

Summary
The intracellular target of diphtheria toxin is a modified histidine residue, diphthamide, in the translation elongation factor, eEF2. This enigmatic modification occurs in all eukaryotes, and is produced in yeast by the action of five gene products, DPH1 to DPH5. Sequence homologues of these genes are present in all sequenced eukaryotic genomes and in higher eukaryotes there is functional evidence for DPH1, 2, 3, and 5 acting in diphthamide biosynthesis. We have identified a mouse mutant in the remaining gene, Dph4. Cells derived from homozygous mutant embryos lack the diphthamide modification of EF2 and are resistant to killing by diphtheria toxin. Reporter-tagged DPH4 protein localizes to the cytoskeleton, in contrast to the localization of DPH1, and consistent with evidence that DPH4 is not part of a proposed complex containing DPH1, 2 and 3. Mice homozygous for the mutation are retarded in growth and development and almost always die before birth. Those that survive long enough have preaxial polydactyly, a duplication of digit 1 of the hind foot. This same defect is seen in embryos homozygous for mutation of DPH1, suggesting that lack of diphthamide on eEF2 could result in translational failure of specific proteins, rather than a generalized translation downregulation.
INTRODUCTION
Diphthamide is an enigmatic posttranslational modification occurring on a single amino acid residue of a single protein in the eukaryotic proteome. Diphthamide modification is present in all eukaryotic organisms, where it is restricted to a histidine residue of the translation elongation factor 2, eEF2 (position 715 in mammals and 699 in yeast) (Robinson et al, 1974; van Nesset al 1980). There is also evidence of a diphthamide modification, or a precursor of diphthamide, in the analogous elongation factor of archaebacteria (Pappenheimer et al, 1983). However, it is absent from EFG, the eubacterial orthologue of eEF2. The modification produces the target of diphtheria toxin (DT) and Pseudomonas aeruginosa exotoxin A (ETA). The DT and ETA toxins catalyse the transfer of ADP-ribose from NAD+ to the diphthamide residue, inactivating eEF2 and causing a lethal blockage in protein synthesis, reviewed in (Collier, 2001).
The normal function of diphthamide is not entirely clear. The modified histidine is located at the tip of a domain loop within the eEF2 structure that is proposed to mimic the anticodon loop of tRNA (Jorgenson et al, 2005; Jorgenson et al 2006; Ortiz et al, 2006). It has been suggested that this loop, bearing the diphthamide modification, stabilizes the tRNA anticodon-mRNA codon interaction and is necessary to maintain the translation reading frame. Analysis of cells lacking diphthamide shows that unmodified eEF2 results in an increase of -1 frameshifting during translation (Ortiz et al, 2006).
Diphthamide biosynthesis has been well characterized in yeast, and has been shown genetically to require the action of five proteins, DPH1-5 (Chen et al, 1985; Liu et al, 2004). Functional homologues of four of these, DPH1, 2, 3 and 5 have been identified in mammals, by identification of mutant CHO cells, in which they have been called respectively CG-4, -3 −2 and −1 (Liu and Leppla, 2003; Moehring and Moehring, 1979; Moehring et al, 1980; Moehring et al, 1984; Nobukuni et al, 2005). Diphthamide-deficient yeast strains have allowed analysis of the pathway, which proves to be a complex, multi-step process (Liu et al, 2004). The first step involves the transfer of a 3-amino-3-carboxypropyl moiety from S-adenosylmethionine (AdoMet) to the C-2 imidazole of the target histidine to produce an intermediate structure, and requires DPH1 to 4. Subsequently, trimethylation of the intermediate by an AdoMet-dependent methyltransferase produces diphthine, a process requiring DPH5 (Mattheakis et al, 1992). The final step is the amidation of the side chain carboxyl group of diphthine by an ATP-dependent enzyme, resulting in diphthamide. No mutations have been identified in this final step as diphthine can also be ADP-ribosylated by DT and ETA, although at a slower rate than diphthamide (Moehring and Moehring, 1979) meaning that genetic selection cannot be used, and an ATP-dependent amidation enzyme is still to be identified (Liu et al, 2004).
The yeast Dph4 gene has an orthologue detectable in mammalian genome sequences, but no functional data is available. Furthermore, no mutations corresponding to Dph4 have been found in CHO cells. Here we describe the identification of a mutation in the mouse orthologue of Dph4, from a novel genetic screen for lethal mutations. The protein product of Dph4 contains a J-domain, which is the hallmark of the Dnaj or Hsp40 protein family, suggesting that it acts as an Hsp70 co-chaperone. Homozygous mutation of Dph4 results in absence of diphthamide on eEF2 in embryos and resistance to diphtheria toxin in mutant cells derived from them. The homozygous mutation is generally lethal by embryonic day 14 (E14), and embryos have retarded growth from as early as E9. However, if the homozygous embryos survive long enough to begin digit formation, the majority have a distinctive polydactyly phenotype on their hind limbs, similar to embryos mutant in Dph1.
