

Abstract
A 27-year-old woman of Moldavian origin presented at the age of 15 with progressive proximal limb weakness and painful cramps in her calf muscles. Clinical examination revealed prominent muscle weakness in proximal muscles of the lower extremities and distal anterior compartment of legs, and mild weakness in shoulder girdle muscles. In addition, she had marked calf hypertrophy, muscle atrophy involving the anterior and posterior compartments of the thighs, and the distal anterior compartment of legs, as well as mild scapular winging and hyperlordosis. A muscle biopsy taken from the biceps brachii showed mild dystrophic changes, absent vacuoles, and abundant lobulated fibers. Immunofluorescence and Western blot assays demonstrated complete telethonin deficiency. Molecular analysis revealed a homozygous Trp25X mutation in the telethonin (TCAP) gene resulting in termination of transcription at an early point. Four families from Brazil with telethonin deficiency have previously been reported and classified as LGMD2G, but the actual frequency of this disease is unknown. With this current identification of a case outside the Brazilian population, telethonin mutation-associated LGMD should be considered worldwide.
Keywords: LGMD 2G, Telethonin, TCAP mutation, Europe
1. Introduction
Moreira et al. [1,2] reported a relatively mild form of autosomal recessive limb-girdle muscular dystrophy (LGMD) in three interrelated Brazilian families and mapped this syndrome to chromosome 17q11-12. They subsequently discovered a causative mutation in the sarcomeric protein telethonin [2], and demonstrated telethonin deficiency in the muscles of the affected patients [3]. Telethonin is a 19-kDa protein exclusively expressed in adult skeletal and cardiac muscle [4,5]. Located at the Z-disc, telethonin provides binding sites to link titin and other Z-disc-associated proteins during sarcomere assembly. Recent protein interaction studies have shown that the N-terminal region of titin interacts with the N-terminal region of telethonin (residues 1–53), making this part of telethonin critically important for proper formation in the Z-disk structures [6,7]. Telethonin is encoded by the TCAP gene which encompasses two exons and codes for 167 amino acids. Mutations in the telethonin gene are responsible for LGMD type 2G [1–3] and a small subset of hypertrophic and dilated cardiomyopathies [8–10]. Furthermore, a TCAP mutation has recently been reported in a patient suffering from intestinal pseudo-obstruction [11].
2. Materials and methods
2.1. Muscle CT scan
Muscle imaging study was performed using a helical CT scanner (HiSpeed NX/iPRO, GE Medical Systems Milwaukee, WI). Axial images were obtained at the pelvis, mid-thigh and mid-calf levels.
2.2. Muscle biopsy
A muscle biopsy taken from left biceps brachii was immediately frozen in liquid nitrogen chilled isopentane and processed by routine histological and histochemical techniques and for ultrastructural examination using standard methods.
2.3. Immunofluorescence and immunohistochemistry
Unfixed cryostat sections 6-μm-thick were processed for immunohistochemical analysis by using the streptavidin–biotin Super Sensitive™ IHC detection system (BioGenex, San Ramon, CA, USA). Antibodies against dystrophin (dys 1, dys 2 and dys 3), spectrin, utrophin, α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan, δ-sarcoglycan and dysferlin (all from Novocastra, Newcastle Upon Tyne, UK) were used at a dilution of 1:10, 1:20, 1:10, 1:100, 1:10, 1:200, 1:100, 1:100, 1:10, and 1:10, respectively. For telethonin immunofluorescence, a mouse monoclonal antitelethonin antibody (G-11, Santa Cruz, Quimigen, Madrid, Spain) was used at a dilution of 1:500. The secondary antibody Alexa 488 anti-goat (Molecular Probes, Leyden, Netherlands) was used at a dilution of 1:400. Sections were mounted with Fluorescent Mounting Medium (DakoCytomation), sealed and dried overnight at 4°C.
