

Abstract
Leishmania parasites cause a broad spectrum of clinical disease. Here we report the sequencing of the genomes of two species of Leishmania: Leishmania infantum and Leishmania braziliensis. The comparison of these sequences with the published genome of Leishmania major reveals marked conservation of synteny and identifies only ∼200 genes with a differential distribution between the three species. L. braziliensis, contrary to Leishmania species examined so far, possesses components of a putative RNA-mediated interference pathway, telomere-associated transposable elements and spliced leader–associated SLACS retrotransposons. We show that pseudogene formation and gene loss are the principal forces shaping the different genomes. Genes that are differentially distributed between the species encode proteins implicated in host-pathogen interactions and parasite survival in the macrophage.
Leishmaniasis is an infectious disease that is prevalent in Europe, Africa, Asia and the Americas, killing thousands and debilitating millions of people each year. With 2 million new cases reported annually and 350 million people at risk, infection by the insect-transmitted Leishmania parasite represents an important global health problem for which there is no vaccine and few effective drugs (see TDR Leishmaniasis URL in Methods). At least 20 Leishmania species infect humans, and the spectrum of diseases that they cause can be categorized broadly into three types: (i) visceral Leishmaniasis, the most serious form in which parasites leave the inoculation site and proliferate in liver, spleen and bone marrow, resulting in host immunosuppression and ultimately death in the absence of treatment; (ii) cutaneous Leishmaniasis, in which parasites remain at the site of infection and cause localized long-term ulceration; and (iii) mucocutaneous Leishmaniasis, a chronic destruction of mucosal tissue that develops from the cutaneous disease in less than 5% of affected individuals1. Infections, particularly those caused by visceralizing species, do not necessarily lead to clinical disease: despite the annual incidence of 0.5 million cases of life-threatening disease, most infections remain asymptomatic. Although host genetic variability and specific immune responses, together with the transmitting sandfly vector and environmental factors, are known to influence the outcome of infections2, the main factor that determines clinical presentation is thought to be the species of infecting parasite. For example, the New World parasite L. braziliensis is the causative agent of mucocutaneous Leishmaniasis, whereas the Old World species L. major and L. infantum, which are present in Africa, Europe and Asia, are parasites that cause cutaneous and visceral Leishmaniasis, respectively.
Sequencing the genomes of three kinetoplastid parasitic protozoa, L. major3, Trypanosoma brucei4 (the causative agent of African trypanosomiasis) and Trypanosoma cruzi5 (the causative agent of Chagas disease), previously revealed the preservation of large-scale gene synteny over 200–500 million years6. Despite a conserved core of ∼6,200 trypanosomatid genes, more than 1,000 Leishmania-specific genes have been found, many of which remain uncharacterized. Architecturally, the chromosomes of Leishmania differ from those of the trypanosome species in not having extended subtelomeric regions containing species-specific genes.
Here we have extended these studies to the genomes of two other species, L. infantum (of the subgenus Leishmania Leishmania) and L. braziliensis (of the subgenus Leishmania Viannia), and we compare these genomes with that of L. major. Against a background of conserved gene content, synteny and architecture, we have identified roughly 200 differences at the gene or pseudogene content level, including 78 genes that are restricted to individual species. In particular, the genomes show significant differences to the only other Leishmania genome published (L. major), and there is evidence of the existence of RNA-mediated interference (RNAi) machinery and transposable elements in the genome of the most divergent species, L. braziliensis. These findings suggest that a few species-specific parasite genes are important in pathogenesis, that parasite gene expression levels differ considerably between species (perhaps as a consequence of variation in gene copy number) or that, contrary to expectation, the parasite genome plays only a small part in determining clinical presentation. This study therefore provides a framework for experimentally tractable investigations into the role of a few genes that might influence the tissue-specific expression of disease associated with different Leishmania species.
RESULTS
Genome content and architecture
The L. infantum and L. braziliensis genome sequences were produced by whole-genome shotgun sequencing to five- and sixfold coverage, respectively. Comparative-grade finished sequences were produced by aligning contigs against the reference L. major sequence3 and by using PCR amplification between adjacent contig ends to confirm joins. The resulting assemblies of L. infantum and L. braziliensis contain 470 (N50 contig size of 150,519 bases) and 1,031 contigs (N50 contig size of 57,784 bases), respectively, corresponding to ∼98% of the reference 33-Mb haploid genome size (Table 1). As compared with 8,395 annotated genes in the L. major genome3, we found 8,195 and 8,314 genes in the genomes of L. infantum and L. braziliensis, respectively. Genes were manually annotated systematically, facilitated by the strong codon bias of Leishmania species7, conservation of synteny, and the absence of a significant amount of cis splicing. Thus, despite the lack of functional information for more than 50% of the genes identified, these numbers are likely to reflect closely the true gene complement in these species.
Table 1. Summary of the L. major, L. infantum and L. braziliensis genomes.
| L. major (V5.2) | L. infantum (V2) | L. braziliensis (V2) | |
|---|---|---|---|
| Chromosome number | 36 | 36 | 35 |
| Contigs | 36 | 562 | 1,041 |
| Size (bp) | 32,816,678 | 32,134,935 | 32,005,207 |
| Overall G+C content (%) | 59.7 | 59.3 | 57.76 |
| Coding genes | 8,298 | 8,154 | 8,153 |
| Pseudogenesa | 97 | 41 | 161 |
| Coding G+C content (%) | 62.5 | 62.45 | 60.38 |
Pseudogenes include genes that have in-frame stop codons and/or frameshifts but have other characteristics of coding regions, as assessed by similarity to other genes or by codon bias.
About 3–4% of the predicted proteomes of Leishmania spp. comprise conserved amino acid repeats8, which could potentially have a role in pathogenicity. For example, leucine-rich repeats comprise the largest class and can mediate interactions between the parasite surface and macrophage complement receptor9. DNA repeats comprise ∼9–10% of the three Leishmania spp. genomes, and L. braziliensis has the largest number of these repeats (data not shown).