RESULTS
A Genetic Screen for Lethal Mutations on Mouse Chromosome 2
We established a genetic screen in mice to identify both visible and lethal mutations in the vicinity of the Pax6 gene on chromosome 2. The screen is illustrated in Figure 1a. In brief, mutagenised male mice are crossed with females carrying a point mutation in Pax6, which confers a small-eye phenotype (Pax6Sey-Neu) (Hill et al, 1991). Small-eyed offspring from this cross carry a potentially mutant chromosome 2, opposite the mutant Pax6, and these are crossed again to mice bearing a deletion (Pax6Sey-1H) (Kent et al, 1997) encompassing Pax6 and other neighbouring genes (Figure 1b). Offspring from this cross have four potential genotypes; one class, with the compound Pax6Sey-Neu/Sey-1H genotype dies around birth. Two classes of genotype have small eyes; those carrying either the point mutation or the deletion. The latter animals are phenotypically distinct from the point mutation carriers by virtue of their smaller size and unpigmented spots on the belly and feet and these animals potentially will have a newly induced point mutation in one of the genes spanned by the deletion. If this new mutation has a recessive phenotype it will be revealed by the deletion and observed in these mice. If the phenotype is lethal the deletion class will be absent, and the mutant chromosome can be recovered from the non-small-eyed siblings who will be carriers.
Figure 1.
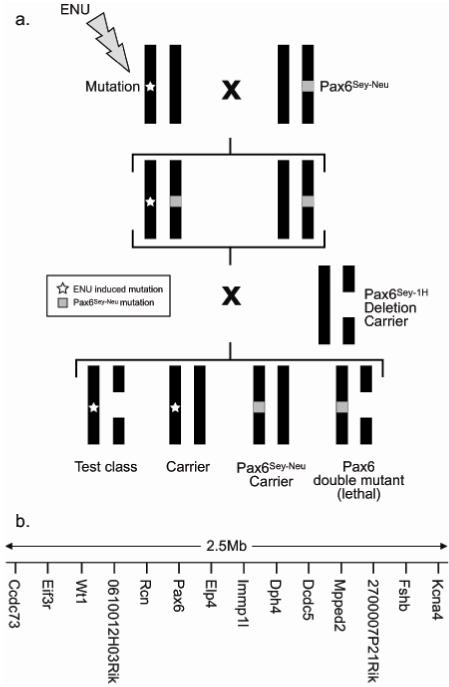
a.) Schematic of genetic cross to screen for mutations in the Pax6 deletion interval. Mutagenised males are crossed with females carrying the Pax6Sey-Neu small eye point mutation. Small eye offspring (a fraction of which will be carrying a novel point mutation in the region) are crossed with mice carrying the Pax6Sey-1H deletion. The four resultant genotypes are shown below.
b.) Gene content in the region encompassed by the Pax6Sey-1H deletion.
We screened 233 pedigrees, each deriving from a single offspring of a mutagenised male. One pedigree produced only a single deletion carrier of 35 offspring in the test generation, and thus likely harbours a lethal mutation in the deletion region. The surviving animal may be due to incomplete penetrance of the mutation or because meiotic recombination has removed the mutation from the non-deletion chromosome. Using microsatellite markers flanking the deletion we were able to follow the mutant chromosome in heterozygotes in subsequent generations. Intercrossing heterozygotes resulted in the birth of 143 offspring, of which only 9 were homozygotes (compared to an expected number of 36; χ2 = 20.02, P <0.001). Six of these did not survive to weaning; the remaining three were small and had an additional digit on one or both hindlimbs (see below). We expected that this line, MUTS1/14, would contain a mutation within the Pax6Sey-1H deletion.
Identification of a Splice Mutation in Dph4
The Pax6Sey-1H deletion is less than 2.5Mb in length and encompasses all or part of 14 genes, from Ccdc73 to Kcna4, which are candidates for the lethal mutation (R. Edgar and S.H. Cross, unpublished results) (Figure 1b). Whilst breeding heterozygous carrier mice we found a recombination within this interval between a mutant and a wild type chromosome, which established a smaller interval of about 1.4 Mb within which the mutation was located. This contained the ten genes from Ccdc73 to Dcdc5. We sequenced the coding exons and splice sites of genes in this interval and found a T to A transversion at the splice donor site of intron 4 of a gene containing a J-domain and a CSL-zinc-finger domain (Figure 2a). Analysis of the sequence databases revealed that this gene is the single mammalian orthologue of the yeast gene, Dph4, required for diphthamide modification of eEF2. We therefore called this mouse gene Dph4.
Figure 2.
