

Abstract
Anthrax lethal toxin (LT) rapidly kills macrophages from certain mouse strains in a mechanism dependent on the breakdown of unknown protein(s) by the proteasome, formation of the Nalp1b (NLRP1b) inflammasome, and subsequent activation of caspase-1. We report that heat shocking LT-sensitive macrophages rapidly protects them against cytolysis by inhibiting caspase-1 activation without upstream effect on LT endocytosis or cleavage of the toxin’s known cytosolic substrates (MEKs). Heat shock protection against LT occurred through a mechanism independent of de novo protein synthesis, HSP90 activity, p38 activation, or proteasome inhibition and was downstream of MEK cleavage and degradation of an unknown substrate by the proteasome. However, the heat shock inhibition of LT-mediated caspase-1 activation was not specific to the Nalp1b (NLRP1b) inflammasome, as heat shock also inhibited Nalp3 (NLRP3) inflammasome-mediated caspase-1 activation in macrophages. We found that heat shock induced pro-caspase-1 association with a large cellular complex which could prevent its activation. Additionally, while heat shocking recombinant caspase-1 did not affect its activity in vitro, lysates from heat shocked cells completely inhibited recombinant active caspase-1 activity. Our results suggest heat shock inhibition of active caspase-1 can occur independently of inflammasome platform, through a titratable factor present within intact, functioning heat shocked cells.
Introduction
Bacillus anthracis lethal toxin (LT) is composed of protective antigen (PA), a receptor binding protein, and lethal factor (LF), a zinc metalloprotease that is responsible for the cleavage of mitogen-activated protein kinase kinsases (MEKs) (Leppla, 2006; Duesbery et al., 1998; Pellizzari et al., 1999 Vitale et al., 2002). Within certain strains of murine macrophages, LT causes a rapid and striking cell death, whereas macrophages from other mouse strains are resistant (Friedlander et al., 1993; Singh et al., 1989). While many of the cellular signals between LT treatment and macrophage death remain to be elucidated, key events required for cell death include the degradation of an unknown protein or proteins by the proteasome and activation of caspase-1 downstream of toxin-induced potassium fluxes (Tang and Leppla, 1999; Boyden and Dietrich, 2006; Wickliffe et al., 2007; Squires et al., 2007). Macrophage sensitivity has been mapped to variations in the gene encoding Nalp1b (also known as NLRP1b), a Nod-like receptor (NLR) protein involved in innate immunity (Boyden and Dietrich, 2006). Nalp family proteins oligomerize in response to numerous bacterial and ‘danger’ stimuli including cytosolic flagellin (Miao et al., 2006), gout-associated uric acid crystals (Martinon, et al., 2006) and microbial toxins (for review, see Freche et al., 2007; Lamkanfi et al., 2007; Petrilli et al., 2007) to trigger the formation of an inflammasome, a cytosolic, multi-protein signaling complex. The inflammasome platform promotes pro-caspase-1 autocleavage and activation, an event essential for LT-induced cell death (Boyden and Dietrich, 2006). Activated caspase-1 p10 and p20 subunits form a heterotetramer, which cleaves IL-1β and IL-18 to their mature forms (Dinarello, 1996; Akita et al., 1997), although this process is not required for LT-induced cell death (Wickliffe et al., 2007). IL-1β and IL-18 are pro-inflammatory cytokines which can trigger the initiation of fever (Dinarello, 1996; Roth et al., 2006). However, little examination has been made of NLR inflammasome systems once inflammation has begun and cells are subjected to conditions of thermal stress.
The classical heat shock response centers around the up-regulation of heat shock proteins (HSPs). These proteins are involved in a multitude of cellular responses, including serving as chaperones for the re-folding of thermally denatured proteins (for review, see Picard, 2002; Rutherford, 2003; Lepock, 2005). HSP transcription is controlled by heat shock factor-1, a regulatory protein that is required for multi-pathogen resistance in Caenorhabditis elegans (McMillan et al., 1998; Singh and Aballay, 2006). The heat shock response may regulate several aspects of innate immunity, as HSPs have been implicated in a variety of molecular models of inflammation and bacterial infection (Tanaka et al., 2007; Tang et al., 2007) and heat-stressed mice and rats were shown to be protected from severe endotoxemia (Hotchkiss et al., 1993; Heidemann et al., 2000). Heat shock has also been linked to differential regulation of signals induced by Toll-like receptors, a group of pathogen-sensing proteins related to NLRs (Zhao et al., 2007; Peng et al., 2006). Therefore, it is not unreasonable to suspect that heat shock may have an impact upon NLR signaling as well.
This study investigates the in vitro effects of thermal stress on the killing of murine macrophages by LT. We show that heat shock rapidly halts LT-induced cell death without any impact on toxin uptake or MEK cleavage, by a mechanism independent of novel protein synthesis, p38 activation, HSP90 activity, or proteasome inhibition. Rather, heat shock prevents the activation of pro-caspase-1 in LT-treated cells, apparently by the sequestration of pro-caspase-1 in a large, inhibitory complex. Additionally, heat-shocked cell lysates strongly inhibit the active caspase-1 heterotetramer in vitro, independent of a specific inflammasome platform. Our results suggest the presence of a titratable, cellular, heat shock-inducible, caspase-1 inhibiting factor. The potential implications of these findings for the regulation of inflammatory responses are discussed.
Results
Heat shock protects against LT-induced cell death in macrophages
RAW 264.7 cells and bone marrow-derived macrophages (BMDMs) from BALB/cJ mice with LT-sensitive macrophages were heat shocked for 2 h prior to LT treatment and were subsequently protected against cell death (Fig. 1 shows data for RAW 264.7 cells). Similar results were obtained whether the cells were incubated at 37°C (data not shown) or at elevated heat shock temperatures during the period of LT treatment. Heme Oxygenase 1 is a marker for thermal shock (Ewing and Maines, 1995) and its up-regulation was used to monitor the induction of a heat shock state (Fig. 1 inset).
Figure 1. Heat shock protects against LT-induced macrophage death.
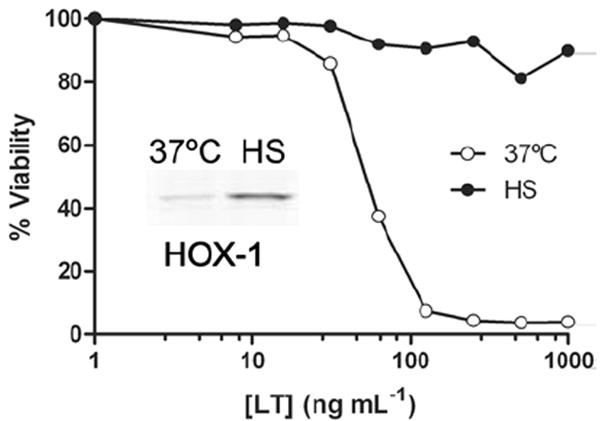
RAW 264.7 cells were treated with LT after 2 h of heat shock and compared to non-heat shocked toxin-treated controls. Cell viability was assessed after 2.5 h of toxin treatment and percent viability was calculated relative to medium-treated cells. Heat shock induction of Heme Oxygenase-1 in cells receiving similar treatment was used to verify effective heat shock (inset).
Heat shock does not prevent MEK cleavage by LT
LF cleaves members of the mitogen-activated protein kinase kinase (MEK) family after its delivery into the cytosol by PA (Duesbery et al., 1998; Pellizzari et al., 1999). We hypothesized that heat shock could interfere with the binding, endocytosis or translocation of LF into the cytosol. Therefore, we examined MEK cleavage under heat shock conditions to see if active LF was properly delivered to the cytosol. In RAW 264.7 and BALB/cJ BMDM cells heat shocked for 2 h before LT treatment, MEK1, MEK2, and MEK3 were cleaved similarly in heat shocked cells and those incubated at 37°C (Fig. 2 shows results for RAW 264.7 cells, similar results obtained with BALB/cJ BMDMs), indicating that heat shock did not deactivate LF or prevent its proper translocation into the cytosol.
Figure 2. Heat shock does not prevent Mek cleavage by LT.
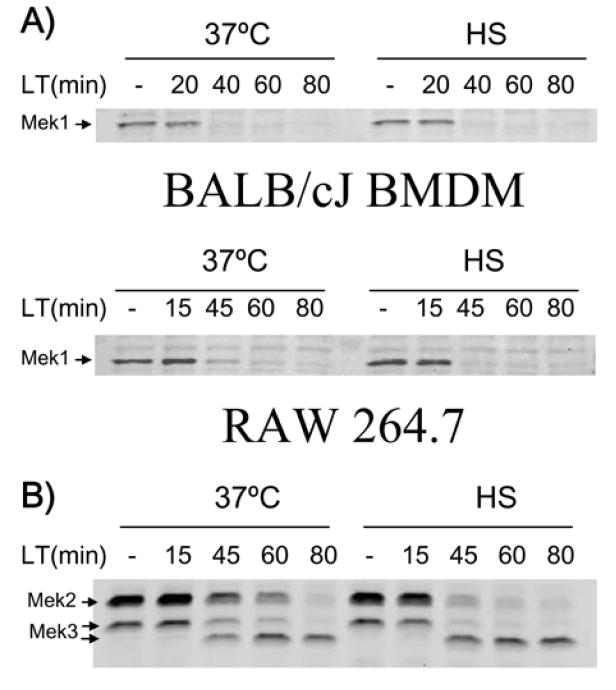
BALB/cJ BMDM and RAW 264.7 cells were heat shocked 2 h prior to LT addition (1 μg mL-1 for 0 to 80 min) and compared to toxin-treated cells at 37°C. Cleavage of Mek1 (A), Mek2, and Mek3 (B, RAW 264.7 only) was monitored at various times after toxin treatment in heat shock or 37°C conditions by Western blot.