Despite an estimated 20–100 million years of separation between the L. Viannia spp. and the L. Leishmania spp. (depending on whether the Leishmania genus was separated by migration events or the breakup of the supercontinent Gondwanda10,11), synteny is conserved for more than 99% of genes between the three genomes. Conservation within coding sequences is also high: the average amino acid identity between L. major and L. infantum is 92%, and the average nucleotide identity is 94% (L. major versus L. braziliensis, 77% and 82%, respectively; L. infantum versus L. braziliensis, 77% and 81%, respectively). On the basis of sequence similarity and chromosome architecture, the New World L. braziliensis is clearly an outlier, consistent with its subgenus classification. L. major and L. infantum both have 36 chromosomes, whereas L. braziliensis, consistent with previous linkage analysis, has only 35 chromosomes owing to an apparent fusion of chromosomes 20 and 34 (ref. 12). Unlike many pathogenic protozoa in which subtelomeres play a central part in generating diversity, directional clusters of ‘housekeeping’ genes extend to within 5 kb of the telomeres.
Sexual reproduction is not an obligatory part of the Leishmania life cycle and may occur only rarely13. Nevertheless, strong selection clearly maintains both the organization and sequence of the Leishmania genomes. A plausible explanation is that there is a spatial constraint on the organization of genes into directional clusters, which are either polycistrons or groups of genes sharing uncharacterized regulatory elements.
Retrotransposons and RNAi
In addition to selection pressure acting against chromosomal rearrangements, Leishmania may lack some of the machinery that generates diversity in other eukaryotes. A lack of transposable elements would favor chromosome stability and is seen in the genomes of L. major and L. infantum. In other kinetoplastid parasites, namely T. brucei and T. cruzi, several classes of transposable elements are present (the non–long terminal repeat (LTR) retrotransposons, ingi/L1Tc and SLACS/CZAR and the LTR retrotransposon VIPER), but the L. major genome has only remnants of ingi/L1Tc-related elements (DIREs), suggesting their loss during evolution of the Leishmania lineage14. Similarly, L. infantum and L. braziliensis also contain the ingi/L1Tc DIREs.
Unexpectedly, we found evidence in L. braziliensis for the site-specific non-LTR retrotransposon SLACS/CZAR, which is associated with tandemly repeated spliced leader sequences in an arrangement similar to that of the SLACS or CZAR element in T. brucei or T. cruzi, respectively15,16. In addition, the telomeres of L. braziliensis contain a family of 20–30 previously unknown DNA transposable elements, each including putative reverse transcriptase, phage integrase (site-specific recombinase) and DNA and/or RNA polymerase domains, which we have called ‘telomere-associated transposable elements (TATEs; Supplementary Fig. 1 online). The TATEs and their bordering regions are highly conserved and are inserted only in the telomeric hexamer repeats at the same relative position (GGG↑TTA). As observed for most mobile elements, a duplicated motif (TT), present on either side of the transposable element, seems to correspond to a target site duplication. Unlike non-LTR retrotransposons, the TATEs do not contain an APE-like endonuclease domain but they do contain a putative integrase-like domain (site-specific recombinase), related to the transposase domains of other transposable elements, that may contribute to the observed telomeric site specificity. The telomeres seem to contain clusters of tandemly arranged TATEs, including short elements probably derived from full-length elements by internal deletions. It has not been possible to determine the precise organization of the TATEs owing to their repetitive nature.
In many eukaryotes, the effects of retrotransposable elements can be regulated through a RNA silencing mechanism such as RNAi. Despite its demonstration and utility in T. brucei17, RNAi has not been detected in other kinetoplastid species including L. major and T. cruzi6,18. Our comparison revealed genes in L. braziliensis that may be involved in the RNAi pathway (Supplementary Fig. 2 online). A hallmark of this pathway in other eukaryotes is Dicer activity, which converts double-stranded RNA (dsRNA) into small interfering RNA (siRNA). A divergent gene (Tb927.8.2370) encoding a Dicer-like protein (TbDcl1) has been described in T. brucei19. The TbDcl1 protein bears the two RNAse III–like domains typical of Dicer and is required for generating siRNA-sized molecules, and its downregulation results in a less efficient RNAi response19. An ortholog of TbDcl1 has not been found in T. cruzi or L. major, trypanosomatids that lack a functional RNAi pathway. L. braziliensis, however, contains a similar gene (LbrM23_V2.0390) that is endowed with two conserved RNAse III domains. Dicer activity could also be carried out by a combination of independent proteins carrying the relevant dsRNA-binding domain, DEAD/H box RNA helicase and RNase III domains. The RNase genes implicated in this complex19 are missing in L. major and L. infantum, but present in the L. braziliensis genome at regions of otherwise conserved synteny between the Leishmania species (Supplementary Table 1 online).
Argonaute, an endonuclease involved in the dsRNA-triggered cleavage of mRNA, is another crucial component of the RNAi machinery and, unlike L. major, L. braziliensis contains an ortholog of the functional argonaute gene (TbAGO1) present in T. brucei. A second gene containing an argonaute PIWI domain (TbPWI1), which was originally identified in T. brucei and has orthologs in both Leishmania and T. cruzi, has been shown not to be involved in the RNAi pathway20. TbAGO1 can be functionally replaced by the human gene encoding Argonaute2, suggesting that TbAGO1 encodes the endonuclease activity required for mRNA target degradation in the trypanosome RNAi pathway21. The L. braziliensis gene contains the typical argonaute domains PAZ and PIWI, the latter of which contains key amino acids essential for TbAGO1 activity22. In addition, the L. braziliensis AGO1 gene encodes an amino-terminal RGG domain, which is present in TbAGO1 and shown to be essential for association with polyribosomes22.
Examination of the syntenic regions on chromosome 11 in L. major and L. infantum revealed remnants of AGO1, suggesting that the RNAi machinery has been lost from the Leishmania subgenus to which they both belong (Supplementary Table 1). In the alternative subgenus L. viannia (which includes L. braziliensis), RNA viruses have been characterized23, however, suggesting that this lineage could have retained RNAi as an antiviral defense mechanism. The RNAi machinery may also have a role in regulating the functions of transposable elements.
Genes differentially distributed between species
So far, only one gene locus has been directly implicated in Leishmania disease tropism. In Leishmania donovani, the causative agent of visceral Leishmaniasis, A2 gene products are required for parasite survival in visceral organs; by contrast, L. major contains only A2 pseudogenes24. Given this precedent, we systematically searched the three genomes in parallel (using ACT software25) for species-specific genes that might contribute to differences in disease presentation, immune response and pathogenicity. Despite the broad differences in disease phenotype, we found that few genes are specific to individual Leishmania species. Table 2 lists those genes that have been ascribed a putative function (the full list is given in Supplementary Table 2 online). We found 5 L. major–specific genes, 26 L. infantum–specific genes and ∼47 L. braziliensis–specific genes, which were distributed throughout the genome (Fig. 1) rather than concentrated in subtelomeric regions or breakpoints of directional gene clusters, as previously observed across kinetoplastid species6. In addition to the 47 genes specific to L. braziliensis, an almost equivalent number of genes are present in L. major and L. infantum but absent or degenerate in L. braziliensis.