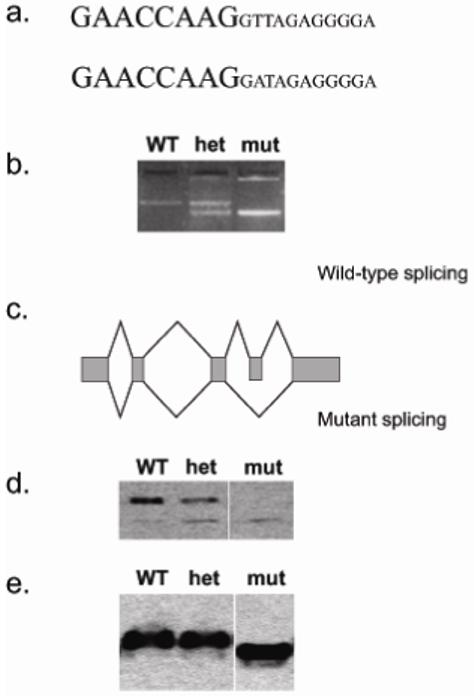
a.) Mutation of Dph4. Upper, wild type (C57BL6/J) sequence. Lower, mutant sequence. Large font, exon 4 sequence. Small font, intron 4 sequence.
b.) Reverse transcriptase- PCR analysis of mRNA from wild type (WT) heterozygous (het) and homozygous mutant (mut) embryos. The wild type allele produces a 246bp fragment, and the mutant allele produces one of 177bp. The heterozygous produces contains both fragments.
c.) Schematic illustrating the splicing pattern of the wild type (upper) and mutant (lower) Dph4 gene.
d.) Western blot to detect DPH4 protein in wild type (WT) heterozygous (het) and homozygous mutant (mut) E12.5 embryos. The lower band is a non-specific reaction of the antiserum, which serves as a loading control. Tracks are from the same gel but lane order has been edited for clarity.
e.) Native gel and western blot, probed to detect eEF2 protein in wild type (WT) heterozygous (het) and homozygous mutant (mut) E12.5 embryos. Mutant protein has a +1 charge shift relative to heterozgote and wild type, and therefore migrates faster. Tracks are from the same gel but lane order has been edited for clarity.
To investigate the consequences of the splice site mutation we performed RT-PCR on cDNA isolated from wild-type, heterozygous and homozygous embryos. Using primers within exons 3 and 5, we could amplify a 246bp cDNA fragment encompassing exon 4 from wild-type mRNA. On amplifying cDNA from homozygous mutant embryos the PCR product was smaller by 69bp, the size of exon 4, and DNA sequencing showed that this exon was absent from the cDNA. Both wild type and mutant PCR products were amplified from heterozygous cDNA. Thus, the ENU-induced mutation results in the skipping of exon 4 in the mutant mRNA. (Figure 2b, c).
We expressed the wild type mouse DPH4 peptide as a fusion protein in E.coli and raised an antibody in rabbits against the DPH4 component. Western blotting of proteins from wild-type embryos probed with this antibody revealed a band of approximately 17 kD, close to the predicted molecular weight of the protein (Figure 2d). The exon skipping resulting from the splice site mutation should cause an in-frame deletion, which would produce a protein internally truncated by 23 amino acids in mutant and heterozygous embryos. However, Western blot analysis of these embryos did not detect a truncated protein (Figure 2d). In particular, in homozygous embryos there was no protein of the predicted size (nor any other novel band). We conclude that the translation product of the internally-deleted Dph4 mRNA must be unstable within the cell and is degraded. The mutant embryos therefore lack not only full length Dph4 protein, but also lack the shortened, mutant version, and presumably lack all Dph4 function.
Loss of Dph4 Results in Lack of Diphthamide Modification of eEF2 and Resistance to Diphtheria Toxin
We anticipated that the homozygous mutant embryos would lack the diphthamide modification of EF2 protein. Diphthamide modification of His715 results in an additional positive charge on eEF2, which can be detected by non-denaturing gel electrophoresis. Protein extracts from wild-type, mutant and heterozygous embryos were separated by native PAGE and probed on Western blots with an antibody against eEF2. Figure 2e shows that EF2 from mutant embryos has a faster electrophoretic mobility, consistent with a charge of -1 relative to the protein from wild type embryos, consistent with lack of diphthamide modification. Notably the eEF2 from heterozygotes had a mobility identical to wild-type consistent with normal presence of diphthamide.
We predicted that eEF2 from mutant cells would be resistant to ADP-ribosylation by diphtheria toxin (DT), and the cells would be resistant to its toxic effects. Mouse cells are normally resistant to DT due to a lack of the surface receptor via which the B-subunit of the toxin gains access to the cell (Naglich et al, 1992; Stenmark et al, 1988). However, if the A-subunit of the toxin (DTA) is transfected into mouse cells it triggers rapid cell death. We isolated embryonic fibroblasts (MEFs) from wild type, mutant and heterozygous embryos, The MEFs were transfected with the firefly luciferase gene, with luciferase plus the DTA gene, or with luciferase plus a vector-only plasmid. Cotransfection with DTA into wild-type or heterozygous cells results in cell death and thus a reduction of luciferase activity (Figure 3). By contrast, cotransfection of DTA has no effect on luciferase activity in mutant cells, indicating that are they lack the diphthamide target of DT (Figure 3).
Figure 3.