Synthesis of heat shock proteins is not required for protection
A number of inducible HSPs have been implicated in the activation of both innate and adaptive immunity (Srivastava, 2002). We postulated that the rapid up-regulation of a heat shock-induced protein could be involved in macrophage protection from LT. Therefore, we tested the effect of the transcription inhibitor actinomycin D and of the translation inhibitors puromycin and cycloheximide to see if the protection afforded by heat shock was prevented by the inhibition of protein synthesis. The activity of these inhibitors was confirmed in other experiments (data not shown). We observed strong heat shock protection from LT with all the inhibitors tested over both a range of LT concentrations and drug doses (Fig. 3), suggesting that this protection does not involve the synthesis of a novel protein. Additionally, the inhibitors did not have any protective effect on LT-mediated cytolysis at 37°C (Fig. 3), indicating that transcriptional and translational responses to LT play no role in cell death.
Figure 3. Protein synthesis is not required for protection.
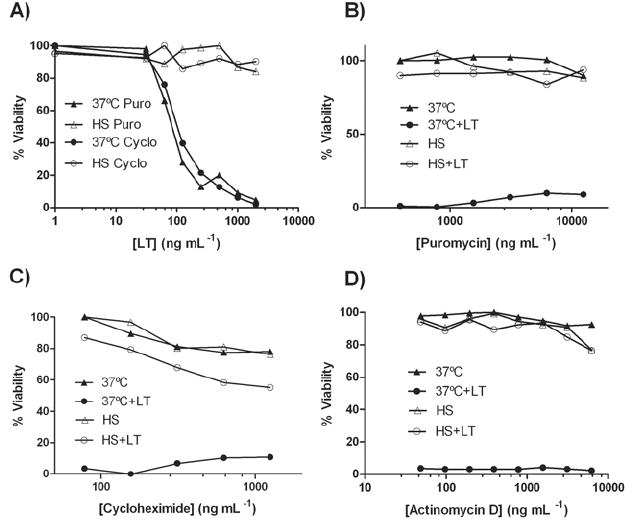
RAW 264.7 cells were treated with the translation inhibitors cycloheximide (312 ng mL-1 in (A) or concentration range in (C)) or puromycin (1.56 μg mL-1 in (A) or concentration range in (B)) for 1 h before a 2 h heat shock treatment. LT was added in a concentration range (A) or at 1 μg mL-1 (B, C) and viability was assessed after 2 h. In (D), cells were incubated with the transcription inhibitor Actinomycin D (1 μg mL-1) for 1 h at 37°C prior to continued incubation at this temperature or a 2 h heat shock. LT was added and viability assessed as above.
p38 inhibition does not reverse heat shock protection
Previous studies have suggested that the activation of p38 MAP kinase is required for protection from LT-induced cell death in C57BL/6J macrophages (Park et al., 2002). Since p38 activation is a general protective stress response in macrophages which has also been shown to occur in response to thermal stress (Aggeli et al., 2002; Maroni et al., 2002), we hypothesized that our heat shock protection could be attributed to the protective effect of activated p38. We found that p38 was indeed phosphorylated in response to our heat shock conditions in both RAW 264.7 cells and BALB/cJ macrophages, and that this activation was lost after 60 min of LT treatment (Fig. 4A). LF cleavage of MEK3 (Fig. 2), the upstream activator of p38, inhibits p38 activation at these later LT treatment times. To test if early p38 activation under heat shock was responsible for protection against LT we inhibited p38 activity using the inhibitor SB203580. Cells pre-treated with this inhibitor were then heat shocked or incubated at 37°C prior to LT treatment. The continued effectiveness of the SB203580 inhibitor under heat shock and 37°C conditions was confirmed by examining the LPS-induced phosphorylation of mitogen-activated protein kinase-activated protein-2 kinase (MAPKAP-2), a direct target of phosphorylated p38. Using an antibody that detects altered migration of this protein in its activated form, we found that MAPKAP-2 was not phosphorylated in cells treated with p38 inhibitor in either 37°C or heat shock conditions (Fig. 4B inset). MAPKAP-2 was not activated at 37°C without LPS treatment (data not shown). Thus, the p38 inhibitor was effective at both 37°C and heat shock conditions, but was unable to reverse the heat shock protection against LT (Fig. 4B). We conclude that p38 activation is not required for heat shock protection. Similar results were obtained with BALB/cJ BMDM cells (data not shown).
Figure 4. p38 inhibition does not reverse heat shock protection.
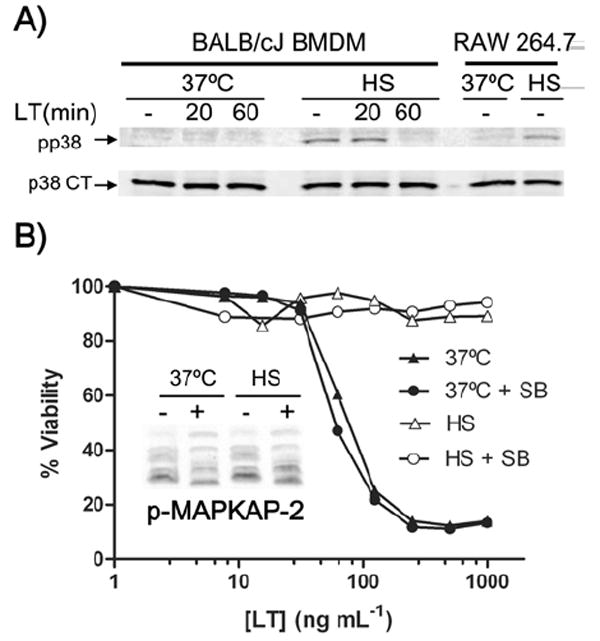
(A) BALB/cJ BMDM and RAW 264.7 cells were heat shocked (2 h) before LT treatment (1 μg mL-1) and compared to non-heat shocked controls. Phosphorylated p38 (upper) and p38 CT (lower) were monitored at various times after toxin treatment. (B) RAW 264.7 cells were incubated for 30 min with SB203580 p38 inhibitor (SB, 26 μM) or media alone (control) at 37°C before a 1 h heat shock incubation. LT was added and cell viability was assessed after 2 h. Phosphorylation of MAPKAP-2 was monitored in cells stimulated with 500 ng mL-1 LPS for 15 min and treated with (+) and without (-) SB203580 as described above to verify effective p38 inhibition (inset).
HSP90 inhibition does not reverse heat shock protection
HSP90 is a constitutively expressed and highly abundant molecular chaperone for a large number of signal transduction proteins in eukaryotic cells, including those involved in the inhibition of apoptosis (Picard, 2002; Bishop et al., 2007). This protein has also been implicated in the translocation of some bacterial toxins from cellular endosomes, but does not play a role in the translocation of LT (Ratts et al., 2003; Haug et al., 2003). Additionally, HSP90 inhibition does not prevent macrophage death via the LT-induced Nalp1b (NLRP1b) inflammasome (Haug et al., 2003; data not shown), contrary to the HSP90 requirement that was previously shown in the Nalp3 (NLRP3) inflammasome system (Mayor et al., 2007). We tested whether HSP90 activity was required for heat shock protection from LT using the potent HSP90 inhibitor geldanamycin. The activity of this inhibitor was confirmed in other experiments (data not shown). Heat shocked cells continued to be protected from LT with a range of inhibitory geldanamycin doses (Fig. 5), showing that HSP90 is not involved in heat shock protection against the toxin.
Figure 5. HSP90 inhibition does not reverse heat shock protection.
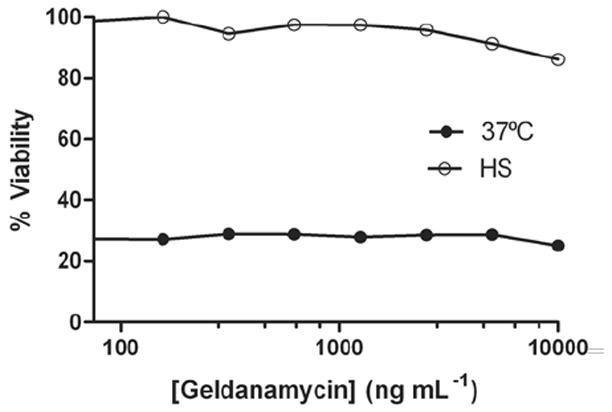
RAW 264.7 cells were incubated with geldanamycin at 37°C for 5 h before being heat shocked or incubated at 37°C for 2 h. LT was added (1 μg mL-1) and cell viability was assessed after 2 h.