Table 2.
Leishmania genes of putative function that vary between speciesa
| Product | L. major | L. infantum | L. braziliensis | T. brucei | T. cruzi (Tc00.1047053…) |
|---|---|---|---|---|---|
| Protein kinase | LmjF01.0750 | LinJ01.0760 | LbrM01_V2.0720 | – | 511585.160 |
| Tagatose-6-phosphate kinase | LmjF02.0030 | LinJ02.0030 | LbrM02_V2.0030 | – | – |
| Aminopeptidase P1 | LmjF02.0040 | LinJ02.0010 | LbrM02_V2.0060 | – | – |
| SLACS | – | – | LbrM02_V2.0550 LbrM02_V2.0720 |
Tb09.211.5015 | – |
| 31-0-demethyl-FK506 methyltransferase | LmjF04.1165 | LinJ04.1185 | LbrM04_V2.1180 | – | – |
| Viscerotropic gene | LmjF05.0240 | LinJ05.0340 | LbrM05_V2.0230 | – | 503583.100 |
| Flavoprotein subunit protein | LmjF07.0800 | LinJ07.0870 | LbrM07_V2.0880 | – | – |
| CFAS, putative | – | LinJ08.0560 | LbrM08_V2.0590 | – | – |
| Argonaute | LmjF11.0570 | LinJ11.0500 | LbrM11_V2.0360 | Tb10.406.0020 | – |
| EF hand protein | LmjF13.1450 | LinJ13.1380 | – | – | – |
| PI3K | LmjF14.0020 | LinJ14.0020 | LbrM14_V2.0020 | – | 508859.90 |
| Carboxypeptidase | LmjF14.0180 | LinJ14.0180 | LbrM14_V2.0180 | – | – |
| Guanine nucleotide-binding protein | LmjF14.0760 | LinJ14.0800 | LbrM14_V2.0740 | – | 510989.30 |
| Flagellar Ca2+-binding protein | LmjF16.0910 | LinJ16.0950 | LbrM16_V2.0920 | Tb08.5H5.30 | 507891.38 |
| Flagellar Ca2+-binding protein | LmjF16.0920 | LinJ16.0960 | LbrM16_V2.0930 | Tb08.5H5.50 | 507891.47 |
| Transporter (sugar) | LmjF18.0040 | LinJ18.0040 | LbrM18_V2.0050 | Tb10.61.2747 | 507993.310 |
| Glycerol uptake protein | LmjF19.1347 | LinJ19.1260 | LbrM19_V2.1570 | Tb10.61.0380 | 511355.40 |
| Phosphate-repressible phosphate permease | – | LinJ20.0040 | LbrM10_V2.0990 | – | – |
| Zn2+-binding phosphatase | LmjF20.1480 | LinJ20.1530 | LbrM20_V2.5730 | Tb927.1.3300 | 510636.50 |
| Methylenetetrahydrofolate dehydrogenase | LmjF22.0340 | LinJ22.0330 | – | – | 511809.20 |
| Phosphoinositide-specific phosphatase C | LmjF22.1680 | LinJ22.1500 | LbrM22_V2.1590 | Tb11.02.3780 | 504149.160 |
| Argininosuccinate synthase | LmjF23.0260 | LinJ23.0300 | LbrM23_V2.0290 | – | – |
| Oxoreductase | LmjF23.0670 | LinJ23.0810 | LbrM23_V2.0770 | – | – |
| RNase III domain gene | – | – | LbrM23_V2.0390 | Tb927.8.2370 | – |
| HASPA | LmjF23.1040,1082, 1088 | LinJ23.1160, 1200 | – | – | – |
| SHERP | LmjF23.1050, 1080, 1086 | LinJ23.1170, 1190 | – | – | – |
| HASPB | LmjF23.1060, 1070 | LinJ23.1180 | – | – | – |
| Transcription elongation factor | LmjF24.0200 | LinJ24.a | LbrM24_V2.0190 | – | 507669.104 |
| Multi-pass transmembrane protein | LmjF24.0700 | LinJ24.0350 | LbrM24_V2.0710 | Tb11.02.3050 | 503789.20 |
| Lysophospholipase | LmjF24.1840 | LinJ10.0030 | LbrM24_V2.1910 | – | – |
| RNase III gene | – | - | LbrM25_V2.1020 | Tb927.3.1230 | – |
| Glutathioneylspermidine synthase | LmjF25.2380 | LinJ25.2500 | LbrM25_V2.1980 | – | 508479.110 |
| Adenine phosphoribosyltransferaseb | – | – | LbrM26_V2.0120 | Tb927.7.1790 | 507519.150 |
| Eukaryotic translation release factor | LmjF27.1710 | LinJ27.1220 | LbrM27_V2.1850 | Tb11.22.0012 | 506127.110 |
| Lipase | LmjF29.1260 | LinJ29.1500 | LbrM29_V2.1340 | Tb927.3.3860 | 504029.21 |
| Multidrug resistance protein | – | LinJ30.1840 | LbrM24_V2.1400 | Tb927.8.2160 | – |
| Triacyglycerol lipase | LmjF31.0830 | LinJ31.0860 | LbrM31_V2.1010 | – | – |
| n-Acyl-l-amino acid amidohydrolaseb | – | LinJ31.1490 | – | – | – |
| p-Nitrophenylphosphataseb | – | LinJ31.3030 | – | – | – |
| Helicase | LmjF32.1590 | LinJ32.1990 | LbrM32_V2.1760 | Tb11.01.6420 | 503677.20 |
| β-Galactofuranosyle transferase | – | – | LbrM20_V2.0480 | – | 504115.30 |
| Aminophospholipid translocase | LmjF34.3220 | LinJ34.2740 | LbrM20.2800 | Tb927.4.1510 | 511003.10 |
| Galactokinase | – | – | LbrM35_V2.3650 | – | – |
| Cysteine peptidase | LmjF35.3910 | LinJ35.4000 | LbrM35_V2.3890 | – | – |
| l-Ribulokinase | LmjF36.0060 | LinJ36.2610 | LbrM36_V2.0100 | – | – |
| Amino acid transporter | – | – | LbrM36_V2.1500 | – | – |
| Phosphatidylinositol/phosphatidylcholine/ SEC14 cytosolic factor |
– | LinJ36.2050 | LbrM36_V2.0690 | – | 510293.20 |
Gene found in the sequencing reads but not assembled into the genome.