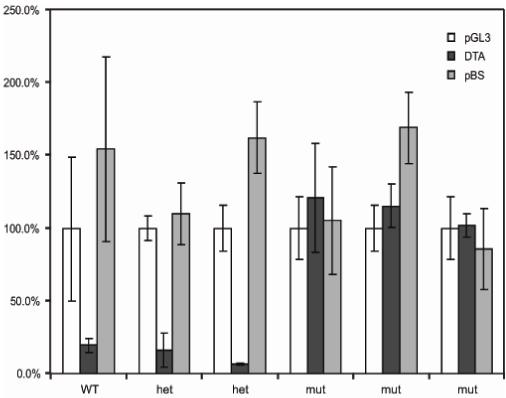
The activity of diphtheria toxin on mouse embryonic fibroblasts. White bars indicate luciferase activity following transfection of pGL3-P luciferase reporter alone into the cells. Black bars show luciferase activity after transfection of the luciferase reporter plus pPGK-DTA at a molar ratio of 4:1. Grey bars are cotransfection control showing luciferase activity following transfection of the luciferase reporter plus pBluescript vector only, at a molar ratio of 4:1. Transfections into six different cell lines are shown representing one wild-type (WT), two heterozygous (het) and three homozygous mutant (mut) genotypes. For each set of transfections the mean of the luciferase reporter alone is normalized to 100%. Error bars are +/- standard deviation.
DPH4 Protein Localises to the Cytoskeleton
The DPH1 and DPH2 proteins interact in mammalian cells (Liu et al, 2004), and there is evidence from yeast that DPH3 also interacts with both of them (Fichtner, 2003), suggesting that there may be a multimeric protein complex which catalyses diphthamide biosynthesis. The mouse DPH1 protein (also known as OVCA1) has been localized to the perinuclear region by transfection of an epitope-tagged gene (Chen and Behringer, 2001). As DPH4 participates in the same biochemical pathway we asked if it colocalised with DPH1. We cotransfected NIH-3T3 cells with a Myc-tagged Dph1 construct and FLAG-tagged Dph4 and localized both proteins by immunofluorescence. Surprisingly, whilst we see DPH1 in a punctuate, perinuclear pattern, in agreement with the published localization, DPH4 appears to localize to the cytoskeleton (Figure 4a-c). To confirm this localization we stained cells transfected with FLAG-tagged Dph4 with the cytoskeletal marker phalloidin, which indicated that the tagged DPH4 does indeed localise to the cytoskeleton (Figure 4d-i).
Figure 4.
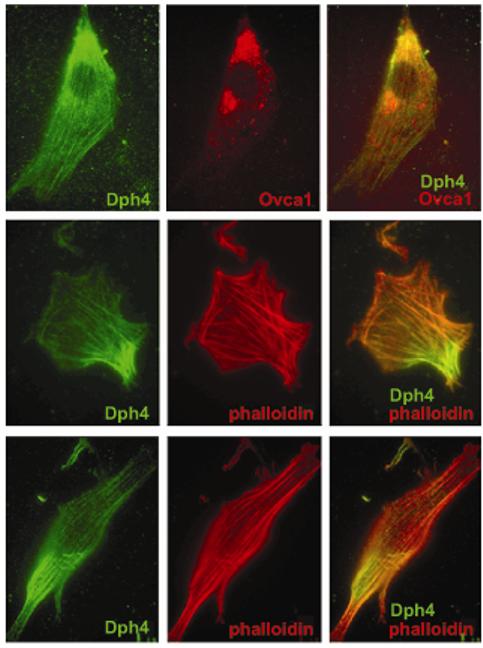
Subcellular localization of DPH4 protein. NIH-3T3 cells transfected with FLAG-tagged DPH4 (all panels) and myc-tagged OVCA1 (upper panels). Cells were subsequently stained with anti-FLAG antibodies to detect DPH4 (green), anti-myc to detect OVCA1 (red, upper panels) or with phalloidin to detect microfilaments (middle and lower panels). Right-hand panels are merged images.
Dph4 Mutant Mice Are Growth Retarded and Have a Specific Digit Defect
Initial observations indicated a deficit in the number of births of homozygous Dph4 mutant embryos, and that the homozygotes were small. Following repeated backcrosses onto a C3H/HeH background homozygotes were no longer born. To investigate this embryonic lethality we collected and genotyped embryos at a range of developmental time points and observed their gross morphology. Table 1 shows that homozygous embryos are present throughout gestation, albeit at lower than expected numbers. Even as early as E10.5 homozygous embryos were smaller than wild type or heterozygous littermates (Figure 5a). This apparent growth deficit persisted and is clearly seen at E11.5 to E13.5 (Figure 5b-d). After E14.5 homozygous embryos could be recovered but all had died at an earlier stage and were beginning to be resorbed.
Table 1.
Genotypes of embryos from an intercross of carriers of the Dph4 mutation on a C3H background
| Embryonic Age | Wild type | Heterozygous | Homozygous |
|---|---|---|---|
| E8.5 | 3 | 10 | 3 |
| E9.5 | 3 | 4 | 1 |
| E10.5 | 10 | 15 | 3 |
| E11.5 | 7 | 12 | 2 |
| E12.5 | 6 | 12 | 3 |
| E13.5 | 10 | 26 | 5 |
| E14.5 and older | 16 | 36 | 9 |
Figure 5.
