

Abstract
Recent literature has highlighted an important role of inflammation in promoting cancer. However, the immune system can also play a central role in protecting the body against cancer as well as infection, although its role in cancer is not well understood. A study published in the September issue of Nature Medicine adds a new twist to the role of inflammation in cancer. Apetoh et al. describe how activation of innate immunity after conventional radiation or chemotherapy can trigger protective antitumor immunity.
Inflammation is a salutary response to insult or injury and an important part of innate immunity; however, chronic inflammation has been linked with the development of cancer. Individuals with ulcerative colitis, a chronic inflammatory disease of the colon, have a 10-fold higher likelihood of developing colorectal carcinoma. Similarly, inflammatory conditions of the liver, such as chronic hepatitis and cirrhosis, are well established risk factors for the development of hepatocellular carcinoma. Recent research has highlighted an important role for inflammation in cancer from the perspective that innate immune cells, such as macrophages, drive malignant progression through the production of proinflammatory mediators such as tumor necrosis factor (TNF)-α and interleukin (IL)-6 (Greten et al., 2004; Maeda et al., 2003; Rakoff-Nahoum et al., 2004). In the context of gastric or colon cancer, the stimulus for activation of the innate immune cells may be provided by chronic infection with Helicobacter pylori or commensal bacteria that access the resident inflammatory cells through a breakdown in the barrier function of the epithelium during carcinogenesis. In cervical cancer and hepatocellular carcinoma, chronic infection with human papilloma virus (HPV) and hepatitis C virus (HCV), respectively, are clearly linked with carcinogenesis. The recent study by Naugler et al., using a mouse model of chemically induced liver cancer, suggests cell injury may also lead to the release of endogenous factors that activate innate immune cells. These authors showed that dead hepatocytes activate liver macrophages (Kupffer cells) through the molecule MyD88, which is an essential adaptor for Toll like receptor (TLR) signaling (Lawrence et al., 2007; Naugler et al., 2007).
The TLRs are pathogen recognition molecules that are hard-wired to trigger activation of innate immunity upon recognition of pathogen-associated molecular patterns (PAMPs). TLRs have an important role in driving the inflammatory response but also in priming adaptive immunity through the activation and maturation of antigen-presenting cells, including dendritic cells (DCs) and macrophages. Apetoh et al. (2007) have revealed an interesting role of inflammation and TLR signaling in cancer therapy. The authors studied the immunostimulatory properties of dying tumor cells after chemotherapy or radiation therapy. Using TLR4 and MyD88-deficient DCs, they show that TLR4 signaling is required for crosspresentation of antigens from apoptotic tumor cells on MHC class I to generate antitumor cytotoxic T cell (CTL) responses. Apetoh et al. also identify a “danger signal” from dying tumor cells, the nuclear protein high-mobility group box 1 protein (HMGB1) that triggers this protective immune response through activation of TLR4. DCs modulate adaptive immunity in response to signals delivered within the local inflammatory milieu; these may be PAMPs or endogenous “danger signals” generated by tissue injury. HMGB1 is released during necrotic cell death and also secreted from activated macrophages, NK cells, and mature myeloid dendritic cells (Lotze and Tracey, 2005). Apetoh et al. show that HMGB1 derived from dying tumor cells regulates antigen crosspresentation by DCs through activation of TLR4. Furthermore, they show that mutation of TLR4 in mice reduces the efficacy of both chemotherapy and radiation therapy. Finally, the authors identify a mutation in the human TLR4 gene that is associated with an increased frequency of metastasis in breast cancer patients after conventional chemotherapy, suggesting that TLR4 signaling may affect clinical outcome in patients.