2.4. Western blotting
Western blot analysis was performed using 10% (for telethonin, α-sarcoglycan, γ-sarcoglycan), 9% (for calpain) or 6% (for dystrophin and dysferlin) SDS–PAGE electrophoresis. Briefly, extracted muscle proteins were transferred to nitrocellulose membrane in a Semi-Dry Transfer System (Bio-Rad, Madrid, Spain). The corresponding membranes were blocked and incubated with the mouse anti-telethonin antibody at a dilution of 1:1000, or with antibodies against dystrophin (dys 1 and dys 2), α-sarcoglycan, and calpain (calp 12a2, and calp 2c4, both from Novocastra, Newcastle Upon Tyne, UK) at a dilution of 1:250, 1:25, 1:50, 1:100, 1:100 and 1:30, respectively. Subsequently, the membranes were washed and then incubated with the corresponding secondary antibody labeled with horseradish peroxidase (Dako, Barcelona, Spain). The protein bands were detected by chemiluminescence ECL method (Amersham Biosciences, Little Chalfont, UK). The myosin band of 205-kDa stained with Coomassie Brilliant Blue R (Sigma) in the post-transfer gel was used as control of protein loading. For comparative purposes samples from eight patients suffering from a variety of muscle disorders (LGMD 2I, n = 1, LGMD 2B, n = 1, BMD, N = 2, myotilinopathy, n = 1, LGMD 2A, n = 2), and a healthy control were processed in parallel.
2.5. Telethonin (TCAP) gene analysis
The patient, her parents and her sibling were screened for the presence of mutations in the telethonin (TCAP) gene. Genomic DNA was extracted from peripheral lymphocytes and used as a template for polymerase chain reaction (PCR) amplification of each theletonin exon with intronic primers. Amplified fragments were purified using QIAquick PCR purification Kit (Qiagen) and directly sequenced using DyeTerminator™ Sequencing Protocol (Applied Biosystems). To determine whether the identified TCAP mutation is a common DNA variation, we screened by sequencing 154 individuals, 96 from Poland and the rest from the UK.
3. Results
3.1. Clinical manifestations
The patient presented at the age of 15 years with bilateral proximal weakness in the lower extremities expressed by progressive difficulty in running, climbing stairs and standing up from a chair. The illness progressed slowly and over the next several years she developed mild weakness in the shoulder muscles. Ten years after the initial symptoms she noted difficulties in performing ankle dorsiflexion. Painful cramps in calf muscles were present from the disease onset. No cardiac or respiratory symptoms were recorded. The patient was born in a Moldavian village of about 4000 inhabitants, and moved to Spain with her brother. Their parents are healthy and apparently unrelated. The patient's brother and son are clinically unaffected and have normal CK levels. No cases of neuromuscular disease have been reported in other family members. On examination, the facial and neck muscles had normal strength. She had marked calf hypertrophy, muscle wasting involving the anterior and posterior compartments of the thighs and distal anterior compartments of legs, and mild scapular winging and hyperlordosis (Fig. 1). There was prominent weakness involving quadriceps (3/5), iliopsoas (4/5), hip adductors (2/5), hip abductors (0/5), knee flexors (4/5), anterior tibialis (2/5), great toe (0/5) and finger extensors (1/5). In the upper extremities weakness was restricted to the humeral rotators (4/5) and biceps brachii (4/5). The patient was able to walk on heels, but not on toes. Achilles tendons were tightened; deep tendon reflexes were hypoactive and sensation was intact. CK level was at 1.826 U/L (normal < 167 U/L). EKG and echocardiography were normal; EMG examination was consistent with myopathy. Muscle CT scan performed at the age of 24 showed marked hypodensity of the glutei, biceps femoris, semimbranosus, semitendinosus and quadriceps muscles contrasting with marked hypertrophy of the sartorius and a relative preservation of the adductor longus. At mid-calf level, there was a selective involvement of anterior tibialis muscle, more pronounced in the left leg (Fig. 2). Neurological exam and CK levels in the patient's 26-year-old brother were normal.
Fig. 1.
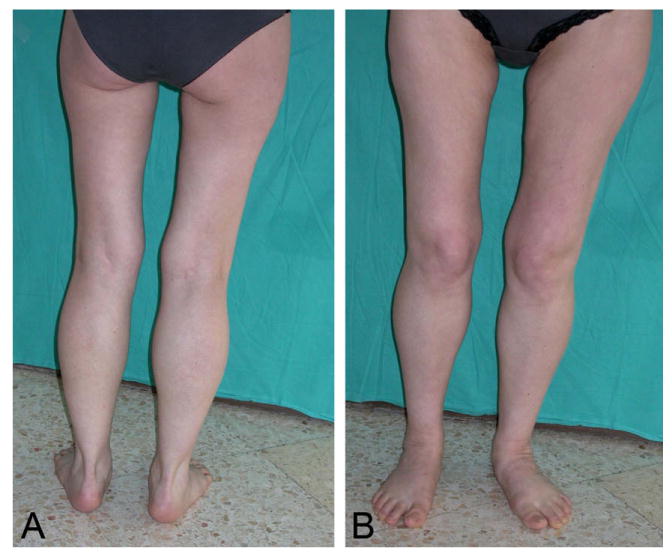
LGMD2G patient with TCAP Trp25X mutation. Atrophy of the anterior and posterior thigh muscles and anterior tibialis, marked calf hypertrophy.