Heat shock prevents LT-induced activation of caspase-1 in cells
Caspase-1 activation by the Nalp1b (NLRP1b) inflammasome is known to be required for LT-induced macrophage cell death (Boyden and Dietrich, 2006). To determine whether heat shock affected events upstream or downstream of caspase-1 activation, we compared the levels of the activated p10 subunit of caspase-1 in LPS-primed BALB/cJ BMDMs that were treated with LT under heat shock or 37°C conditions. In contrast to cells at 37°C, no active p10 caspase-1 was observed in the heat shocked cells following LT treatment (Fig. 6A). IL-1β cleavage by caspase-1 was also monitored in the same cells and no mature IL-1β was observed in heat shocked samples (Fig. 6B), confirming the absence of activated caspase-1 under heat shock conditions. Identical caspase-1 cleavage results were also obtained from BALB/cJ BMDMs that were not LPS-primed (data not shown).
Figure 6. Heat shock prevents caspase-1 activation by LT, but does not inhibit caspase-1 activity.
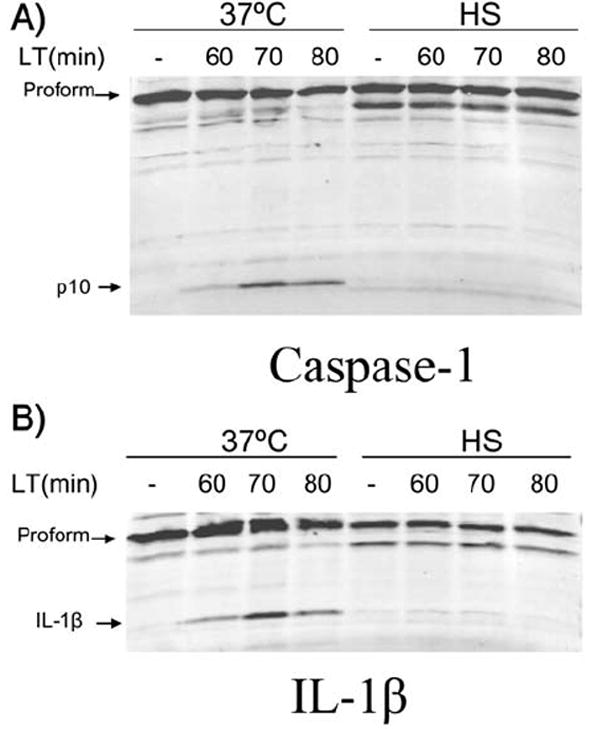
LPS-primed BALB/cJ BMDM (500 ng mL-1) were heat shocked (2 h) before LT treatment (1 μg mL-1) and compared to non-heat shock controls remaining at 37°C. Cell lysates were probed for caspase-1 p10 (A) or IL-1β (B) by Western blot.
Late heat shock protects against LT
We have previously established a timeline of the known cellular events involved in LT-induced cell death in murine macrophages (Wickliffe et al., 2007). We next asked how rapidly and how late heat shock could protect cells from LT. BALB/cJ BMDMs were given LT and then heat shocked immediately (at t=0 min) or moved to heat shock conditions after various durations of LT incubation at 37°C (between t=45 min and t=75 min). After t=110 min, viability was assessed. We determined the amount of activated caspase-1 at the time when cells were moved to heat shock by lysing a parallel set of identically treated samples at every shift time point at 37°C. Additionally, we assessed the levels of continued caspase activation in a third set of similarly treated samples after the move to heat shock.
We observed that no heat shock incubation before LT treatment was necessary for heat shock protection against the toxin. In fact, both BMDMs and RAW 264.7 cells could be heat shocked well after LT addition and be protected from LT-induced death, especially if heat shock was applied before the active p10 subunit was first observed (Fig. 7B shows results for BALB/cJ BMDMs). In parallel, we determined that heat shock applied before the initiation of caspase-1 activation (i.e. at t=0 min or t=45 min) completely prevented subsequent caspase-1 activation (Fig. 7A, compare 1st and 2nd lanes with 9th and 8th lanes). However, heat shock applied after p10 was observed (i.e. at t=65 min or 75 min) resulted in levels of active p10 comparable to that found in cells that were not heat shocked (Fig. 7A, compare 6th and 7th lanes to 5th lane). These results suggest that heat shock prevents caspase-1 activation by the Nalp1b (NLRP1b) inflammasome, but once caspase-1 activation is initiated, heat shock is ineffective. In parallel, heat shock applied as late as 65 min and 75 min does appear to slow down the death event (Fig. 7B), but is unable to provide complete protection once some caspase-1 has been activated. This may be because at late times the cell death-inducing events downstream of caspase-1 have already occurred.
Figure 7. Late heat shock protects against LTi.
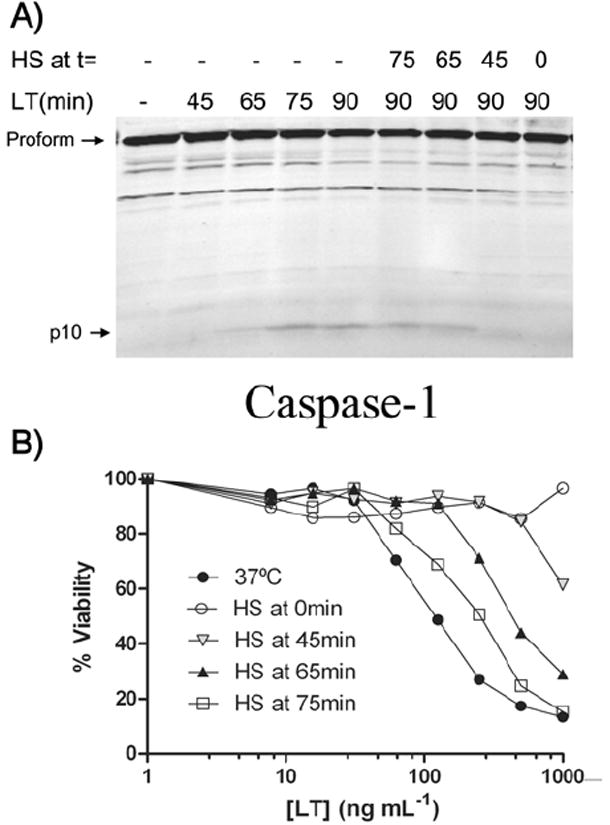
(A) At t=0 min, LT (1 μg mL-1) was added to BALB/cJ BMDMs. Cells were moved to heat shock conditions between 0 and 75 min later, and lysates were made after 0 to 90 min of LT treatment to monitor activated caspase-1 p10. (B) BALB/cJ BMDMs given identical heat shock treatments in parallel to the samples in (A) and a range of LT doses were assessed for viability 110 min after LT treatment.
Heat shock does not inhibit proteasome activity
We and others have previously demonstrated that proteasome activity upstream of caspase-1 activation is necessary for LT-induced cell death, and that proteasome inhibition provides protection from LT (Wickliffe et al., 2007; Squires et al., 2007). As proteasome inhibition has sometimes been associated with the induction of a cellular heat shock response (Bush et al., 1997; Kim et al., 1999; Lee and Goldberg, 1998) and, conversely, heat shock has been associated with differential proteasomal processing (Pajonk et al., 2005; Kuckelkorn et al., 2000; Kelly et al., 2007; Luo et al., 2000), we asked whether our observed heat shock protection acted through proteasome inhibition. We first compared the levels of ubiquitinated proteins in LT-treated or untreated macrophages at 37°C, in the presence of the proteasome inhibitor lactacystin, or under conditions of heat shock. A LT-mediated decrease in ubiquitinated proteins in macrophages has been previously reported (Alileche et al., 2006; Salles et al., 2003), although we observed no difference in ubiquitination when cells were treated with LT (Fig. 8A), possibly because previous studies had examined cells at late times, after lysis from LT had already occurred. As expected, there was a large build-up of ubiquitinated proteins in cells treated with lactacystin (Fig. 8A). We also observed a moderate increase in ubiquinated proteins in the heat shocked cells compared to 37°C cells, suggestive of either inhibition of the proteasome under heat shock or an increase in protein ubiquitination in heat shocked cells, with potentially no change in proteasome function. Both of these phenomena have been previously reported (Pajonk et al., 2005; Paraq et al., 1987). To distinguish between these possibilities, we investigated proteasome activity in cells directly using a cell-permeable proteasome substrate activated to luminesce only after cleavage by the proteasome. Lactacystin was used as a positive control for proteasome inhibition in these studies. We found that the proteasome was not inhibited in heat shocked cells, and that proteasomal processing was actually markedly increased under those conditions (Fig. 8B), much in the manner previously shown for the ER unfolded protein stress response to heat shock (Kelly et al., 2007). The apparent increase in cleavage of the proteasome substrate under heat shock conditions may have been due to changes in its uptake and delivery. In both heat shocked and 37°C cells, the lactacystin control treatment resulted in a significant decrease in luminescence as was expected from proteasome inhibition. From these data, we conclude that heat shock does not inhibit proteasome activity, and therefore that heat shock protection from LT is not a result of proteasome inhibition.