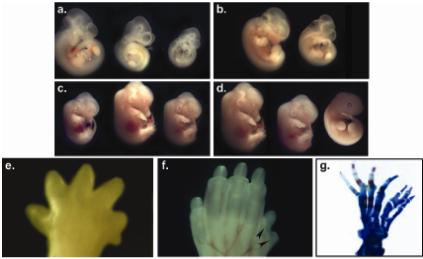
The developmental consequences of Dph4 mutation.
a-d. Dph4 homozyogus mutants are small in size.
a. E10.5 embryos; left, wild type embryo; centre and right, two homozygous mutant embryos.
b. E11.5 embryos; left, wild-type; right, homozygous mutants.
c. E12.5 and E13.5 embryos; left, E12.5 wild-type embryo; centre, E13.5 wild-type embryo; right E13.5 homozyogus mutant embryo, to illustrate the ∼1 day developmental delay.
d. E13.5 embryos; left, wild type; centre and right, two homozygous mutant embryos.
e-g. Dph4 homozyogus mutants have a preaxial polydactyly.
e. E14.5 homozygous embryo hindfoot
f. E16.5 homozygous embryo hindfoot
g. stain
We introduced the mutation onto an outbred genetic background. After intercrossing on this background mutant embryos were recovered at a higher frequency, and survived longer (data not shown). Embryos that survived long enough to initiate digit formation showed a specific defect in the hindlimbs. At early stages of digit development, one or more additional preaxial digits were seen (Figure 5e). These additional digits persisted through to later stages (Figure 5f). Skeletal preparation of these digits from a rare neonatal homozygote showed in this case both a total duplication of digit 1, in addition to a partial digit 1 duplication (Figure 5g).
DISCUSSION
Five yeast genes have been identified, following saturation mutagenesis, as required for diphthamide biosynthesis, (Liu et al, 2004) and all five have sequence orthologues in mouse and humans. Mutations in four of these have been selected in CHO cells (Liu and Leppla, 2003; Liu et al, 2004; Moehring and Moehring, 1979; Moehring et al, 1980; Moehring et al, 1984; Nobukuni et al, 2005), and two have been subject to targeted mutagenesis in mouse (Chen and Behringer, 2004; Liu et al, 2006). Mutations in Dph4 have not been reported to date in higher eukaryotes, and direct functional evidence for its role has been absent. We have presented a mouse mutation in Dph4 and functional evidence that the gene product is required for diphthamide biosynthesis.
eEF2 protein is essential for polypeptide elongation in protein synthesis, forming a complex with GTP and the ribosomes and catalysing the GTP dependent translocation of peptidyl-tRNA from the aminoacyl (A) site to the peptidyl (P) site on the ribosome (Spahn et al, 2004). The function of diphthamide within eEF2 is not well understood. However, recent work has shown a requirement for diphthamide in the maintenance of translational fidelity. Structural studies have suggested a possible interaction between diphthamide and two universally conserved adenine residues on the ribosomal RNA that are essential for tRNA recognition at the A-site in the small ribosomal subunit. These two adenines undergo a conformational change, switching from a stacked (closed) position in the absence of tRNA to a flipped out (open) position in the presence of cognate tRNA. JØrgensen et al (2005) proposed that diphthamide may be required for the stabilisation of the stacked position of the adenines, and therefore be essential for maintaining the correct reading frame during translocation across the ribosome. Studies using yeast mutant strains that lack either DPH2 or DPH5 show an increase in -1 frameshifting (Ortiz et al, 2006).
Biochemical studies indicate that DPH1-4 catalyse the first step in dipthamide biosynthesis, the transfer of 3-amino-3-carboxypropyl from S-adenosylmethionine to the histidine. DPH5 catalyses the subsequent trimethylation of the intermediate. DPH1 and 2 are related, but not redundant proteins that physically interact in yeast and mammalian cells (Liu et al, 2004). Large scale interaction studies in yeast have found mutual interactions between DPH1, 2, and 3, and between all of these three and eEF2. DPH5 in addition also interacts with eEF2 (Collins et al, 2007; Gavin et al, 2006; Krogan et al, 2006). However, DPH4 has not been found to interact with any of these components. Our finding that at least some DPH4 localises to the cytoskeleton, unlike DPH1, supports the model that a complex of DPH1-3 directly interacts with eEF2 to promote the first stage of diphthamide synthesis. DPH5 may also separately bind eEF2 during the second stage. The role of DPH4 is not clear. DPH4 has a bipartite structure consisting of a J-domain and a so-called CSL zinc finger. The CSL finger motif is found in only one other protein, DPH3, in which it comprises almost the entire molecule, which makes it surprising that DPH4 does not appear to bind to DPH1 or 2. The J-domain is the crucial feature of the Dnaj, or Hsp40, family of proteins, which act as Hsp70 co-chaperones (Walsh et al, 2004). The J-domain interacts with Hsp70 and regulates its activity, inducing ATPase activity and altering substrate binding. Previous work on mouse DPH4, as mmDjc7, indicates that the J-domain is indeed functional in that it is able to stimulate the ATPase activity of the Hsp70 proteins Hsc70 and BiP (Kroczynska and Blond, 2001). Thus DPH4 is likely to be a molecular chaperone and enable protein folding. Whether the substrate is one or more of DPH1-3, eEF2, or indeed is another still to be identified substrate remains to be established. It is notable that in mammalian cells some fraction of eEF2, along with other translation elongation factors, appears to be localized to the actin cytoskeleton (Bektas et al, 1994; Bektas et al, 1998; Shestakova, 1991) and this colocalisation may permit at least transient interaction with DPH4.