Gene diversification after duplication.
Pseudogenes are indicated in boldface; coding genes, without boldface. Table 1 identifies those genes with a putative function that are differently distributed between the three Leishmania species. The full list of genes, including those encoding hypothetical proteins, is given in Supplementary Table 2.
Figure 1.

Chromosome 32 of L. major showing the positions of genes with a differential distribution between the three Leishmania species analyzed. The organization of chromosome 32 is shown schematically; both strands containing long, non-overlapping gene clusters2. Genes that are restricted to only one or two of the three Leishmania species are not concentrated in the subtelomeric regions or at the breakpoint between polycistronic transcription units, as seen in other kinetoplastid parasites5, but are distributed more evenly along the chromosome. Most gene differences are a result of pseudogene formation rather than insertion or deletion of new sequences.
Given 20–100 million years of divergence within the Leishmania genus, the small number of species-specific differences in gene content is unexpected. For example, more than 1,000 genes differ between the human infective Plasmodium falciparum and the rodent malarial species26, which may have diverged over a similar timescale because the mouse and human lineages diverged from their common ancestor 75 million years ago27.
We found no obvious breaks in synteny or evidence that translocations or segmental duplications have served to generate lineage-specific diversity in Leishmania. We did, however, find clear instances where tandem duplication, followed by diversification, accounts for species-specific differences; for example, copies of a hydrolase gene in L. infantum (LinJ31.3030) and an adenine phosphoribosyltransferase gene in L. braziliensis (LbrM26_V2.0120) seem to have arisen and diverged from an adjacent gene. Larger tandem gene arrays are a characteristic feature of all kinetoplastid parasite genomes6, facilitating increased protein expression in the absence of gene regulation by transcription initiation. Although correctly assembling highly repetitive regions is technically difficult from randomly sequenced DNA, the depth of assembled reads provides an indication of the number of repeat units present in specific regions. The largest family of surface-expressed protein genes in Leishmania, the amastins, are specifically expressed by intracellular parasites in the host28. In L. major, the largest amastin array (comprising 21 out of 54 amastin genes) is interspersed with repeat units of the unrelated tuzin genes that encode proteins of unknown function. Although similar in organization, the amastin-tuzin array seems to be reduced in size by at least half in L. braziliensis (on the basis of the depth of coverage of reads across this repeat region). By contrast, the surface-expressed GP63 zinc metalloproteinases, which function in host cell binding and parasite protection from complement-mediated lysis29, are encoded by a repeated gene cluster that seems to be enlarged fourfold in L. braziliensis as compared with L. major or L. infantum.
A major determinant of lineage-specific differences in gene content seems to be pseudogene formation. The species specificity of ∼80% of the genes listed in Table 2 and Supplementary Table 2 can be attributed to the deterioration of an existing coding sequence in the two other species: in each case, there is a degenerate sequence in the corresponding region of synteny in the species that lacks the ‘functional’ gene. This observation contrasts with an analysis of other kinetoplastid species, where gene insertions or substitutions were found more commonly to generate genus-specific sequences6.
We identified 23 pseudogenes, present in all three species, that show little degeneracy, suggesting that they have become pseudogenes recently or are under positive selection (Supplementary Table 2). In addition, they are interrupted by both frameshifts and in-frame stop codons in different positions across the three species (Fig. 2), indicating that they have arisen independently three times in the Leishmania lineage. Strong codon bias, a feature of Leishmania coding sequences, and sequence similarity are maintained in each pseudogene, and in-frame UAG or UAA stop codons are present in almost all, thereby ruling out translation through selenocysteine incorporation, a process that has been described in Leishmania30. For several pseudogenes, non-degenerate orthologs were identified in T. brucei and T. cruzi. Functions could be conceptually ascribed, on the basis of sequence similarity, to 12 pseudogenes, and in many cases relate to housekeeping (for example, carboxypeptidase, phosphoglycerate kinase, oxidoreductase, glutamamyl carboxypeptidase, aminoacyclase, epsilon-adaptin and beta-adaptin).
Figure 2.
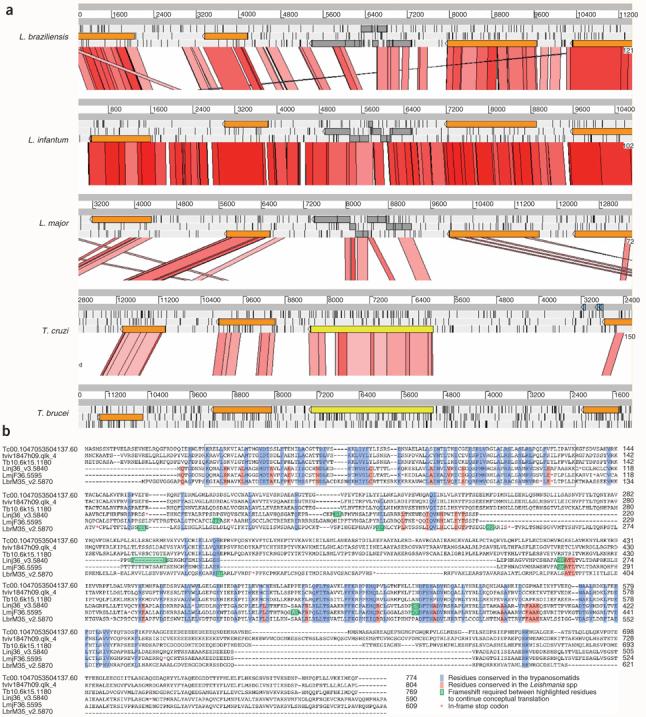
Conserved pseudogenes in Leishmania species. Many Leishmania genes present in all three species retain sequence conservation but have frameshifts and/or in-frame stop codons. Some of these pseudogenes have intact syntenic orthologs in other kinetoplastids. (a) Comparison, using the sequence tool ACT, of a region of conserved synteny containing orthologs of the beta-adaptin 4 gene (gray/yellow) and the adjacent syntenic genes (brown) from L. major, L. infantum, L. braziliensis, T. cruzi and T. brucei. Gray bars represent the forward and reverse strands of DNA. The red-pink lines between sequences represent sequence similarity (tBLASTx). Each of the Leishmania orthologs of the beta-adaptin 4 gene (gray) contains several frameshifts and stop codons, whereas the two trypanosome species have uninterrupted intact copies (yellow). Gene prediction of the Leishmania pseudogenes was done by using similarity and codon bias. (b) Alignment of amino acid sequences from the three Leishmania species with their orthologs in T. cruzi, T. brucei and Trypanosoma vivax, showing that there are conserved domains across all species. The N-terminal β-adaptin domain (boxed region) shows conservation between all six species, and the most conserved residues correspond to residues that are restricted in higher eukaryotes.