Figure 8. Heat shock does not inhibit proteasome activity.
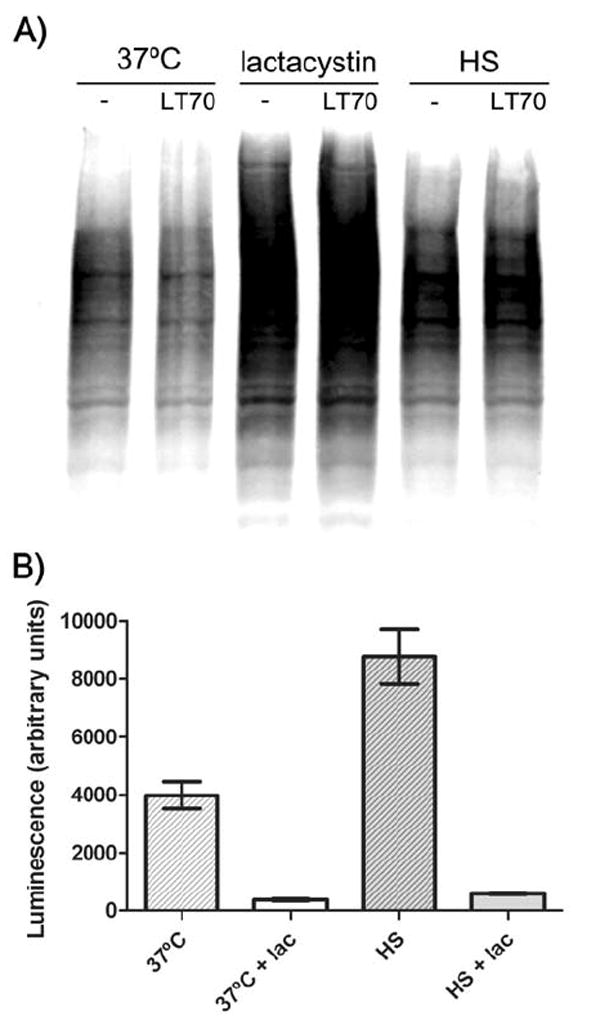
(A) RAW 264.7 cells were heat shocked for 50 min or treated with lactacystin (25 μM) for 10 min. LT was added (1 μg mL-1) and after 70 min cells were lysed and protein ubiquitination monitored by Western blot. (B) RAW 264.7 cells were heat shocked or incubated at 37°C for 1.5 h before a 20 min lactacystin treatment (25 μM). Cells were brought to room temperature and proteasome activity assayed using a cell permeable luminescent substrate as described in Experimental procedures.
Heat shock prevents nigericin-induced caspase-1 activation by the Nalp3 inflammasome
We next sought to determine if heat shock can prevent the activation of caspase-1 by other inflammasome protein complexes. Nigericin has been shown to rapidly induce the Nalp3 (NLRP3) inflammasome in BMDMs, resulting in the activation of caspase-1 and release of IL-1β (Mariathasan et al., 2006). Because nigericin also requires an LPS co-signal and TLR4 signaling is affected by heat shock conditions (Data not shown; Zhao et al., 2006), BALB/cJ BMDM cells were LPS pre-treated at 37°C prior to heat shock incubation and subsequent nigericin treatment. There was a distinct absence of active caspase-1 and mature IL-1β in the heat shocked, nigericin-treated cell lysates (Fig. 9). This result could suggest that heat shock can prevent the activation by the Nalp3 (NLRP3) inflammasome and may affect a component necessary for the induction of multiple inflammasome complexes. However, as the precise timing and mechanism of LPS/nigericin activation of the Nalp3 (NLRP3) inflammasome is poorly understood, it is also possible that the lack of activated caspase-1 was due to altered TLR signaling in heat shock conditions, as reflected by heat shock’s inhibitory effect on LPS-mediated IκB-α breakdown (data not shown).
Figure 9. Heat shock prevents caspase-1 activation by nigericin.
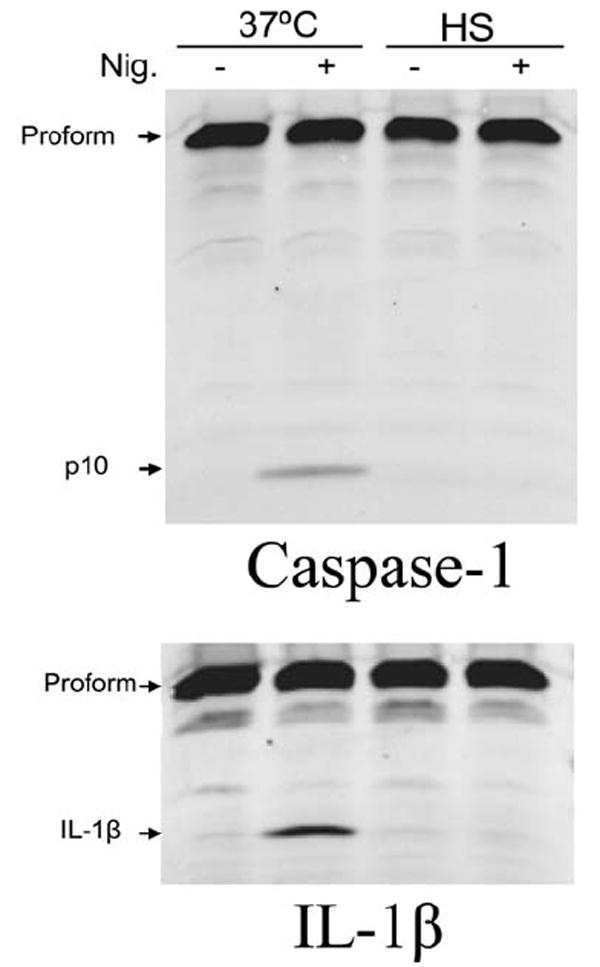
BALB/cJ BMDM cells were treated with LPS (500 ng mL-1) for 2 h at 37°C prior to a 1 h incubation in heat shock or 37°C conditions. Heat shocked cells were moved back to 37°C for an additional 30 min, prior to treatment with nigericin (30 μM, 20 min) and caspase-1 p10 and IL-1β were monitored by Western blot.
Whole cell lysates from heat shocked cells inhibit caspase-1 activity
We tested the hypothesis that higher temperatures could alter the proteolytic activity of caspase-1. Sucrose lysates of LPS-treated RAW 264.7 cells which contained highly up-regulated, uncleaved IL-1β were treated for 1 h with recombinant caspase-1 at 37°C or under heat shock. Samples were then analyzed for IL-1β cleavage. We found that caspase-1 effectively cleaved IL-1β to its mature form in both heat shock and 37°C conditions (Fig. 10A), even when the enzyme and substrate were pre-heat shocked for extended periods before mixing (data not shown). This implies that a high temperature does not interfere directly with the proteolytic activity of activated caspase-1.
Figure 10. Whole cell lysates from heat shocked cells inhibit caspase-1 activity.