Given the ubiquity of the diphthamide modification it is perhaps surprising that ablation of its synthesis is not cell lethal. More remarkable still is that mouse embryos that lack diphthamide synthesis, although retarded in growth, nevertheless undergo developmental processes. Mouse embryos lacking Dph3 appear to be more severely affected than those lacking Dph1 (Liu et al, 2006) or Dhp4. It is possible that DPH3 has an additional cellular function that results in earlier lethality. In yeast, Dph3 null mutants are more severely affected than nulls in the other genes in the pathway. Indeed yeast Dph3 (first identified as Kti11 and required for sensitivity to the Kluveromyces lactis toxin zymocin) is one of a number of genes needed to produce specific modifications of uridines in the wobble positions of tRNA anticodons (Huang et al, 2005; Lu et al, 2005).
The phenotype of our Dph4 mutant is similar to the Dph1 knockout mice, with developmental delay of about 1 day and prenatal lethality. We have shown a strong effect of genetic background such that when outbred, occasional mice can survive beyond birth to maturity and fertility. It is very striking that both Dph1 and Dph4 mutants have a very similar preaxial polydactyly phenotype. It remains to be determined whether this is due to a defect in translation of a specific developmental factor or factors, or whether the overall growth retardation of the embryos results in a mismatch in timing of developmental events which leads to the limb phenotype.
The DPH1 gene was originally identified as a gene commonly deleted in human ovarian cancer, and mice heterozygous for a mutation of Dph1 have an increased incidence of various tumours (Chen and Behringer, 2004). We have not observed tumours in mice heterozygous for the Dph4 mutation, nor have tumours been reported in Dph3 heterozygous mice. It is possible that Dph1 has additional functions which act as tumour suppressors.
MATERIALS AND METHODS
Mouse mutagenesis and husbandry
Animal studies were carried out under the guidance issued by the Medical Research Council in “Responsibility in the Use of Animals in Medical Research” (July 1993) and licenced by the Home Office under the Animals (Scientific Procedures) Act 1986. Male BALB/c mice were dosed with ethylnitrosourea (ENU) as described (Nolan et al, 2000) to mutagenise the spermatogonial stem cells. Following recovery of fertility they were mated to Pax6Sey-Neu females and subsequently bred as in Figure 1a. The mutant BALB/c chromosomal segment was followed through matings using the microsatellite markers D2Mit42 and D2Mit58 which flank the region deleted in Pax6Sey-1H.
DNA sequencing
Candidate exons were amplified using primers designed to permit sequencing of the whole exon plus the consensus splice sites. BALB/c DNA was also sequenced to allow identification of SNPs compared to the reference C57BL6/J sequence.
Reverse transcriptase-PCR
RNA was isolated from mouse embryos using the RNAgents Total RNA Isolation System (Promega). RT-PCR was carried out using the Access RT-PCR System (Promega) using the following primers from exons 3 and 5 of Dph4: CAAAGTGCAGATGTGCCA and GGAGACAGTGTATTTCCCACC. Annealing was at 60°.
Production of antiserum
A DPH4-GST fusion protein was made by cloning Dph4 cDNA into pGEX-KG and expression induced by IPTG in BL21 cells (Guan and Dixon, 1991). Following extraction by sonication the fusion protein was bound to glutathione beads (GS4B Sepharose: Amersham Biosciences), washed and DPH4 liberated by cleavage with thrombin. The purified protein was injected into rabbits by Diagnostics Scotland to generate antiserum.
Embryo protein analysis
Individual E10.5 embryos were disrupted by pipetting in modified RIPA buffer followed by centrifugation to remove debris. The isolated proteins were either run on standard SDS-PAGE or on native (non-denaturing) PAGE, using precast 4-12% tris-glycine gels (Novex). Proteins were transferred to Hybond-P by electrophoretic blotting. The western blots were probed with the whole antiserum raised against DPH4 or a goat antibody against eEF2 (Santa Cruz, sc13004), both at dilution of 1 in 500. Antibody reaction was detected using horse radish peroxidase conjugated secondary antibodies: NA934VS goat anti-rabbit (Amersham) at 1 in 10,000 or sc2020, donkey anti-goat, (Santa Cruz) at 1 in 5000. Visualisation used ECL-Plus (Amersham).
Mouse embryonic fibroblasts
MEFs were isolated by dissecting individual E13.5 embryos to remove the head and internal organs, incubating in dispase and dispersing cells using a syringe needle. DNA was isolated from the extraembryonic membranes of each embryo and sequenced to determine the Dph4 genotype. The cells were cultured in DMEM with 10% foetal calf serum.
Diphtheria toxin sensitivity assay
DNA was introduced into MEFs by electroporation using the Digital Bio Microporator, with parameters recommended by the manufacturers. The pGL3-P luciferase reporter plasmid (Promega) was introduced into about 4 × 104 cells per electroporation. To assay the sensitivity to diphtheria toxin, the DTA subunit gene, driven by the PGK promoter was co-electroporated with the pGL3-P at a molar ratio approximately 3:1. Control co-electroporations used pBluescript vector at the same molar ratio. Following overnight incubation luciferase activity was measured using the Luciferase Assay System (Promega) and a the Lumat LB9507 luminometer (Berthold). Electroporations and luciferase assays were performed in triplicate.