Of ∼200 genes with a differential distribution between Leishmania species, the functions of only 34% could be annotated on the basis of sequence similarity or protein domain searches (Table 2 and Supplementary Table 2). Some gene products have similarity to proteins of unknown function in different organisms, whereas others are unique to the Leishmania species analyzed. Not surprisingly, a single candidate that might explain the different disease tropisms of the individual species did not emerge; however, many significant gene differences were identified.
One gene in L. infantum, which has become a pseudogene in L. braziliensis but seems to be absent from L. major, encodes a putative phosphatidylinositol or phosphatidylcholine transfer protein (PITP), SEC14 cytosolic factor. An apparently intact ortholog is present in T. cruzi but not in T. brucei. Although the precise role of this protein is unknown, it has been implicated in the budding of secretory vesicles from the trans-Golgi network31 and could therefore influence cell-surface molecule expression in L. infantum, affecting host-parasite interactions as a result.
Another L. infantum gene, which is a pseudogene in the other Leishmania species and T. brucei (but not in T. cruzi), encodes a putative phosphatidylinositol 3-kinase (PI3K). This PI3K has the remnants of a Ras-binding domain, a C2 lipid-binding domain, and accessory and catalytic domains reminiscent of class I PI3Ks present in other eukaryotes, including Dictyostelium discoideum, yeast and mammals. The only true PI3K identified in trypanosomatids so far is VPS34, a class III PI3K present in T. brucei32. Orthologs of VPS34 are present in all Leishmania species, but the L. infantum–specific class I PI3K is novel. Evidence suggests that PI3Ks and PITPs can work synergistically at the trans-Golgi to facilitate vesicle budding33 but, given the properties of class I PI3Ks in other systems and the large number of downstream effectors, the L. infantum PI3K might influence as yet unidentified processes that may have an impact on parasite tropism.
Another L. infantum–specific gene encodes glutathionylspermidine synthase (GspS), which is required for synthesis of the unusual thiol trypanothione that functions in protecting the parasite against oxidative stress. Although both GspS and trypanothione synthetase (TryS) are required to generate trypanothione in the related organism Crithidia fasciculata, a broad specificity trypanothione synthetase substitutes for both GspS and TryS in T. brucei and T. cruzi34. The gene encoding TryS in L. major is also sufficient to generate trypanothione, although a GspS pseudogene is also present in the genome35 and, with only four mutations, could be the result of a recent acquisition. Despite a much greater predicted period of separation, the L. braziliensis genome also has a clearly identifiable GspS pseudogene (with approximately ten mutations) with highly conserved domains.
Cyclopropane fatty acids (CFAs), although rare in eukaryotes, are common plasma membrane components in some bacteria and have been previously detected in lipid extracts from some but not all Leishmania species36. Consistent with that analysis, a single gene encoding cyclopropane fatty acyl phospholipid synthase (CFAS) is present in both L. infantum and L. braziliensis but not in L. major. In bacteria, cyclopropanation by CFAS requires S-adenosyl methionine (as a methylene donor) in a modification predicted to maintain the integrity of the plasma membrane—an important factor in the innate immune response to Mycobacterium tuberculosis infection37. The Leishmania CFAS gene is most similar to its bacterial homologs, and strong phylogenetic evidence (Supplementary Fig. 3 online) suggests that the Leishmania lineage acquired this gene by horizontal transfer (and secondary loss from L. major). Given that neither the enzyme nor its fatty acid modification are present in humans, CFAS is a putative chemotherapeutic target for the most severe form of leishmaniasis. In addition, the presence of this gene in some species but not others may explain published experimental data38 on the effects of the S-adenosyl methionine analog sinefungin, a compound with known antiparasitic properties. This drug inhibits the growth of L. donovani parasites (which are closely related to L. infantum and also have a CFAS gene) but has little effect on L. major38.
A notable absence from the L. braziliensis genome is the multigene HASP/SHERP locus, which encodes the HASP family of hydrophilic acylated surface proteins (expressed exclusively in infective stages of L. major and L. donovani) and the vector-stage–specific SHERP protein39. Although deletion of this region in L. major does not influence virulence, its overexpression causes increased sensitivity to complement-mediated parasite lysis and reduced viability in host macrophages40.
Gene evolution
In addition to the small number of species-specific and differentially distributed genes, other genetic factors are likely to define the differences between the species. We therefore searched for genes with signatures of positive selection as an indicator that they may be involved in host-pathogen interactions (Methods). Those genes with the highest ratios of non-synonymous to synonymous mutations (dN/dS) were, for the most part, involved in undefined biological processes (Supplementary Table 3 online). We found, however, that ∼8% of genes seem to be evolving at different rates between the three Leishmania species (Supplementary Table 4 online) and are involved in a spectrum of core processes (including transport, biopolymer metabolism, cellular metabolism, lipid metabolism and RNA metabolism), which might influence parasite survival in the host and disease outcome (Supplementary Table 5 online).
DISCUSSION
Comparisons of the complete genomes of three species of Leishmania have revealed a greater extent of synteny and similarity than would be expected, given their predicted period of separation. Contrary to previous comparisons of distantly related kinetoplastid genomes, gene loss and pseudogene formation are the principal factors shaping the Leishmania genomes. We have found little evidence of lineage-specific genetic acquisition accounting for differences between these parasite species.
