immediate/Early Response to Trypanosoma cruzi Infection Involves Minimal Modulation of Host Cell Transcription. Journal of Biological Chemistry. 2002;277(1):639–644. doi: 10.1074/jbc.M109037200. [DOI] [PubMed] [Google Scholar]
- Doornaert B, Leblond V, Planus E, Galiacy S, Laurent VM, Gras G, Isabey D, Lafuma C. Time course of actin cytoskeleton stiffness and matrix adhesion molecules in human bronchial epithelial cell cultures. Exp Cell Res. 2003;287(2):199–208. doi: 10.1016/s0014-4827(03)00114-9. [DOI] [PubMed] [Google Scholar]
- Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Taback NA, Millet EJ, Fredberg JJ. Time scale and other invariants of integrative mechanical behavior in living cells. Phys Rev E Stat Nonlin Soft Matter Phys. 2003;68(4 Pt 1):041914. doi: 10.1103/PhysRevE.68.041914. [DOI] [PubMed] [Google Scholar]
- Goldberg MT, Han YP, Yan C, Shaw MC, Garner WL. TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: an implication for abnormal wound healing. J Invest Dermatol. 2007;127(11):2645–2655. doi: 10.1038/sj.jid.5700890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves MF, Umezawa ES, Katzin AM, de Souza W, Alves MJ, Zingales B, Colli W. Trypanosoma cruzi: shedding of surface antigens as membrane vesicles. Exp Parasitol. 1991;72(1):43–53. doi: 10.1016/0014-4894(91)90119-h. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Juliano RL. SIGNAL TRANSDUCTION BY CELL ADHESION RECEPTORS AND THE CYTOSKELETON: Functions of Integrins, Cadherins, Selectins, and Immunoglobulin-Superfamily Members. Annual Review of Pharmacology and Toxicology. 2002;42(1):283–323. doi: 10.1146/annurev.pharmtox.42.090401.151133. [DOI] [PubMed] [Google Scholar]
- Karamichos D, Lakshman N, Petroll WM. Regulation of corneal fibroblast morphology and collagen reorganization by extracellular matrix mechanical properties. Invest Ophthalmol Vis Sci. 2007;48(11):5030–5037. doi: 10.1167/iovs.07-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatiwala CB, Peyton SR, Putnam AJ. Intrinsic mechanical properties of the extracellular matrix affect the behavior of pre-osteoblastic MC3T3-E1 cells. Am J Physiol Cell Physiol. 2006;290(6):C1640–1650. doi: 10.1152/ajpcell.00455.2005. [DOI] [PubMed] [Google Scholar]
- Kirchhoff LV. American Trypanosomiasis (Chagas' Disease) Gastroenterology Clinics of North America. 1996;25:517. doi: 10.1016/s0889-8553(05)70261-2. [DOI] [PubMed] [Google Scholar]
- Lang P, Gesbert F, Delespine-Carmagant M, Stancou R, Pouchelet M, Bertoglia J. Protein kinase A phosphorylation of RhoA mediates that morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15:510–519. [PMC free article] [PubMed] [Google Scholar]
- Lenormand G, Bursac P, Butler JP, Fredberg JJ. Out-of-equilibrium dynamics in the cytoskeleton of the living cell. Phys Rev E Stat Nonlin Soft Matter Phys. 2007a;76(4 Pt 1):041901. doi: 10.1103/PhysRevE.76.041901. [DOI] [PubMed] [Google Scholar]
- Lenormand G, Chopin J, Bursac P, Fredberg JJ, Butler JP. Directional memory and caged dynamics in cytoskeletal remodelling. Biochem Biophys Res Commun. 2007b;360(4):797–801. doi: 10.1016/j.bbrc.2007.05.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung LY, Tian D, Brangwynne CP, Weitz DA, Tschumperlin DJ. A new microrheometric approach reveals individual and cooperative roles for TGF-beta1 and IL-1beta in fibroblast-mediated stiffening of collagen gels. Faseb J. 2007;21(9):2064–2073. doi: 10.1096/fj.06-7510com. [DOI] [PubMed] [Google Scholar]
- Ley V, Robbins ES, Nussenzweig V, Andrews NW. The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the pH of acidic compartments. J Exp Med. 1990;171(2):401–413. doi: 10.1084/jem.171.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low HP, Paulin JJ, Keith CH. Trypanosoma cruzi infection of BSC-1 fibroblast cells causes cytoskeletal disruption and changes in intracellular calcium levels. The Journal of Protozoology. 1992;39(4):463–470. doi: 10.1111/j.1550-7408.1992.tb04833.x. [DOI] [PubMed] [Google Scholar]
- Martens JC, Radmacher M. Softening of the actin cytoskeleton by inhibition of myosin II. Eur. J. Physiol. 2008;456:95–100. doi: 10.1007/s00424-007-0419-8. [DOI] [PubMed] [Google Scholar]
- Melo TG, Almeida DS, de Meirelles MdNSL, Pereira MCS. Trypanosoma cruzi infection disrupts vinculin costameres in cardiomyocytes. European Journal of Cell Biology. 2004;83:531–540. doi: 10.1078/0171-9335-00419. [DOI] [PubMed] [Google Scholar]