Immunofluorescence
Subcellular localization of tagged proteins was carried out in NIH-3T3 cells, following transfection with lipofectamine 2000 (Invitrogen). FLAG-tagged DPH4 was generated by cloning the cDNA into p3xFLAG-CMV10 (Sigma). OVCA1 tagged with myc was a gift of Chun-Ming Chen (Chen and Behringer, 2001). Following transfection the tagged proteins were detected by anti-FLAG mouse monoclonal F-3165 (Sigma) at 1 in 200, followed by anti-mouse Texas Red (Stratech) or Alexa-Fluor conjugated anti-myc, sc789 (Santa Cruz) at 1 in 100.
Embryos
Embryos were dissected into chilled PBS before fixation overnight in 4% PFA. Extraembryonic membranes or tails were used as source of DNA for genotyping. Skeletal samples were prepared and stained with Alcian Blue and Alizarin Red as described (http://empress.har.mrc.ac.uk/).
ACKNOWLEDGEMENTS
This work was supported by the UK Medical Research Council.
We would like to thank Darcy Butts and Emily Pritchard for additional work on this project. Thanks also to Cathy Abbott for helpful comments on the manuscript and to Craig Nicol for help with the figures.
REFERENCES
- Bektas M, Nurten R, Gurel Z, Sayers Z, Bermek E. Interactions of eukaryotic elongation factor 2 with actin: a possible link between protein synthetic machinery and cytoskeleton. FEBS Lett. 1994;356:89–93. doi: 10.1016/0014-5793(94)01239-3. [DOI] [PubMed] [Google Scholar]
- Bektas M, Nurten R, Sayers Z, Bermek E. Interactions of elongation factor 2 with the cytoskeleton and interference with DNase I binding to actin. Eur.J.Biochem. 1998;256:142–147. doi: 10.1046/j.1432-1327.1998.2560142.x. [DOI] [PubMed] [Google Scholar]
- Chen CM, Behringer RR. Cloning, structure, and expression of the mouse Ovca1 gene. Biochem.Biophys.Res.Commun. 2001;286:1019–1026. doi: 10.1006/bbrc.2001.5488. [DOI] [PubMed] [Google Scholar]
- Chen CM, Behringer RR. Ovca1 regulates cell proliferation, embryonic development, and tumorigenesis. Genes Dev. 2004;18:320–332. doi: 10.1101/gad.1162204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Bodley JW, Livingston DM. Diphtheria toxin-resistant mutants of Saccharomyces cerevisiae. Mol.Cell Biol. 1985;5:3357–3360. doi: 10.1128/mcb.5.12.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier RJ. Understanding the mode of action of diphtheria toxin: a perspective on progress during the 20th century. Toxicon. 2001;39:1793–1803. doi: 10.1016/s0041-0101(01)00165-9. [DOI] [PubMed] [Google Scholar]
- Collins SR, Kemmeren P, Zhao XC, Greenblatt JF, Spencer F, Holstege FC, Weissman JS, Krogan NJ. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol.Cell Proteomics. 2007;6:439–450. doi: 10.1074/mcp.M600381-MCP200. [DOI] [PubMed] [Google Scholar]
- Fichtner L, Jablonowski D, Schierhorn A, Kitamoto HK, Stark MJ, Schaffrath R. Elongator’s toxin-target (TOT) function is nuclear localization sequence dependent and suppressed by post-translational modification. Mol.Microbiol. 2003;49:1297–1307. doi: 10.1046/j.1365-2958.2003.03632.x. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal.Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Hill RE, Favor J, Hogan BL, Ton CC, Saunders GF, Hanson IM, Prosser J, Jordan T, Hastie ND, van,Heyningen V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991;354:522–525. doi: 10.1038/354522a0. [DOI] [PubMed] [Google Scholar]
- Huang B, Johansson MJ, Bystrom AS. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11:424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen R, Merrill AR, Yates SP, Marquez VE, Schwan AL, Boesen T, Andersen GR. Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature. 2005;436:979–984. doi: 10.1038/nature03871. [DOI] [PubMed] [Google Scholar]
- Jorgensen R, Merrill AR, Andersen GR. The life and death of translation elongation factor 2. Biochem. Soc. Trans. 2006;34:1–6. doi: 10.1042/BST20060001. [DOI] [PubMed] [Google Scholar]
- Kent J, Lee M, Schedl A, Boyle S, Fantes J, Powell M, Rushmere N, Abbott C, van Heyningen V, Bickmore WA. The reticulocalbin gene maps to the WAGR region in human and to the Small eye Harwell deletion in mouse. Genomics. 1997;42:260–267. doi: 10.1006/geno.1997.4706. [DOI] [PubMed] [Google Scholar]