Given our poor understanding of the way in which different human-infective species of the Leishmania genus cause diverse clinical disease, the identification of only a few differentially distributed parasite genes should facilitate timely experimental verification of their role in disease development. In addition, the unexpected identification of a putative RNAi pathway increases the likelihood that the findings from the three genome projects can be translated into insights into gene function. The potential to manipulate gene expression by RNAi, perhaps by using a tetracycline-inducible promoter system (as demonstrated in L. donovani41), may be especially useful to complement the classical ‘two-step gene knockout’ strategy for disruption of Leishmania gene function42. Identification of a few genes that are either species-specific or under positive selective pressure provides a comprehensive and manageable resource to target efforts in identifying parasite factors that influence infection. Conversely, factors that are unique to the Leishmania genus but common to all species may be used as potential drug targets or vaccine candidates.
METHODS
DNA preparation
Details of the sequenced L. major strain have been published3. L. infantum JPCM5 (MCAN/ES/98/LLM-877)43 and L. (Viannia) braziliensis M2904 (MHOM/BR/75M2904)44 were the strains selected for analysis here. The L. infantum JPC (MCAN/ES/98/LLM-724) strain, from which the JPCM5 clone used in the sequencing project was derived, was isolated in the WHO Collaborating Centre for Leishmaniasis, ISCIII, Madrid, Spain, from the spleen of a naturally infected dog residing in the area in 1998 (ref. 43). The parasites were tested for virulence by inoculation into hamsters: parasites were recovered from the spleen 15 weeks after infection. The parasites also infected the human U937 macrophage cell line and the dog DH82 macrophage cell line43.
L. (Viannia) braziliensis clone LB2904 (MHOM/BR/75M2904) is a reference strain from Evandro Chagas Institute, Belém, Brazil. This strain was isolated by direct culture from a lesion on the right side of the thorax of a man who had been performing survey work in Serra dos Carajás, Brazilian Amazonia. The LB2904 clone is infective in hamsters and BALB/c mice and can be genetically transfected and cloned on plates. The L. infantum and L. braziliensis strains used are available on request from D.F.S. or J.C.M., and A.K.C., respectively.
Sequencing
The following methodology for sequencing, assembly, finishing and annotation applies to both L. infantum and L. braziliensis. A whole-genome shotgun strategy was used and produced roughly sixfold coverage of the whole genome from plasmid clones containing small fragments of up to 4 kb inserted into the pUC19 vector (Sanger Institute). Problems associated with high G+C sequence were addressed by optimizing the sequencing mixture (a 4:1 ratio of standard Big Dye terminator mix and dGTP Big Dye mix with the addition of dimethylsulfoxide). Sequence reads were assembled with PHRED/PHRAP on the basis of overlapping sequence and were edited in a GAP4 database45. The quality of the reads for both projects was similar: 91.5% of L. infantum and 92.7% of L. braziliensis bases had a quality score (derived from the PHRED score generated by GAP4; ref. 45) >70 (P = 1.0−7). In comparison, in the finished genome of L. major 96.8% of bases exceeded this value.
Regions containing repeat sequences or with an unexpected read depth were manually inspected. We used positional information from sequenced read-pairs to help to resolve the orientation and position of contigs. Pre-finishing used an automated in-house software program (Auto-Prefinish) to identify primers and clones for additional sequencing to close physical and sequence gaps by oligo-walking. In addition, end sequences from a L. braziliensis fosmid library (4–5-fold clone coverage) were produced to provide paired-read information from 40-kb inserts. The assembled contigs were iteratively ordered and orientated by alignment to the L. major genome sequence and by manual checking. In particular, we re-examined regions with apparent breaks in synteny for potential mis-assembly errors or genuine breaks. Information from orientated read-pairs, together with additional sequencing from selected large insert clones, was used to resolve potential mis-assemblies. Version 2 of the L. infantum and L. braziliensis genomes were used for the subsequent analyses reported here.
Annotation
Manual annotation of the L. major genome3 was transferred to the assembled genomes of both L. infantum and L. braziliensis on the basis of BLASTp matches and positional information by using an in-house Perl script. Gene models were manually inspected and further edited, where appropriate, with Artemis software46. New gene models were identified by using a combination of CodonUsage47 and Hexamer48, and by visualizing tBLASTx comparisons of regions with conserved synteny using ACT software25. We compared protein sequences against the non-redundant protein database UniProt and an in-house kinetoplastid-only database. Repetitive regions can largely account for small discrepancies in apparent sequence coverage and gene number.
Evolutionary analysis
For the dN/dS analysis, three-way positional orthologs were identified by a combination of reciprocal BLAST and manual curation of conserved synteny regions. Codon-based alignments were produced by using codeml from the PAML package49 and the settings: model = 0 (one dN/dS estimate over whole tree) for the dN/dStree estimates, and model = 1 (one dN/dS estimate for each branch of tree) for the dN/dSbranch estimates, with the assumption that orthologous rates were equivalent. dN/dS estimates were considered significantly different between species if 2(lnLmodel1 – lnLmodel0) > 5.911 (5% χ2 critical value with 2 d.f.). Genes with dN/dS > 5, or 2(lnLmodel1 – lnLmodel0) ≤ 0 were excluded from further analysis. Mann-Whitney tests were used to determine whether groups of genes had significantly higher or lower dN/dS values as compared with all other genes. A Kruskall-Wallis test was used to determine whether differences in dN/dSbranch values were significant between species for genes grouped by gene ontology category.
For repeat sequences, genome-wide searches were undertaken with RepSeq8 to identify amino acid repeats. We used RepeatScout50 and RepeatMasker to identify nucleic acid repeats.
CFAS phylogeny
The CFAS gene was identified as a potential lateral transfer by similarity searching (BLASTp) against the GenBank non-redundant protein database using the L. infantum CFAS sequence as query. To assemble the data set for phylogenetic analysis, all sequences with an e-value of <10−30 were downloaded. Note that, although eukaryotes were not specifically excluded from this process, none of the eukaryotic sequences in GenBank, which includes the completely sequenced genomes of Trypanosoma cruzi and Trypanosoma brucei, met the e-value cut-off criterion.
Sequences were aligned with MUSCLE using default parameters. Regions of poor alignment where homology could not be ascertained with confidence were identified by eye and excluded. We conducted preliminary analyses of all sequences by unweighted parsimony using PAUP. The data set was narrowed down through successive rounds of analysis and sequence removal to obtain a final subset of sequences that were broadly representative of the full data set.