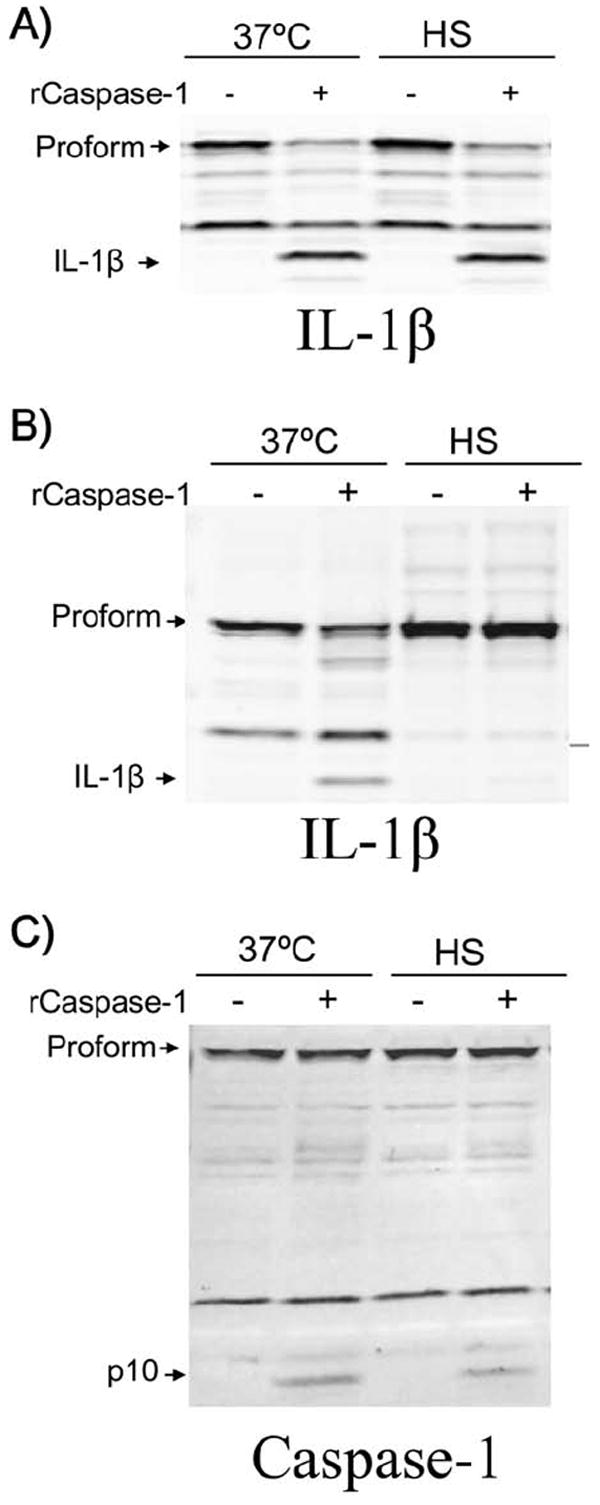
(A) Sucrose lysates from LPS-treated (1 μg mL-1 for 2 h) RAW 264.7 cells were mixed with recombinant caspase-1 (1 unit/40μL) or H2O, and incubated at 37°C or heat shocked for 1h. IL-1β cleavage was monitored by Western blot. (B) RAW 264.7 cells were LPS treated as above before a 1 h heat shock or 37°C incubation. Sucrose lysates were made and mixed with recombinant caspase-1 or H2O as above at 37°C and examined for IL-1β cleavage (B) or caspase-1 (C) by SDS electrophoresis Western blotting.
We next asked whether a component in heat shocked, intact cells was necessary for inhibition of caspase-1 activity. In this scenario, heat shock could initiate a signaling event, relocalization, protein modification or other event requiring whole, live cells, which could alter the proteolytic activity of caspase-1. To this end, we heat shocked LPS pre-treated RAW 264.7 cells for 1 h before making sucrose lysates. These lysates were then treated with recombinant active caspase-1 at 37°C for 1 h and examined for IL-1β cleavage (Fig. 10B). We observed a complete absence of IL-1β and IL-18 (data not shown) processing within lysates from heat shocked cells, even though we had added recombinant, active caspase-1. To address the hypothesis that the recombinant caspase-1 was degraded in these lysates, we examined the levels of caspase-1 within the same samples (Fig. 10C). We observed that a clear caspase-1 p10 band was present in all samples receiving the recombinant active caspase-1 (Fig. 10C), suggesting that the lack of IL-1β cleavage was not due to caspase-1 breakdown. No protein synthesis is required for this recombinant caspase-1 inhibition, as identical cleavage patterns were observed when cells were treated with translation inhibitors puromycin and cycloheximide before heat shock (data not shown). As this inhibition was independent of the Nalp1b inflammasome platform, we were not surprised to find identical patterns of inhibition when using heat shock lysates from LT-resistant macrophages (data not shown).
The titratable nature of the heat shock-induced inhibitory activity was shown by combining lysates from 37°C and heat shocked cells in different ratios before incubation with the recombinant caspase-1 (Fig 11A), suggesting the presence of a factor that non-enzymatically binds or modifies caspase-1. We hypothesized that this factor could be a protein or protein complex that binds and inactivates caspase-1. Therefore, we examined recombinant caspase-1 treated with heat shock or 37°C lysates by native gel in order to observe any differential migration of protein complexes. The diffuse band representing recombinant caspase-1 migrated similarly in the 37°C and heat shock samples (Fig. 11B, upper). It is possible that the recombinant caspase-1 heterotetramer in heat shock lysates is inhibited by a modification or binding event that is too small to be detected on these gels.
Figure 11. Caspase-1 inhibition by lysates of heat shocked cells is titratable.
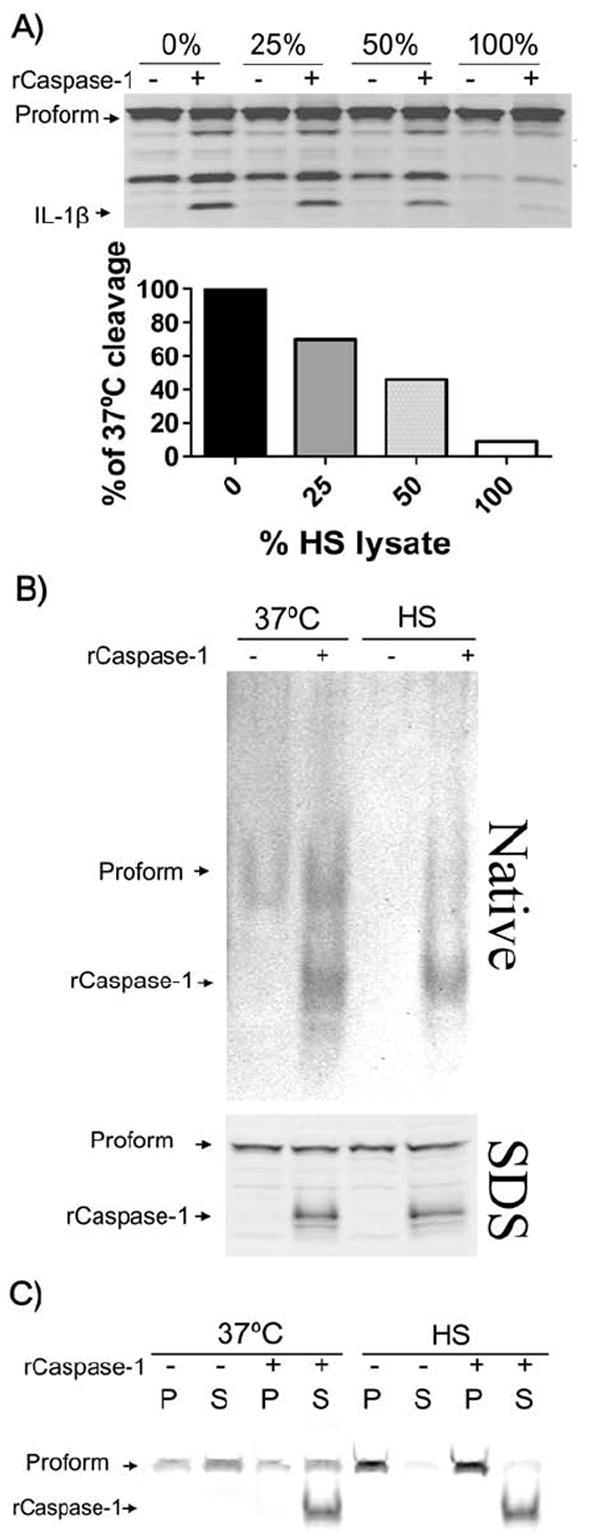
(A) RAW 264.7 cells were LPS-treated (1 μg mL-1 for 2 h) and subsequently heat shocked for 1 h. Sucrose lysates of 37°C and heat shock cells were combined in various ratios (% heat shock lysate, vol/vol) before the addition of recombinant caspase-1 (1 unit/40μL). After a 1 h incubation at 37°C, samples were examined for IL-1β cleavage. Semi-quantitative analysis of IL-1β cleavage band intensity is shown below. (B) RAW 264.7 cells were LPS treated as above before a 1 h heat shock or 37°C incubation. Sucrose lysates of the cells were mixed with recombinant caspase-1 or H2O at 37°C and examined for caspase-1 by native gel electrophoresis (upper) and SDS-PAGE (lower) by Western blotting. (C) RAW 264.7 cell lysates prepared as above were mixed with native gel loading dye with β-mercaptoethanol and centrifuged at 16,000 × G for 10 min. Pellets (P) and supernatants (S) were examined for caspase-1 by SDS-PAGE and Western blotting.
We also observed that while endogenous pro-caspase-1 was present in both of the lanes with 37°C lysates on a native gel, independent of the addition of recombinant active caspase-1, there was no observable endogenous pro-caspase-1 band in the heat shock lysates (Fig. 11B, upper). However, heat shock samples clearly showed the presence of pro-caspase-1 when subjected to SDS electrophoresis (Fig. 11B, lower). This suggests that endogenous pro-caspase-1 is not broken down, but may be part of a large protein complex in heat shocked cells.
Centrifugation of native lysates showed that although pro-caspase-1 was found in both the pellet and supernatant of lysates made from cells at 37°C, the lysates from heat shocked cells contained pro-caspase-1 almost entirely in the pellet fraction alone, confirming that pro-caspase-1 was part of a larger complex (Fig. 11C). Thus, it appears that heat shock induces the binding of endogenous pro-caspase-1 to other cellular protein/s which may prevent caspase-1 activation by inflammasomes. Additionally, when we tested whether heating 37°C lysates could relocalize pro-caspase-1 to this larger complex we found no difference between heated and unheated samples (data not shown). This indicated that the sequestration of pro-caspase-1 by heat shock also requires an event within intact cells, as observed for inhibition of active recombinant caspase-1.