The concept that activation of innate immunity could promote a protective host response to malignancy is not new. Dr. William Coley, in the late 19th century, applied this philosophy to treat cancer and developed what became known as Coley's vaccine, or Coley's toxin, which was in fact a soup of killed Streptococcus pyogenes and Serratia marcescens (Coley, 1894). Coley extrapolated the association of postoperative infections with improved clinical outcome. The approach is still used today in the form of bacillus Calmette-Guerin (BCG) for treatment of bladder cancer (Bassi, 2002). Researchers have tried in vain to identify the “active” component of both Coley's vaccine and BCG for treatment of cancer; however, it is likely these agents trigger an innate immune response through multiple TLRs, which may provide both direct tumoricidal activity and a platform for the development of antitumor immunity.
The major antigen-presenting cells present in tumors are macrophages, which in certain cases may account for as much as 50% of the tumor mass; however, often it is not possible to detect an adaptive immune response to tumor antigens. There is increasing evidence that tumor associated macrophages (TAMs) express animmunosuppressive phenotype and display several protumoral functions, including promotion of angiogenesis and matrix remodeling (Balkwill et al., 2005; Pollard, 2004). Although usually rare, DCs have been detected in several tumor types, but DCs in tumors have been shown to express an immature phenotype and therefore to have low immunostimulatory properties (Mantovani et al., 2002). Both DCs and macrophages have the ability to pick up tumor antigens for crosspresentation on MHC class I molecules (Ardavin et al., 2004). However, the phenotype of TAMs and intratumoral DCs has been suggested to promote tolerance through production of immune-suppressive factors rather than prime a protective immune response (Mantovani et al., 2002). The data from Apetoh et al. suggest tumor cell death after radiation or chemotherapy leads to the release of endogenous factors (HMGB1) that are able to “reset” the innate immune system and provide prolonged antitumor protection through priming of CTLs (Figure 1). This information may be exploited therapeutically with the use of adjuvants to enhance immunogenicity of conventional cancer therapy.
Figure 1. HMGB1: An Endogenous Adjuvant for Antitumor Immune Responses.
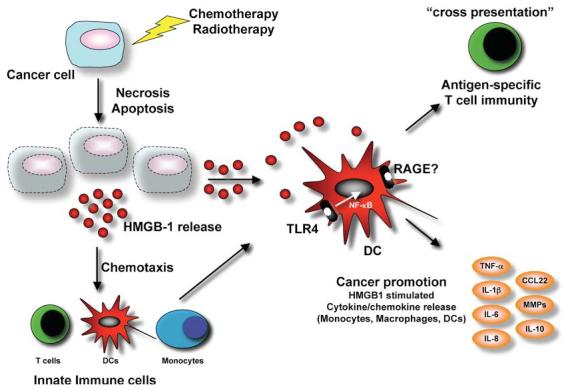
Damage of cancer cells (radiation, chemotherapy) leads to the release of signal molecules, e.g., HMGB1, but also other chemokines and cytokines from the necrotic and apoptotic cells. HMGB1 act as a chemoatractant for monocytes, T cells, and dendritic cells to the tumor microenvironment. Additionally, HMGB1 can bind to its main receptor RAGE, receptor for advanced glycation end products, but also interacts with TLR4 signaling. Activation of TLR4 by HMGB1 on dendritic cells induces crosspresentation and the generation of antigen-specific T cells. Activated DC or macrophages may also release tumor-promoting proinflammatory cytokines upon TLR4:HMGB1 activation.
So, this is the big question: what's the difference between inflammation that drives cancer progression and inflammation that inhibits tumor growth?
REFERENCES
- Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Nat. Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- Ardavin C, Amigorena S, Reis e Sousa C. Immunity. 2004;20:17–23. doi: 10.1016/s1074-7613(03)00352-2. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Charles KA, Mantovani A. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Bassi P. Surg. Oncol. 2002;11:77–83. doi: 10.1016/s0960-7404(02)00008-7. [DOI] [PubMed] [Google Scholar]
- Coley WB. Trans. Am. Surg. Assn. 1894;12:183–212. [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Hagemann T, Balkwill F. Science. 2007;317:5834–5835. doi: 10.1126/science.1146052. [DOI] [PubMed] [Google Scholar]
- Lotze M, Tracey K. Nat. Rev. Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Trends Immunol. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Nat. Rev. Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]