The final tree was derived by bayesian inference using a mixture of amino acid models. Alignment positions were weighting according to evolutionary rate by using a four-category γ-distribution with the shape parameter α calculated by the program on the basis of a neighbor-joining tree. Analyses consisted of two sets of four chains run for 600,000 generations with results saved every 1,000 generations. Analyses were run until both sets of chains converged (split frequency = 0.007), and tree topology and posterior probabilities were calculated after discarding a 25% burn-in (150 trees). The tree topology was further tested with 100 replicates of maximum likelihood bootstrapping by the program PhyML using a JTT substitution model with a four-category γ-distribution and with the shape parameter α calculated by the program.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the support of the Wellcome Trust Sanger Institute core sequencing and informatics groups. We thank N. Goldman (European Bioinformatics Institute) for advice on the evolutionary analysis, C. Hertz-Fowler for help in constructing the figures, J. Shaw for his help in selecting the strain for the L. braziliensis genome sequencing project and D. Harper for quality scores on the sequencing projects. This study was funded by the Wellcome Trust through its support of the Pathogen Sequencing Unit at the Wellcome Trust Sanger Institute. L.O.B. and J.C.R. were recipients of Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) fellowships. D.P.D. was supported by a postgraduate studentship from the Biotechnology and Biological Sciences Research Council. J.C.R. received financial support from the UNICEF/UNDP/WORLD BANK/WHO Special Programme for Research and Training in Tropical Diseases (TDR).
Footnotes
Accession codes European Molecular Biology Laboratory (EMBL): L. infantum chromosomes 1–36, AM502219 to AM502254; L. braziliensis chromosomes 1–35, AM494938 to AM494972.
URLs The L. infantum and L. braziliensis genome sequencing reads, quality files and annotated consensus sequences can be accessed from the following FTP sites: ftp://ftp.sanger.ac.uk/pub/pathogens/L_infantum/, ftp://ftp.sanger.ac.uk/pub/pathogens/L_braziliensis/. The fully annotated genomes for all three species of Leishmania are also available for searching, viewing and downloading at the GeneDB database (http://www.genedb.org). Other URLs: MUSCLE, http://phylogenomics.berkeley.edu/cgi-bin/muscle/input_muscle.py; PAUP, http://www.molecularevolution.org/software/paup*/; PhyML, http://atgc.lirmm.fr/phyml/; pUC19 vector information, http://www.sanger.ac.uk/Teams/Team53/psub/sequences/pUC19.shtml; RepeatMasker, http://www.repeatmasker.org/; TDR Leishmaniasis URL, http://www.who.int/tdr/diseases/leish.
Supplementary information is available on the Nature Genetics website.
COMPETING INTERESTS STATEMENT The authors declare no competing financial interests.
Published online at http://www.nature.com/naturegenetics
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Marsden PD. Mucosal leishmaniasis (‘espundia’ Escomel, 1911) Trans. R. Soc. Trop. Med. Hyg. 1986;80:859–876. doi: 10.1016/0035-9203(86)90243-9. [DOI] [PubMed] [Google Scholar]
- 2.Lipoldova M, Demant P. Genetic susceptibility to infectious disease: lessons from mouse models of leishmaniasis. Nat. Rev. Genet. 2006;7:294–305. doi: 10.1038/nrg1832. [DOI] [PubMed] [Google Scholar]
- 3.Ivens AC, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berriman M, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 5.El-Sayed NM, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 6.El-Sayed NM, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- 7.Myler PJ, et al. Leishmania major Friedlin chromosome 1 has an unusual distribution of protein-coding genes. Proc. Natl. Acad. Sci. USA. 1999;96:2902–2906. doi: 10.1073/pnas.96.6.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Depledge DP, et al. A database of amino acid repeats present in lower eukaryotic pathogens. BMC Bioinformatics. 2007;8:122. doi: 10.1186/1471-2105-8-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kedzierski L, et al. A leucine-rich repeat motif of Leishmania parasite surface antigen 2 binds to macrophages through the complement receptor 3. J. Immunol. 2004;172:4902–4906. doi: 10.4049/jimmunol.172.8.4902. [DOI] [PubMed] [Google Scholar]
- 10.Kerr SF. Molecular trees of trypanosomes incongruent with fossil records of hosts. Mem. Inst. Oswaldo Cruz. 2006;101:25–30. doi: 10.1590/s0074-02762006000100006. [DOI] [PubMed] [Google Scholar]
- 11.Momen H, Cupolillo E. Speculations on the origin and evolution of the genus Leishmania. Mem. Inst. Oswaldo Cruz. 2000;95:583–588. doi: 10.1590/s0074-02762000000400023. [DOI] [PubMed] [Google Scholar]
- 12.Britto C, et al. Conserved linkage groups associated with large-scale chromosomal rearrangements between Old World and New World Leishmania genomes. Gene. 1998;222:107–117. doi: 10.1016/s0378-1119(98)00472-7. [DOI] [PubMed] [Google Scholar]
- 13.Victoir K, Dujardin JC. How to succeed in parasitic life without sex? Asking Leishmania. Trends Parasitol. 2002;18:81–85. doi: 10.1016/s1471-4922(01)02199-7. [DOI] [PubMed] [Google Scholar]
- 14.Bringaud F, et al. Evolution of non-LTR retrotransposons in the trypanosomatid genomes: Leishmania major has lost the active elements. Mol. Biochem. Parasitol. 2006;145:158–170. doi: 10.1016/j.molbiopara.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Aksoy S, Williams S, Chang S, Richards FF. SLACS retrotransposon from Trypanosoma brucei gambiense is similar to mammalian LINEs. Nucleic Acids Res. 1990;18:785–792. doi: 10.1093/nar/18.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villanueva MS, Williams SP, Beard CB, Richards FF, Aksoy S. A new member of a family of site-specific retrotransposons is present in the spliced leader RNA genes of Trypanosoma cruzi. Mol. Cell. Biol. 1991;11:6139–6148. doi: 10.1128/mcb.11.12.6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ngo H, Tschudi C, Gull K, Ullu E. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA. 1998;95:14687–14692. doi: 10.1073/pnas.95.25.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson KA, Beverley SM. Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Mol. Biochem. Parasitol. 2003;128:217–228. doi: 10.1016/s0166-6851(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 19.Shi H, Tschudi C, Ullu E. An unusual Dicer-like1 protein fuels the RNA interference pathway in Trypanosoma brucei. RNA. 2006;12:2063–2072. doi: 10.1261/rna.246906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durand-Dubief M, Kohl L, Bastin P. Efficiency and specificity of RNA interference generated by intra- and intermolecular double stranded RNA in Trypanosoma brucei. Mol. Biochem. Parasitol. 2003;129:11–21. doi: 10.1016/s0166-6851(03)00071-9. [DOI] [PubMed] [Google Scholar]