- Kroczynska B, Blond SY. Cloning and characterization of a new soluble murine J-domain protein that stimulates BiP, Hsc70 and DnaK ATPase activity with different efficiencies. Gene. 2001;273:267–274. doi: 10.1016/s0378-1119(01)00583-2. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O’Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Liu S, Leppla SH. Retroviral insertional mutagenesis identifies a small protein required for synthesis of diphthamide, the target of bacterial ADP-ribosylating toxins. Mol.Cell. 2003;12:603–613. doi: 10.1016/j.molcel.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Liu S, Milne GT, Kuremsky JG, Fink GR, Leppla SH. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol.Cell Biol. 2004;24:9487–9497. doi: 10.1128/MCB.24.21.9487-9497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wiggins JF, Sreenath T, Kulkarni AB, Ward JM, Leppla SH. Dph3, a small protein required for diphthamide biosynthesis, is essential in mouse development. Mol.Cell Biol. 2006;26:3835–3841. doi: 10.1128/MCB.26.10.3835-3841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Huang B, Esberg A, Johansson MJ, Bystrom AS. The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA. 2005;11:1648–1654. doi: 10.1261/rna.2172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheakis LC, Shen WH, Collier RJ. DPH5, a methyltransferase gene required for diphthamide biosynthesis in Saccharomyces cerevisiae. Mol.Cell Biol. 1992;12:4026–4037. doi: 10.1128/mcb.12.9.4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring JM, Moehring TJ. Characterization of the diphtheria toxin-resistance system in Chinese hamster ovary cells. Somatic.Cell Genet. 1979;5:453–468. doi: 10.1007/BF01538880. [DOI] [PubMed] [Google Scholar]
- Moehring JM, Moehring TJ, Danley DE. Posttranslational modification of elongation factor 2 in diphtheria-toxin-resistant mutants of CHO-K1 cells. Proc.Natl.Acad.Sci.U.S.A. 1980;77:1010–1014. doi: 10.1073/pnas.77.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring TJ, Danley DE, Moehring JM. In vitro biosynthesis of diphthamide, studied with mutant Chinese hamster ovary cells resistant to diphtheria toxin. Mol.Cell Biol. 1984;4:642–650. doi: 10.1128/mcb.4.4.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell. 1992;69:1051–1061. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- Nobukuni Y, Kohno K, Miyagawa K. Gene trap mutagenesis-based forward genetic approach reveals that the tumor suppressor OVCA1 is a component of the biosynthetic pathway of diphthamide on elongation factor 2. J.Biol.Chem. 2005;280:10572–10577. doi: 10.1074/jbc.M413017200. [DOI] [PubMed] [Google Scholar]
- Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, Washbourne R, Hough T, Greenaway S, Hewitt M, Liu X, McCormack S, Pickford K, Selley R, Wells C, Tymowska-Lalanne Z, Roby P, Glenister P, Thornton C, Thaung C, Stevenson JA, Arkell R, Mburu P, Hardisty R, Kiernan A, Erven A, Steel KP, Voegeling S, Guenet JL, Nickols C, Sadri R, Nasse M, Isaacs A, Davies K, Browne M, Fisher EM, Martin J, Rastan S, Brown SD, Hunter J. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat.Genet. 2000;25:440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- Ortiz PA, Ulloque R, Kihara GK, Zheng H, Kinzy TG. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J.Biol.Chem. 2006;281:32639–32648. doi: 10.1074/jbc.M607076200. [DOI] [PubMed] [Google Scholar]
- Pappenheimer AM, Jr., Dunlop PC, Adolph KW, Bodley JW. Occurrence of diphthamide in archaebacteria. J.Bacteriol. 1983;153:1342–1347. doi: 10.1128/jb.153.3.1342-1347.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson EA, Henriksen O, Maxwell ES. Elongation factor 2. Amino acid sequence at the site of adenosine diphosphate ribosylation. J.Biol.Chem. 1974;249:5088–5093. [PubMed] [Google Scholar]
- Shestakova EA, Motuz LP, Minin AA, Gelfand VI, Gavrilova LP. Some of eukaryotic elongation factor 2 is colocalized with actin microfilament bundles in mouse embryo fibroblasts. Cell Biol.Int.Rep. 1991;15:75–84. doi: 10.1016/0309-1651(91)90084-v. [DOI] [PubMed] [Google Scholar]
- Spahn CM, Gomez-Lorenzo MG, Grassucci RA, Jorgensen R, Andersen GR, Beckmann R, Penczek PA, Ballesta JP, Frank J. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 2004;23:1008–1019. doi: 10.1038/sj.emboj.7600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Olsnes S, Sandvig K. Requirement of specific receptors for efficient translocation of diphtheria toxin A fragment across the plasma membrane. J.Biol.Chem. 1988;263:13449–13455. [PubMed] [Google Scholar]
- Van Ness BG, Howard JB, Bodley JW. ADP-ribosylation of elongation factor 2 by diphtheria toxin. Isolation and properties of the novel ribosyl-amino acid and its hydrolysis products. J.Biol.Chem. 1980;255:10717–10720. [PubMed] [Google Scholar]
- Walsh P, Bursac D, Law YC, Cyr D, Lithgow T. The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Rep. 2004;5:567–571. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]