Discussion
The general phenomenon of heat shock has been widely studied and documented, but its molecular and cellular effects on immunity are only beginning to be explored. Some studies have shown heat shock conditions to protect from endotoxemia in vivo (Hotchkiss et al., 1993; Heidemann et al., 2000) and to affect macrophage and dendritic cell activation (for review, see Ostberg and Repasky, 2006; Srivastava, 2002), TLR signaling (Zhao et al., 2006), cytokine secretion (Peng et al., 2006; Tanaka et al., 2007), and T-cell responses to MHC-presented antigens (Polla et al., 2006; Blachere et al., 1997). To date, there is little published data on the effects of heat shock on NLR signaling, inflammasome formation, or caspase-1 activity.
In this study, we found that heat shock can rapidly prevent anthrax LT-induced macrophage death by inhibiting caspase-1 activation, an event known to be required for cell death from LT (Boyden and Dietrich, 2006; Cordoba-Rodriguez et al., 2004). This inhibition of capsase-1 activation was also observed for the nigericin-induced Nalp3 (NLRP3) inflammasome, and may be due to binding of pro-caspase-1 to a large, inhibitory complex. Independently, heat shock was found to induce a titratable cellular factor, signaling, or modification event that inhibits the active caspase-1 heterotetramer without binding to a large complex. This factor could also be a general cellular condition, such as an alteration of the redox state of the cell. Previous studies (Cruz et al., 2007; Dostert et al., 2008) as well as a recent paper have implicated reactive oxygen species in the regulation of caspase-1 activity (Meissner et al., 2008). Experiments are currently underway to assess the redox state of cells following heat shock as well as possible modifications of caspase-1 in this context. Previous work has provided preliminary evidence that thermal shock can alter the redox state of cells (Jackson et al., 2006; Murapa et al., 2007)
Although the majority of studies of the heat shock response focus on HSP synthesis and activity, we found that de novo production of proteins was not important to heat shock protection from LT or for LT-mediated cell death. Other events induced by heat shock and previously shown to induce protective cellular responses, including p38 activation and the recruitment of HSP90, were also not involved in heat shock protection from LT. The breakdown of one or more cellular proteins by the proteasome is a specific requirement for the formation of the LT-induced Nalp1b (NLRP1b) inflamasome (Wickliffe et al., 2007; Squires et al., 2007). Previous studies have suggested that heat shock can either increase (Luo et al., 2000; Meinander et al., 2007; Smith et al., 2005) or decrease (Kuckelkorn et al., 2000; Pajonk et al., 2005) proteasomal degradation in mammalian cells. We found that proteasome activity was increased in macrophages after heat shock and therefore heat shock protection from LT was not a result of general proteasome inhibition.
When we extended our study of heat shock to the better characterized Nalp3 (NLRP3) inflammasome, we observed that caspase-1 and IL-1β were strongly activated in response to nigericin at 37°C but not in heat shock conditions. This was suggestive of an inhibition of pro-caspase-1 activation within the Nalp3 pathway, similar to that observed for the Nalp1b inflammasome. However, it was uncertain if this inhibition was due to heat shock interference with the LPS co-signal required for Nalp3 inflammasome activation.
Our in vitro studies show that recombinant caspase-1 activity is directly inhibited by a titratable factor in heat shocked cell lysates, suggesting that heat shock can also inhibit activated caspase-1, independent of the NLR inflammasome system of activation. We therefore expect our findings to apply to all inflammasome systems in addition to other caspase-1-related events in murine macrophages. Further studies are needed to see if a similar inhibitory activity also exists in human cells.
The precise mechanism of caspase-1 inhibition under heat shock remains to be determined. The events involved may include protein modification events (such as phosphorylation), altered ion fluxes, the production of reactive oxygen species, or the relocalization or altered conformation of a protein, among other possibilities. We hypothesize that within the intact cell system heat shock mediated events alter pro-caspase-1 binding in a complex which prevents its activation by the inflammasome. Separately, heat shock mediated factors can also inhibit the active caspase-1 heterotetramer independent of any effects on pro-caspase-1. However, an intact cellular structure is required for the cellular modifications or signaling events leading to heat shock inhibition of the caspase-1 heterotetramer, as heating the bulk cell lysate components does not inhibit caspase-1 activity.
Caspase-1 inactivation by heat shock could represent a regulatory pathway for the IL-1β inflammatory cascade. If a similar inhibitory effect is also shown for the activation of numerous other inflammasome systems that lead to the IL-1β response (see Lamkanfi et al., 2007 for review), thermal heat shock may represent a general negative feedback mechanism for regulating IL-1β mediated inflammation, possibly extending to the febrile response induced by this cytokine in vivo. Although there have been previous studies that have remarked on the in vitro inhibition of IL-1β production in heat shock conditions (Kappel et al., 1991; Velasco et al., 1990; Boneberg and Hartung, 2003), no study has linked this phenomenon to an inhibition of caspase-1 activity and to date, no correlation between in vitro heat shock regulation and in vivo events has been found. In vivo studies in LT-treated mice have shown a unique and rapid IL-1β release seen only in mice with LT-sensitive macrophages (Moayeri et al., 2004), which is rapidly shut down before any indications of malaise in animals. We are currently investigating the possibility of a temperature-based shutdown of the IL-1β response in these animals.
In summary, we have discovered that heat shock inhibits the activation of pro-caspase-1 by the Nalp1b (NLRP1b) and Nalp3 (NLRP3) inflammasomes while also directly inhibiting activated caspase-1 independent of any inflammasome platform. The identification of the cellular events and proteins involved in the shutdown of the caspase-1 pathway will have substantial ramifications in the study of the continually expanding set of inflammatory conditions which are known to act through caspase-1 (for review, see Wilmanski et al., 2008).
Experimental Procedures
Materials
Protective antigen (PA) and lethal factor (LF) were purified in our laboratory as described previously (Park and Leppla, 2000; Ramirez et al, 2002; Varughese et al, 1998). Concentrations of LT given for each experiment correspond to the concentration of each toxin component (i.e. 1 μg mL-1 LT has 1 μg mL-1 PA and 1 μg mL-1 LF). Anti-MEK1 NT (444942) antibody, ultra-pure LPS, and nigericin were purchased from Calbiochem (San Diego, CA). Caspase-1 p10 (sc-514), Mek2 NT (sc-524), Mek 3 NT (sc-959), p38 CT (sc-535-G), and Heme Oxygenase 1 (HOX) (sc-10789) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-p38 (4631) antibody was from Cell Signaling Technology (Danvers, MA). Anti-ubiquitin (Z0458) antibody was from Dako (Glostrup, Denmark). Anti-IL-1β (AF-401-NA) was from R& D Systems (Minneapolis, MN). Anti-goat infrared dye secondary antibody (#605-731-125) was from Rockland (Gilbertsville, PA). Anti-rabbit infrared dye secondary antibody was from Licor Biosciences (Lincoln, NE) or from Rockland Immunochemicals Inc. (Gilbertsville, PA). The p38 inhibitor SB203580 was purchased from Invitrogen (Carlsbad, CA). Puromycin, cycloheximide and LPS were obtained from Sigma-Aldrich (St. Louis, MO). Geldanamycin was purchased from Stressgen (San Diego, CA). Active recombinant caspase-1 was purchased from MBL International (Woburn, MA). Proteasome-Glo Cell-Based Assay was purchased from Promega (Madison, WI) and used as directed by manufacturer protocol with slight modification. Briefly, after heat shock and lactacystin treatments, all cells were equilibrated to room temperature before a 1:1 addition of the Proteasome-Glo substrate. Cells were shaken and allowed to equilibrate for 10 min before luminescence was measured.
Cell Culture
RAW 264.7 and L929 mouse fibroblast cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 10 mM Hepes, and 50 μg mL-1 gentamicin (all obtained from Invitrogen, Carlsbad, CA) at 37°C in 5% CO2. Bone marrow-derived macrophages (BMDMs) were cultured from BALB/cJ mice (Jackson Laboratories, Bar Harbor, ME) in complete DMEM (as described above) with 30% L929 cell culture supernatant. Bone marrow cells were grown for 7-9 days to allow time for differentiation before use in assays. All experiments involving animals were performed under protocols approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Heat shock
Cells were heat shocked in a cell culture incubator at 5% CO2. Because of incubator variability, heat shock temperatures ranged from 41C-45C for experiments performed on different days.