Fig. 2.
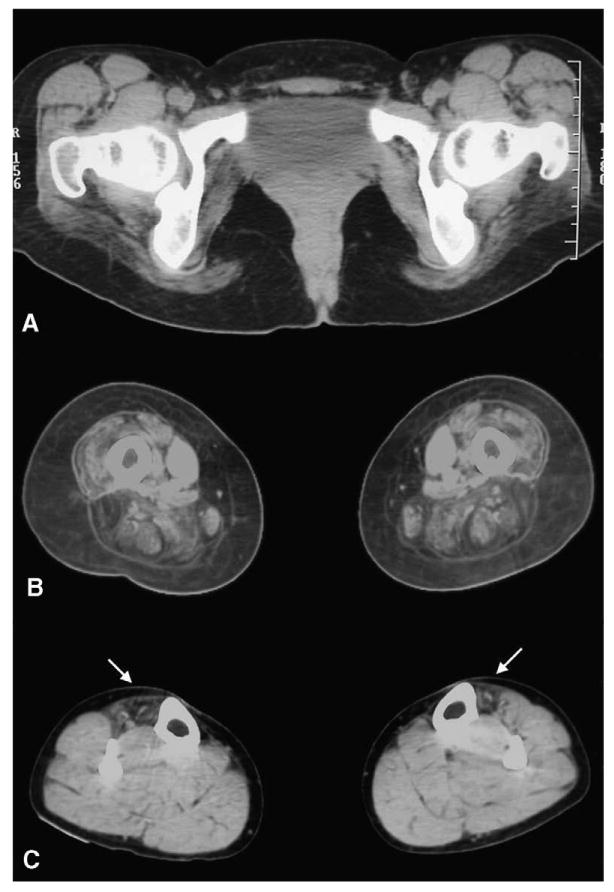
Muscle CT images at pelvic (A), mid-thigh (B), and mid-calf levels. There is marked involvement of the gluteus maximus and anterior and posterior compartment muscles of the thigh, while adductor longus is preserved and sartorius is markedly hypertrophic. At mid-calf level, anterior tibialis are selectively involved (arrows).
3.2. Myopathology, immunofluorescence and Western blot analysis
Histopathological study revealed a wide variation in the fiber size ranging from 20 to 100 μm with large numbers of hypotrophic rounded fibers of type I, central nuclei and mild increase in endomysial connective tissue. Rimmed vacuoles were not observed, and there was no muscle fiber necrosis or regeneration (Fig. 3A). On oxidative stain reactions, the atrophic type I fibers appeared lobulated, whereas other fibers showed areas of irregular staining (Fig. 3B). ATPase reaction revealed type I fiber predominance. Under electron microscopy, the overall sarcomeric structure was preserved and only minor areas of myofibrillary disorganization were seen. Immunofluorescence (Fig. 3C and D) and Western blot (Fig. 4) assays revealed lack of telethonin in skeletal muscle. Dystrophin, the four sarcoglycans and dysferlin were preserved both on immunohistochemistry and Western blots. Furthermore, Western blot to calpain revealed normal band densities.
Fig. 3.
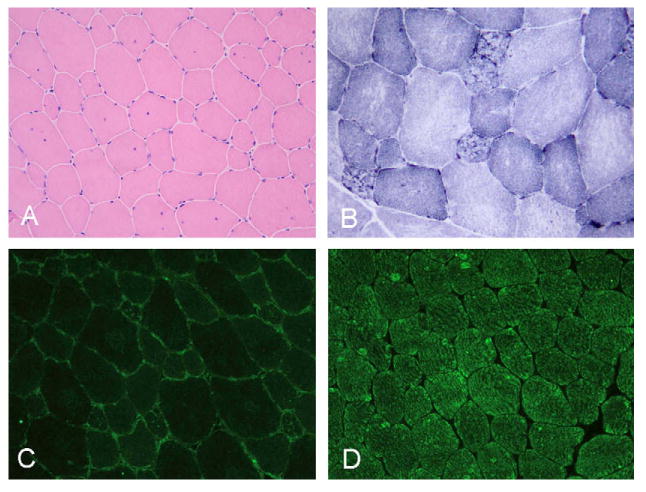
Muscle biopsy showing (A) wide variation in the fiber size with internal nuclei and mild endomysial fibrosis. On NADH reaction with (B) most of the atrophic fibers are lobulated. Lack of telethonin (C) as compared with normal sarcomeric labeling in the control (D). A, C and D 200×; B 400×.