- 21.Shi H, Tschudi C, Ullu E. Functional replacement of Trypanosoma brucei Argonaute by the human slicer Argonaute2. RNA. 2006;12:943–947. doi: 10.1261/rna.20806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi H, Ullu E, Tschudi C. Function of the trypanosome Argonaute 1 protein in RNA interference requires the N-terminal RGG domain and arginine 735 in the Piwi domain. J. Biol. Chem. 2004;279:49889–49893. doi: 10.1074/jbc.M409280200. [DOI] [PubMed] [Google Scholar]
- 23.Stuart KD, Weeks R, Guilbride L, Myler PJ. Molecular organization of Leishmania RNA virus 1. Proc. Natl. Acad. Sci. USA. 1992;89:8596–8600. doi: 10.1073/pnas.89.18.8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang WW, et al. Comparison of the A2 gene locus in Leishmania donovani and Leishmania major and its control over cutaneous infection. J. Biol. Chem. 2003;278:35508–35515. doi: 10.1074/jbc.M305030200. [DOI] [PubMed] [Google Scholar]
- 25.Carver TJ, et al. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 26.Kooij TW, et al. A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PLoS Pathog. 2005;1:e44. doi: 10.1371/journal.ppat.0010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waterston RH, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 28.Rochette A, et al. Characterization and developmental gene regulation of a large gene family encoding amastin surface proteins in Leishmania spp. Mol. Biochem. Parasitol. 2005;140:205–220. doi: 10.1016/j.molbiopara.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Yao C, Donelson JE, Wilson ME. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol. Biochem. Parasitol. 2003;132:1–16. doi: 10.1016/s0166-6851(03)00211-1. [DOI] [PubMed] [Google Scholar]
- 30.Cassago A, et al. Identification of Leishmania selenoproteins and SECIS element. Mol. Biochem. Parasitol. 2006 doi: 10.1016/j.molbiopara.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Bankaitis VA, Malehorn DE, Emr SD, Greene R. The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J. Cell Biol. 1989;108:1271–1281. doi: 10.1083/jcb.108.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall BS, et al. TbVps34, the trypanosome orthologue of Vps34, is required for Golgi complex segregation. J. Biol. Chem. 2006;281:27600–27612. doi: 10.1074/jbc.M602183200. [DOI] [PubMed] [Google Scholar]
- 33.Jones SM, Alb JG, Jr., Phillips SE, Bankaitis VA, Howell KE. A phosphatidylinositol 3-kinase and phosphatidylinositol transfer protein act synergistically in formation of constitutive transport vesicles from the trans-Golgi network. J. Biol. Chem. 1998;273:10349–10354. doi: 10.1074/jbc.273.17.10349. [DOI] [PubMed] [Google Scholar]
- 34.Oza SL, Tetaud E, Ariyanayagam MR, Warnon SS, Fairlamb AH. A single enzyme catalyses formation of Trypanothione from glutathione and spermidine in Trypanosoma cruzi. J. Biol. Chem. 2002;277:35853–35861. doi: 10.1074/jbc.M204403200. [DOI] [PubMed] [Google Scholar]
- 35.Oza SL, Shaw MP, Wyllie S, Fairlamb AH. Trypanothione biosynthesis in Leishmania major. Mol. Biochem. Parasitol. 2005;139:107–116. doi: 10.1016/j.molbiopara.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 36.Beach DH, Holz GG, Jr., Anekwe GE. Lipids of Leishmania promastigotes. J. Parasitol. 1979;65:201–216. [PubMed] [Google Scholar]
- 37.Rao V, Fujiwara N, Porcelli SA, Glickman MS. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J. Exp. Med. 2005;201:535–543. doi: 10.1084/jem.20041668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bachrach U, et al. Inhibitory activity of sinefungin and SIBA (5′-deoxy-5′-S-isobutylthio-adenosine) on the growth of promastigotes and amastigotes of different species of Leishmania. FEBS Lett. 1980;121:287–291. doi: 10.1016/0014-5793(80)80364-4. [DOI] [PubMed] [Google Scholar]
- 39.Knuepfer E, Stierhof YD, McKean PG, Smith DF. Characterization of a differentially expressed protein that shows an unusual localization to intracellular membranes in Leishmania major. Biochem. J. 2001;356:335–344. doi: 10.1042/0264-6021:3560335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKean PG, Denny PW, Knuepfer E, Keen JK, Smith DF. Phenotypic changes associated with deletion and overexpression of a stage-regulated gene family in Leishmania. Cell. Microbiol. 2001;3:511–523. doi: 10.1046/j.1462-5822.2001.00135.x. [DOI] [PubMed] [Google Scholar]
- 41.Yan S, et al. A low-background inducible promoter system in Leishmania donovani. Mol. Biochem. Parasitol. 2002;119:217–223. doi: 10.1016/s0166-6851(01)00418-2. [DOI] [PubMed] [Google Scholar]
- 42.Cruz A, Coburn CM, Beverley SM. Double targeted gene replacement for creating null mutants. Proc. Natl. Acad. Sci. USA. 1991;88:7170–7174. doi: 10.1073/pnas.88.16.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denise H, et al. Studies on the CPA cysteine peptidase in the Leishmania infantum genome strain JPCM5. BMC Mol. Biol. 2006;7:42. doi: 10.1186/1471-2199-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurentino EC, et al. A survey of Leishmania braziliensis genome by shotgun sequencing. Mol. Biochem. Parasitol. 2004;137:81–86. doi: 10.1016/j.molbiopara.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Bonfield JK, Smith K, Staden R. A new DNA sequence assembly program. Nucleic Acids Res. 1995;23:4992–4999. doi: 10.1093/nar/23.24.4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 47.Staden R, McLachlan AD. Codon preference and its use in identifying protein coding regions in long DNA sequences. Nucleic Acids Res. 1982;10:141–156. doi: 10.1093/nar/10.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Claverie JM, Sauvaget I, Bougueleret L. K-tuple frequency analysis: from intron/exon discrimination to T-cell epitope mapping. Methods Enzymol. 1990;183:237–252. doi: 10.1016/0076-6879(90)83017-4. [DOI] [PubMed] [Google Scholar]
- 49.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 50.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.