Cytotoxicity assays
RAW 264.7 and BALB/cJ BMDM cells were grown in 96-well plates to 90% confluence. In assays involving transcription, translation, and HSP90 inhibitors, cells were pre-treated for 1-2.5 h at 37°C with a dose range of each inhibitor as follows: Actinomycin D (0-6250 ng mL-1); Cycloheximide (0-625 ng mL-1); Puromycin (0-3125 ng mL-1); and Geldanamycin (0-10,000 nM). All cells were then treated with 1 μg mL-1 LT for 2.5 h and viability was assessed by addition of 0.5 mg mL-1 MTT [3-(4,5-Dimethyl-2-Thiazolyl)-2,5-Diphenyl Tetrazolium Bromide] (USB Corporation, Cleveland, OH). Following 45 min of incubation with MTT dye, cell culture medium was removed and cells were dissolved with 100 μL/well of 0.5% SDS, 25 mM HCl in 90% isopropanol. A microplate reader was used to read the A570 of each well, and percent viabilities were calculated relative to untreated controls. In all other assays, cells were treated with a dose range of LT (0-1000 ng mL-1), incubated for 2-3 h (as described in the figure legends) and stained with MTT as described above.
MEK, caspase-1, and IL-1β cleavage, HOX-1 up-regulation, p38 and MAPKAP-2 phosphorylation, and protein ubiquitination assays
RAW 264.7 and BALB/cJ BMDM cells grown in supplemented DMEM at indicated temperatures were treated with 1 μg mL-1 LT for various times. Where indicated, cells were first primed with 500 ng mL-1 LPS prior to toxin addition. At the end of each experiment, cells were washed three times with ice-cold PBS before lysates were prepared with RIPA lysis buffer (1% Nonidet-P40, 0.5% sodium deoxycholate, and 0.1% SDS in PBS) containing EDTA-free Complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Lysate protein concentrations were quantified using a BCA protein assay (Pierce, Rockford, IL), and equal amounts of protein were loaded for SDS-PAGE. Western blotting was performed using anti-MEK1 NT (1:7500), anti-MEK2 NT (1:2000), anti-MEK3 NT (1:2000), anti-Heme Oxygenase 1 (1:500), anti-phospho-p38 (1:1000), anti-p38 CT (1:1000), anti-ubiquitin (1:2000), anti-caspase-1 (1:250), and anti-IL-1β (1:700). Primary antibodies were detected using IR-dye-conjugated secondary antibodies (Licor Biosciences anti-rabbit, 1:25,000; Rockland Immunochemicals Inc. anti-rabbit, 1:5000; Rockland Immunochemicals Inc. anti-goat, 1:15,000) and the Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE).
In vitro caspase-1 and IL-1β assays
RAW 264.7 cells were grown in supplemented DMEM and treated with ultra-pure LPS (1 μg mL-1) for 2 h. After heat shock incubations, cells were washed three times with ice-cold PBS before preparing sucrose lysates (250 mM sucrose, 3 mM imidazole). Lysates were passed through a 29G needle 30X prior to protein quantification by BCA assay. Lysates were combined with recombinant caspase-1 (1 unit/40 μL, MBL) in 3 mM NaCl and incubated for 1 h before SDS or native electrophoresis. Native gel electrophoresis loading dye was 80 mM Tris, pH 7.5, 10% glycerol, and 0.02% bromophenol blue, with or without 5% β-mercaptoethanol. Western blotting was performed using anti-caspase-1 (1:250), and anti-IL-1β (1:700). Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE) and accompanying application software (version 2.0) were used integrate band intensity for a semi-quantitative measure of protein abundance.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases. We thank Devorah Crown for help with bone marrow isolation and Rasem Fattah for toxin preparation. We also thank a reviewer for insightful suggestions.
Footnotes
Publisher's Disclaimer: This is an Accepted Work that has been peer-reviewed and approved for publication in the Cellular Microbiology, but has yet to undergo copy-editing and proof correction. See http://www.blackwell-synergy.com/loi/cmi for details. Please cite this article as a “Postprint”; doi: 10.1111/j.1462-5822.2008.01220.x
References
- Aggeli I-K, Gaitanaki C, Lazou A, Beis I. Hyperosmotic and thermal stresses activate p38-MAPK in the perfused amphibian heart. J Exp Biol. 2002;205:443–454. doi: 10.1242/jeb.205.4.443. [DOI] [PubMed] [Google Scholar]
- Akita K, Ohtsuki T, Nukada Y, Tanimoto T, Namba M, Okura T, Takakura-Yamamoto R, Torigoe K, Gu Y, Su MS, Fujii M, Satoh-Itoh M, Yamamoto K, Kohno K, Ikeda M, Kurimoto M. Involvement of caspase-1 and caspase-3 in the production and processing of mature human interleukin 18 in monocytic THP.1 cells. J Biol Chem. 1997;272:26595–26603. doi: 10.1074/jbc.272.42.26595. [DOI] [PubMed] [Google Scholar]
- Alileche A, Squires RC, Muehlbauer SM, Lisanti MP, Borjatsch J. Mitochondrial impairment is a critical event in anthrax lethal toxin-induced cytolysis of murine macrophages. Cell Cycle. 2006;5:100–106. doi: 10.4161/cc.5.1.2283. [DOI] [PubMed] [Google Scholar]
- Bishop SC, Burlison JA, Blagg B. Hsp90: A Novel Target for the Disruption of Multiple Signaling Cascades. Curr Cancer Drug Targets. 2007;7:369–388. doi: 10.2174/156800907780809778. [DOI] [PubMed] [Google Scholar]
- Blachere NE, Li Z, Chandawarkar RY, Suto R, Jaikaria NS, Basu S, Udono H, Srivastava PK. Heat shock protein-peptide complexes, reconstituted in vitro, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J Exp Med. 1997;186:1315–1322. doi: 10.1084/jem.186.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneberg EM, Hartung T. Febrile temperatures attenuate IL-1β release by inhibiting proteolytic processing of the proform and influence Th1/Th2 balance by favoring Th2 cytokines. J Immunol. 2003;171:664–668. doi: 10.4049/jimmunol.171.2.664. [DOI] [PubMed] [Google Scholar]
- Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- Bush KT, Goldberg AL, Nigam SK. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem. 1997;272:9086–9092. doi: 10.1074/jbc.272.14.9086. [DOI] [PubMed] [Google Scholar]
- Cordoba-Rodriguez R, Fang H, Lankford CSR, Frucht DM. Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1β and IL-18. J Biol Chem. 2004;279:20563–20566. doi: 10.1074/jbc.C300539200. [DOI] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura ALM, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude G. Proteolytic Inactivation of MAP-Kinase-Kinase by Anthrax Lethal Factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- Ewing JF, Maines MD. Distribution of constitutive (HO-2) and heat-inducible (HO-1) heme oxygenase isozymes in rat tests: HO-2 displays stage-specific expression in germ cells. Endocrinology. 1995;136:2294–2302. doi: 10.1210/endo.136.5.7720678. [DOI] [PubMed] [Google Scholar]
- Freche B, Reig N, van der Goot FG. The role of the inflammasome in cellular responses to toxins and bacterial effectors. Semin Immunopathol. 2007;29:249–260. doi: 10.1007/s00281-007-0085-0. [DOI] [PubMed] [Google Scholar]
- Friedlander AM, Bhatnagar R, Leppla SH, Johnson L, Singh Y. Characterization of Macrophage Sensitivity and Resistance to Anthrax Lethal Toxin. Infect Immun. 1993;61:245–252. doi: 10.1128/iai.61.1.245-252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug G, Leemhuis J, Tiemann D, Meyer DK, Aktories K, Barth H. The host cell chaperone Hsp90 is essential for translocation of the binary Clostridium botulinum C2 toxin into the cytosol. J Biol Chem. 2003;278:32266–32274. doi: 10.1074/jbc.M303980200. [DOI] [PubMed] [Google Scholar]
- Heidemann SM, Lomo L, Ofenstein JP, Sarnaik AP. The effect of heat on cytokine production in rat endotoxemia. Crit Care Med. 2000;28:1465–1468. doi: 10.1097/00003246-200005000-00035. [DOI] [PubMed] [Google Scholar]
- Hotchkiss R, Nunnally I, Lindquist S, Taulien J, Perdrizet G, Karl I. Hyperthermia protects mice against the lethal effects of endotoxin. Am J Physiol Regulatory Integrative Comp Physiol. 1993;265:1447–1457. doi: 10.1152/ajpregu.1993.265.6.R1447. [DOI] [PubMed] [Google Scholar]
- Jackson IL, Batinic-Haberle I, Sonveaux P, Dewhirst MW, Vujaskovic Z. ROS production and angiogenic regulation by macrophages in response to heat therapy. Int J Hyperthermia. 2006;22:263–273. doi: 10.1080/02656730600594027. [DOI] [PubMed] [Google Scholar]
- Kappel M, Diamant M, Hansen MB, Klokker M, Pedersen BK. Effects of in vitro hyperthermia on the proliferative response of blood mononuclear cell subsets, and detection of interleukins 1 and 6, tumour necrosis factor-alpha and interferon-gamma. Immunol. 1991;73:304–308. [PMC free article] [PubMed] [Google Scholar]
- Kelly SM, VanSlyke JK, Musil LS. Regulation of ubiquitin-proteasome system-mediated degradation by cytosolic stress. Mol Biol Cell. 2007;18:4279–4291. doi: 10.1091/mbc.E07-05-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim SH, Li GC. Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Comm. 1999;254:264–268. doi: 10.1006/bbrc.1998.9840. [DOI] [PubMed] [Google Scholar]