Fig. 4.
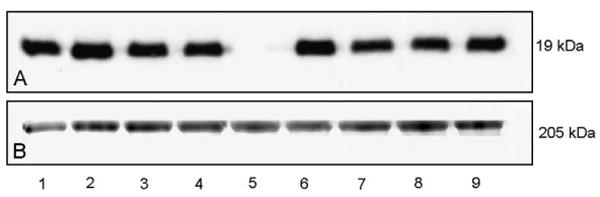
Western blotting of muscle protein extracts to telethonin in patient with LGMD 2G (A, lane 5) compared with patients suffering from LGMD2I (A, lane 1), LGMD2B (A, lane 2), Becker muscular dystrophy (A, lanes 3 and 4), myotilinopathy (A, lane 6), LGMD 2 A (A, lanes 7 and 8), and a healthy control (A, lane 9). Coomassie blue stain (B) is used as protein loading control.
3.3. Molecular studies
Analysis of the patient's TCAP gene sequences identified a TGG-to-TGA substitution at codon 25 of exon 1 predicted to replace triptophan at this position with a stop signal (Trp25X) and result in a truncated telethonin molecule which terminates at position 25 instead of normal 167 (GenBank Accession No. EU257805). This change was present on both chromosomes (Fig. 5). The early termination of transcription was confirmed by the absence of muscle telethonin reactivity: the epitope to which the used antibody is directed is located at the C-terminal part of telethonin, amino acids 58–167 (Santa Cruz Biotechnology, Inc., http://www.scbt.com/). Both patient's parents and her brother were heterozygous for this mutation. The affected region of the TCAP gene is highly conserved in the evolution (data not shown) and no irregularities in this region were detected in 154 control individuals. This suggests that TCAP Trp25X is a pathogenic myopathy-causing mutation. DNA sequence analysis ruled out mutations in the FKRP gene.
Fig. 5.
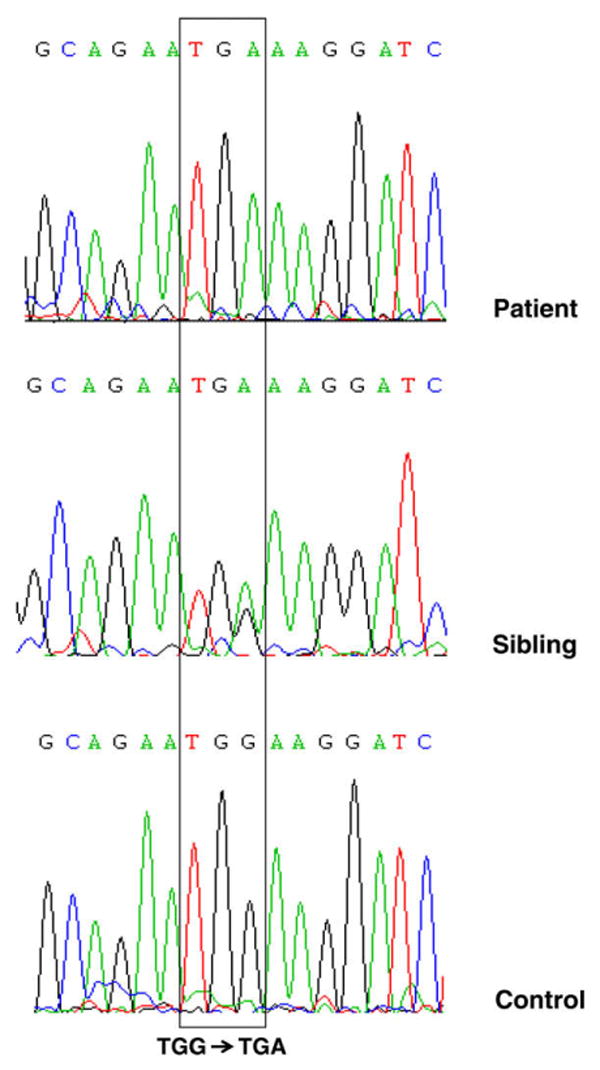
Nucleotide sequences of telethonin (TCAP) gene in a patient with LGMD 2G, her brother and a control individual. A G-to-A transition changes TCAP codon 25 sequence from TGG to TGA and results in a predicted substitution of tryptophan with Stop, thus terminating transcription of telethonin at this early point. The patient shows the mutation on both chromosomes and is therefore homozygous for this mutation; the unaffected sibling is heterozygous.