- Melo TG, Almeida DS, de Meirelles MdNSL, Pereira MCS. Disarray of sarcomeric alpha-actinin in cardiomyocytes infected by Trypanosoma cruzi. Parasitology. 2006;133:171–178. doi: 10.1017/S0031182006000011. [DOI] [PubMed] [Google Scholar]
- Mijailovich SM, Kojic M, Zivkovic M, Fabry B, Fredberg JJ. A finite element model of cell deformation during magnetic bead twisting. J Appl Physiol. 2002;93(4):1429–1436. doi: 10.1152/japplphysiol.00255.2002. [DOI] [PubMed] [Google Scholar]
- Murthy KS. Signaling for contraction and relaxation in smooth muscle of the gut. Ann. Rev. Physiol. 2006;68:345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- Pereira MCS, Costa M, Filho CC, de Meirelles MdNSL. Myofibrillar breakdown and cytoskeletal alterations in heart muscle cells during invasion by Trypansoma cruzi: immunological and ultrastructural study. Journal of Submicroscopic Cytology and Pathology. 1993;25(4):559–569. [PubMed] [Google Scholar]
- Puig-de-Morales M, Millet E, Fabry B, Navajas D, Wang N, Butler JP, Fredberg JJ. Cytoskeletal mechanics in adherent human airway smooth muscle cells: probe specificity and scaling of protein-protein dynamics. Am J Physiol Cell Physiol. 2004;287(3):C643–654. doi: 10.1152/ajpcell.00070.2004. [DOI] [PubMed] [Google Scholar]
- Qi J, Fox AM, Alexopoulos LG, Chi L, Bynum D, Guilak F, Banes AJ. IL-1beta decreases the elastic modulus of human tenocytes. J Appl Physiol. 2006;101(1):189–195. doi: 10.1152/japplphysiol.01128.2005. [DOI] [PubMed] [Google Scholar]
- Ramachandran C, Satpathy M, Mehta D, Srinivas SP. Forskolin induces myosin light chain dephosphorylation in bovine trabecular meshwork cells. Curr. Eye Res. 2008;33:169–176. doi: 10.1080/02713680701837067. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Martinez I, Chung A, Berlot CH, Andrews NW. cAMP Regulates Ca2+− dependent Exocytosis of Lysosomes and Lysosome-mediated Cell Invasion by Trypanosomes. J Biol Chem. 1999;274(24):16754–16759. doi: 10.1074/jbc.274.24.16754. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Rioult M, Ora A, Andrews N. A trypanosome-soluble factor induces IP3 formation, intracellular Ca2+ mobilization and microfilament rearrangement in host cells. J Cell Biol. 1995;129(5):1263–1273. doi: 10.1083/jcb.129.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfstein J, Schmitz V, Morandi V, Capella MMA, Lima APCA, Morrot A, Juliano L, Muller-Esterl W. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B2 receptors. J Exp Med. 2000;192(9):1289–1300. doi: 10.1084/jem.192.9.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J. 2007;93(12):4453–4461. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieux I, Nathanson M, Andrews N. Role in host cell invasion of Trypanosoma cruzi-induced cytosolic-free Ca2+ transients. J Exp Med. 1994;179(3):1017–1022. doi: 10.1084/jem.179.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieux I, Webster P, Ravesloot J, Boron W, Lunn JA, Heuser JE, Andrews NW. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71(7):1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- Unnikrishnan M, Burliegh BA. Inhibition of host connective tissue growth factor expression: a novel Trypanosoma cruzi-mediated response. FASEB Journal. 2004;18(14):1625–1635. doi: 10.1096/fj.04-1554com. [DOI] [PubMed] [Google Scholar]
- Wilkowsky SE, Barbieri MA, Stahl P, Isola ELD. Trypanosoma cruzi: Phosphatidylinositol 3-Kinase and Protein Kinase B Activation Is Associated with Parasite Invasion. Experimental Cell Research. 2001;264(2):211–218. doi: 10.1006/excr.2000.5123. [DOI] [PubMed] [Google Scholar]
- Woolsey AM, Burleigh BA. Host cell actin polymerization is required for cellular retention of Trypanosoma cruzi and early association with endosomal/lysosomal compartments. Cellular Microbiology. 2004;6(9):829–838. doi: 10.1111/j.1462-5822.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- Woolsey AM, Sunwoo L, Petersen CA, Brachmann SM, Cantley LC, Burleigh BA. Novel PI 3-kinase-dependent mechanisms of trypanosome invasion and vacuole maturation. J Cell Sci. 2003;116(17):3611–3622. doi: 10.1242/jcs.00666. [DOI] [PubMed] [Google Scholar]
- Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60(1):24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- Yoshida N. Molecular basis of mammalian cell invasion by Trypanosoma cruzi. An Acad Bras Cienc. 2006;78(1):87–111. doi: 10.1590/s0001-37652006000100010. [DOI] [PubMed] [Google Scholar]