- Kuckelkorn U, Knuehl C, Boes-Fabian B, Drung I, Kloetzel P-M. The effect of heat shock on 20S/26S proteasomes. Biol Chem. 2000;381:1017–1023. doi: 10.1515/BC.2000.125. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Kanneganti T-D, Franchi L, Nunez G. Caspase-1 inflammasomes in infection and inflammation. J Leukoc Biol. 2007;82:220–225. doi: 10.1189/jlb.1206756. [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:30–38. doi: 10.1128/mcb.18.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepock JR. How do cells respond to their thermal environment? Int J Hyperthermia. 2005;21:681–687. doi: 10.1080/02656730500307298. [DOI] [PubMed] [Google Scholar]
- Leppla SH. Bacillus anthracis toxins. In: Alouf JE, Popoff MR, editors. The Comprehensive Sourcebook of Bacterial Protein Toxins. Burlington, MA: Academic Press; 2006. pp. 323–347. [Google Scholar]
- Luo G-J, Sun X, Hasselgren P-O. Hyperthermia stimulates energy-proteasome-dependent protein degradation in cultured myotubes. Am J Physiol Regulatory Integrative Comp Physiol. 2000;278:749–756. doi: 10.1152/ajpregu.2000.278.3.R749. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Maroni P, Bendinelli P, Zuccorononno C, Schiaffonati L, Piccoletti R. Cellular signaling after in vivo heat shock in the liver. Cell Biol Internatl. 2000;24:145–152. doi: 10.1006/cbir.1999.0493. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrelli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol. 2007;8:497–503. doi: 10.1038/ni1459. [DOI] [PubMed] [Google Scholar]
- McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- Meinander A, Soderstrom TS, Kaunisto A, Poukkula M, Sistonen L, Eriksson JE. Fever-like hyperthermia controls T lymphocyte persistence by inducing degradation of cellular FLIPshort. J Immunol. 2007;178:3944–3953. doi: 10.4049/jimmunol.178.6.3944. [DOI] [PubMed] [Google Scholar]
- Meissner F, Molawi K, Zychlinsky A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol. 2008 doi: 10.1038/ni.1633. published online. [DOI] [PubMed] [Google Scholar]
- Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nature Immunology. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- Moayeri M, Martinez NW, Wiggins J, Young HA, Leppla SH. Mouse susceptibility to anthrax lethal toxin is influenced by genetic factors in addition to those controlling macrophage sensitivity. Infect Immun. 2004;72:4439–4447. doi: 10.1128/IAI.72.8.4439-4447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murapa P, Gandhapudi S, Skaggs HS, Sarge KD, Woodward JG. Physiological fever temperature induces a protective stress response in T lymphocyes mediated by heat shock factor-1 (HSF1) J Immunol. 2007;179:8305–8312. doi: 10.4049/jimmunol.179.12.8305. [DOI] [PubMed] [Google Scholar]
- Ostberg JR, Repasky EA. Emerging evidence indicates that physiologically relevant thermal stress regulates dendritic cell function. Cancer Immunol Immunother. 2006;55:292–298. doi: 10.1007/s00262-005-0689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajonk F, van Ophoven A, McBride WH. Hyperthermia-induced proteasome inhibition and loss of androgen receptor expression in human prostate cancer cells. Cancer Res. 2005;65:4836–4843. doi: 10.1158/0008-5472.CAN-03-2749. [DOI] [PubMed] [Google Scholar]
- Paraq HA, Raboy B, Kulka RG. Effect of heat shock on protein degradation in mammalian cells: involvement of the ubiquitin system. EMBO J. 1987;6:55–61. doi: 10.1002/j.1460-2075.1987.tb04718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–2050. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- Park S, Leppla SH. Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr Purif. 2000;18:293–302. doi: 10.1006/prep.2000.1208. [DOI] [PubMed] [Google Scholar]
- Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNγ-induced release of NO and TNFα. FEBS Lett. 1999;462:199–204. doi: 10.1016/s0014-5793(99)01502-1. [DOI] [PubMed] [Google Scholar]
- Peng JC, Hyde C, Pai S, O’Sullivan BJ, Nielsen LK, Thomas R. Monocyte-derived DC primed with TLR agonists secrete IL-12p70 in a CD40-dependent manner under hyperthermic conditions. J Immunother. 2006;29:606–615. doi: 10.1097/01.cji.0000211308.82997.4e. [DOI] [PubMed] [Google Scholar]
- Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Picard D. Heat-shock protein 90, a chaperone for folding and regulation. Cell Mol Life Sci. 2002;59:1640–1648. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polla BS, Gabert F, Peyrusse BM-N, Jacquier-Sarlin MR. Increased proteolysis of diphtheria toxin by human monocytes after heat shock: a subsidiary role for heat-shock protein 70 in antigen processing. Immunology. 2006;120:230–241. doi: 10.1111/j.1365-2567.2006.02494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Leppla SH, Schneerson R, Shiloach J. Production, recovery and immunogenicity of the protective antigen from a recombinant strain of Bacillus anthracis. J Ind Microbiol Biotechnol. 2002;28:232–238. doi: 10.1038/sj/jim/7000239. [DOI] [PubMed] [Google Scholar]
- Ratts R, Zeng H, Berg EA, Blue C, McComb ME, Costello CE, vanderSpek JC, Murphy JR. The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J Cell Biol. 2003;160:1139–1150. doi: 10.1083/jcb.200210028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J, Rummel C, Barth SW, Gerstberger R, Hubschle T. Molecular aspects of fever and hyperthermia. Neurol Clin. 2006;24:421–439. doi: 10.1016/j.ncl.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Rutherford SL. Between genotype and phenotype: protein chaperones and evolvability. Nature Genetics. 2003;4:263–274. doi: 10.1038/nrg1041. [DOI] [PubMed] [Google Scholar]
- Salles II, Tucker AE, Voth DE, Ballard JG. Toxin-induced resistance in Bacillus anthracis lethal toxin-treated macrophages. PNAS. 2003;100:12426–12431. doi: 10.1073/pnas.2134042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Aballay A. Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. PNAS. 2006;103:13092–13097. doi: 10.1073/pnas.0604050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh Y, Leppla SH, Bhatnagar R, Friedlander AM. Internalization and processing of Bacillus anthracis lethal toxin by toxin-sensitive and –resistant cells. J Biol Chem. 1989;264:11099–11102. [PubMed] [Google Scholar]
- Smith HJ, Khal J, Tisdale MJ. Downregulation of ubiquitin-dependent protein degradation in murine myotubes during hyperthermia by eicosapentaenoic acid. Biochem Biophys Res Commun. 2005;332:83–88. doi: 10.1016/j.bbrc.2005.04.097. [DOI] [PubMed] [Google Scholar]
- Squires R, Muehlbauer S, Brojatsch J. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J Biol Chem. 2007;282:34260–34267. doi: 10.1074/jbc.M705687200. [DOI] [PubMed] [Google Scholar]
- Srivastava P. Roles of heat-shock protein in innate and adaptive immunity. Nat Rev Immunol. 2002;2:185–194. doi: 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- Tanaka K-I, Namba T, Arai Y, Fujimoto M, Adachi H, Sobue G, Takeuchi K, Nakai A, Mizushima T. Genetic evidence for a protective role for heat shock factor 1 and heat shock protein 70 against colitis. J Biol Chem. 2007;282:23240–23252. doi: 10.1074/jbc.M704081200. [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Xiao W, Wang H, Calderwood SK, Xiao X. The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proinflammatory function in macrophages. J Immunol. 2007;179:1236–1244. doi: 10.4049/jimmunol.179.2.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Leppla SH. Proteasome activity is required for anthrax lethal toxin to kill macrophages. Infect Immun. 1999;67:3055–3060. doi: 10.1128/iai.67.6.3055-3060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varughese M, Chi A, Teixeira A, Nicholls PJ, Keith JM, Leppla SH. Internalization of a Bacillus anthracis protective antigen-c-Myc fusion protein mediated by cell surface anti-c-Myc antibodies. Mol Med. 1998;4:87–95. [PMC free article] [PubMed] [Google Scholar]
- Velasco S, Tarlow M, Olsen K, Shay JW, McCracken GH, Nisen PD. Temperature-dependent modulation of lipopolysaccaride-induced interleukin-1β and tumor necrosis factor α expression in cultured human astroglial cells by dexamethasone and indomethacin. J Clin Invest. 1990;87:1674–1680. doi: 10.1172/JCI115184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2007;10:332–343. doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmanski JM, Petnick-Ocwieja T, Kobayashi KS. NLR proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukoc Biol. 2008;83:13–30. doi: 10.1189/jlb.0607402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, An H, Zhou J, Xu H, Yu Y, Cao X. Hyperthermia differentially regulates TLR4 and TLR2-mediated innate immune response. Immunol Lett. 2006;108:137–142. doi: 10.1016/j.imlet.2006.11.008. [DOI] [PubMed] [Google Scholar]