4. Discussion
We have identified a patient with 2G type autosomal recessive limb-girdle muscular dystrophy associated with a novel telethonin mutation. Even though LGMD 2G is considered a very rare disorder, the results of immunohistochemical and Western blot studies showing normal expression of dystrophin, sarcoglycans, dysferlin and calpain, the lack of mutations in FKRP gene, and the clinical phenotype of the patient showing limb-girdle and anterior tibialis muscle weakness and calf hypertrophy prompted us to investigate her for telethonin deficiency. This is the first report of LGMD2G on the European continent. Previously reported LGMD2G patients presented between 2 and 15 years of age with muscle weakness in the four limbs and calf hypertrophy. Patients had marked weakness of distal anterior leg muscles, in some cases already present from the onset of illness. Cardiomyopathy was suspected in three cases [1,2]. The patient reported in this communication developed proximal lower limb weakness and marked calf hypertrophy at the age of 15 years. She also complained of frequent painful calf cramps, a feature not reported in previous descriptions of LGMD2G. Detailed studies of this patient established the distribution of skeletal muscle involvement as severe wasting of the glutei and thigh muscles in the anterior and posterior compartments, selective atrophy of the anterior tibialis, and mild involvement of limb-girdle muscles, a pattern generally different from that observed in other forms of LGMD [12,13] and distal myopathies [14]. Muscle biopsies performed in previously reported LGMD2G patients showed mild dystrophic changes, type I fiber atrophy and complete telethonin deficiency. In 4 of 6 patients studied, rimmed vacuoles were observed, although in a small proportion of fibers. Yet, only in one patient were vacuolar changes prominent [3]. Similar pathological abnormalities, but without vacuoles, were observed in our patient. Significantly, we found a large number of lobulated fibers, a feature frequently observed in other forms of muscular dystrophy, especially in LGMD2A and LGMD2B [13]. Clinical and myopathological features characteristic of LGMD2G therefore overlap with other forms of recessive LGMD. The predominant involvement of pelvic girdle muscles with calf hypertrophy observed in LGMD2G resembles the phenotype of some LGMD2I patients. The lack of respiratory or cardiac involvement, which is frequently seen in LGMD2I, and the severe atrophy of the quadriceps and anterior tibialis muscles observed in LGMD2G help to differentiate the two conditions [12,13]. Prominent quadriceps and anterior tibialis involvement and mild scapular winging distinguish LGMD2G from LGMD2A patients. Finally, lack of involvement of posterior calf muscles distinguishes LGMD2G from the distal anterior myopathy due to dysferlin deficiency [15]. Even with all these distinguishing signs, a definitive diagnosis of LGMD2G cannot be based on clinical grounds alone. Immunohistochemical, Western blot and molecular studies are necessary in each case of autosomal recessive LGMD. However, the presence of telethonin in muscle biopsies is not routinely investigated in patients suffering from LGMD [16,17]. Therefore, since as we show here, telethonin deficiency exists outside Brazil, appropriate diagnostic investigations are indicated. This study confirms the existence and a wider than expected geographic distribution of LGMD2G.
The frequency of TCAP Trp25X mutation among the Moldavian population is currently unknown and requires further investigations. The patient's parents, originating from the same village, were found heterozygous for the mutation; therefore the possibility that they have a common ancestor cannot be ruled out.
Finally, the mechanism by which telethonin deficiency causes LGMD 2G is currently unknown. The TCAP Trp25X mutation found in our patient probably prevents telethonin interaction with titin and other proteins involved in the development and maintenance of sarcomeric structure as well as intracellular signalling processes. Furthermore, it has been recently shown that TCAP knockdown in cultured cells treated with short-interfering RNA alters myogenic differentiation and is associated with decreased expression of Myo and myogenin [18,19]. Further studies are necessary to investigate how telethonin deficiency causes muscular dystrophy.
Acknowledgments
The authors are grateful to the members of the affected family for enthusiastic participation in the study. This research was supported by F.I.S. Grant PI051213 (M.O.) and in part by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health (A.S. and L.G.G.). We thank Tom Yohanann for editorial advice.
References
- 1.Moreira ES, Vainzof M, Marie SK, et al. The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11–12. Am J Hum Genet. 1997;61:151–9. doi: 10.1086/513889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet. 2000;24:163–6. doi: 10.1038/72822. [DOI] [PubMed] [Google Scholar]
- 3.Vainzof M, Moreira ES, Suzuki OT, et al. Telethonin protein expression in neuromuscular disorders. Biochim Biophys Acta. 2002;1588:33–40. doi: 10.1016/s0925-4439(02)00113-8. [DOI] [PubMed] [Google Scholar]
- 4.Valle G, Faulkner G, De Antoni A, et al. Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett. 1997;415:163–8. doi: 10.1016/s0014-5793(97)01108-3. [DOI] [PubMed] [Google Scholar]
- 5.Mason P, Bayol S, Loughna PT. The novel sarcomeric protein telethonin exhibits developmental and functional regulation. Biochem Biophys Res Commun. 1999;257:699–703. doi: 10.1006/bbrc.1999.0531. [DOI] [PubMed] [Google Scholar]
- 6.Mues A, van der Ven FM, Young P, et al. Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Lett. 1998;428:111–4. doi: 10.1016/s0014-5793(98)00501-8. [DOI] [PubMed] [Google Scholar]
- 7.Zou P, Pinotsis N, Lange S, et al. Palindromic assembly of the giant muscle protein titin in the sarcomeric Z-disk. Nature. 2006;439:229–33. doi: 10.1038/nature04343. [DOI] [PubMed] [Google Scholar]
- 8.Knoll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–55. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi T, Arimura T, Itoh-Satoh M, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2192–201. doi: 10.1016/j.jacc.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 10.Bos JM, Poley RN, Ny M, et al. Genotype–phenotype relationships involving hypertrophic cardiomyopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab. 2006;88:78–85. doi: 10.1016/j.ymgme.2005.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzone A, Strege PR, Tester DJ, Bernard CE, Faulkner G, Degiorgio R, et al. A mutation in telethonin alters Nav1.5 function. J Biol Chem. 2008;283:16537–44. doi: 10.1074/jbc.M801744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bönnemann C, Bushby K. The limb-girdle muscular dystrophies. In: Engel AG, Franzini-Armstrong A, editors. Myology. 3rd. McGraw-Hill; 2004. pp. 1077–1121. [Google Scholar]
- 13.Fischer D, Walter MC, Kesper K, et al. Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. J Neurol. 2005;252:538–47. doi: 10.1007/s00415-005-0684-4. [DOI] [PubMed] [Google Scholar]
- 14.Udd B. Molecular biology of distal muscular dystrophies–sarcomeric proteins on top. Biochim Biophys Acta. 2007;1772:145–58. doi: 10.1016/j.bbadis.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Bönnemann C, Bushby K. The limb-girdle muscular dystrophies. In: Engel AG, Franzini-Armstrong A, editors. Myology. 3rd. McGraw-Hill; 2004. pp. 1077–1121. [Google Scholar]
- 16.Illa I, Serrano-Munuera C, Gallardo E, et al. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49:130–4. [PubMed] [Google Scholar]
- 17.Moore SA, Shilling CJ, Westra S, et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol. 2006;65:995–1003. doi: 10.1097/01.jnen.0000235854.77716.6c. [DOI] [PubMed] [Google Scholar]
- 18.Lo HP, Cooper ST, Evesson FJ, et al. Limb-girdle muscular dystrophy Diagnostic evaluation, frequency and clues to pathogenesis. Phys Rev Lett. 2008;18:34–44. doi: 10.1016/j.nmd.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 19.Markert CD, et al. TCAP knockdown by RNA interference inhibits myoblast differentiation in cultured skeletal muscle cells. Neuromusc Disord. 2008;18:413–22. doi: 10.1016/j.nmd.2008.03.010. [DOI] [PubMed] [Google Scholar]