

Abstract
Three synthetic routes were developed for structure activity relationship (SAR) studies of HTS-derived isoquinolinone inhibitor probes for the orphan nuclear receptor steroidogenic factor-1 (NR5A1). Among the new analogs reported herein, 31 and 32 have improved potency, lower cellular toxicity, and improved selectivity compared to the initial HTS-derived leads 1 and 2.
Nuclear receptors are transcription factors that regulate gene expression through the binding of endogenous ligands.i Many nuclear receptors have been extensively studied, and several have proven to be important targets for treatment of a range of human diseases.ii However, natural or unnatural ligands are not known for a sub-set of nuclear receptors, the so-called “orphan” nuclear receptors, which has limited efforts to determine the pharmacology and therapeutic potential of these receptors.1 As part of a broader program to develop an understanding of the pharmacology of relatively unexplored orphan receptors, the Scripps Research Institute's Molecular Library Screening Center has performed high throughput screens of several orphan receptors, among them the steroidogenic factor 1 (SF-1, also known as NR5A1).iii
Steroidogenic factor-1 (SF-1) has been implicated in sex determination during development and in formation of steroidogenic tissues.iv SF-1 is involved in endocrine function throughout life with expression in the pituitary, testes, ovaries, and adrenal gland.v Knockout mice exhibit male to female sex reversal and impaired development of adrenals and gonads.vi Due to the potential role SF-1 plays in regulation of steroid hormone synthesis including adrenal androgen and gonadal testosterone synthesis, selective control of this receptor could result in therapeutic treatment of metastatic prostate cancer.vii Additionally the involvement of SF-1 in energy metabolism suggests relevancy in controlling obesity.viii
Thus, the development of selective small-molecule biological probes of SF-1 is an important objective. Phospholipids have been found in the ligand binding domain of human SF-1ix and recently the first small molecules with the ability to modulate the activity of this transcription factor were described.x
Approximately 65,000 compounds were screened for SF-1 inhibition by the Molecular Library Screening Centers Network (MLSCN) at The Scripps Research Institute.3,xi All initial hits were counter screened against the retinoic acid receptor-related orphan receptor α (RORα), a phylogenetically distant nuclear receptor,2 in order to eliminate promiscuous as well as non-selective compounds. This led to the identification of two mid-nanomolar SF-1 selective inhibitors 1 (PubChem SID 7970631) and 2 (SID 7969543; Figure 1).3,11 Accordingly, isoquinolinones 1 and 2 were selected as starting points for the development of SF-1 small molecule probes. We have developed and report herein three routes for the synthesis of 1 and 2 that enable different aspects of the SAR of this SF-1 inhibitor series to be examined. Among the analogs reported here, 31 and 32 have improved SF-1 inhibitor potency, lower cellular toxicity, and possess improved selectivity compared to the initial leads 1 and 2.
Figure 1.
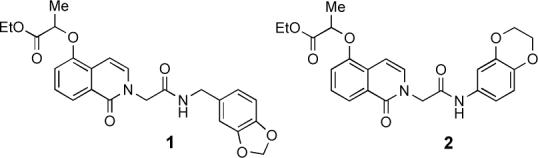
SF-1 inhibitors identified via ultra high throughput screening of the MLSCN library
Our initial strategy for synthesis of analogs of 1 and 2 focused on sequential alkylations of an isoquinolinone core (Scheme 1). Treatment of commercially available 5-hydroxyquinoline with peroxyacetic acid provided N-oxide 4. Heating of the N-oxide 4 in acetic anhydride gave a diacyl derivative that was not isolated but converted directly to 5-hydroxyisoquinolinone 5.xii Selective alkylation of 5 at the phenolic position by using ethyl (±)-2-bromopropionate (NaH, DMF, 25 °C, 80%) gave propionic ester 6. Subsequent N-alkylation of 6 with tert-butyl bromoacetate (Cs2CO3, DMF, 25 °C, 80%) provided diester 7. Acidic deprotection of the tert-butyl ester and subsequent coupling (EDC, DMAP, CH2Cl2) of the derived carboxylic acid with either benzyl amine 8 or aniline 9 provided authentic samples of 1 and 2, which were used to confirm the results of the MLSCN SF-1 inhibitor screen.
Scheme 1.
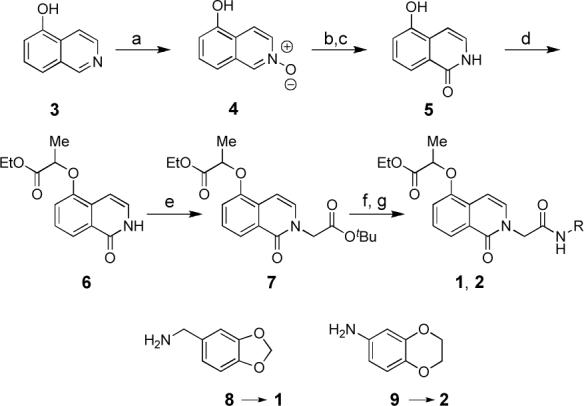
(a) peroxyacetic acid, CH2Cl2 (99%); (b) acetic anhydride, 140°C, 4 h, then concentrate; (c) NaOMe, MeOH (60%, 2 steps); (d) NaH, DMF, 30 min, then ethyl (±)-2-bromopropionate (80%); (e) Cs2CO3, DMF, tert-butyl bromoacetate (80%); (f) 2 M HCl in Et2O and EtOH; (g) EDC, DMAP, CH2Cl2, 8 or 9 (45%, two steps)
While the sequence summarized in Scheme 1 permitted us to synthesize a number of analogs, this route proved less than ideal especially for synthesis of isoquinolinone ether analogs owing to its linear nature. Especially problematic is that the chemoselectivity of alkylations of 5 with agents less reactive than α-halopropionate was poor, with N-alkylation of 5 competing with phenol O-alkylation with alkyl halides. This led us to develop the more convergent second generation synthesis summarized for 14 in Scheme 2.
Scheme 2.
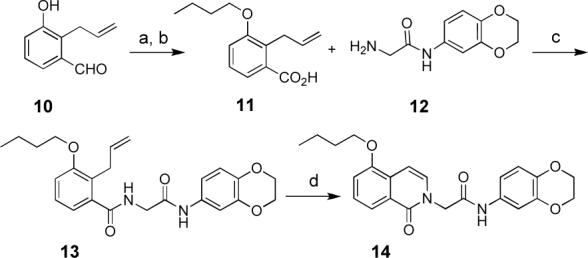
(a) iodobutane, acetone, K2CO3, 60°C, 4 h (60−85%); (b) 1.5 equiv H2CrO4, acetone, 1 h (98%); (c) 9, EDC, DMAP, CH2Cl2 (83%); (d) (i). O3, CH2Cl2, −78°C, (ii) PPh3, (iii). cat. I2, CH2Cl2 (45%).
Aldehyde 10, available from the Claisen rearrangement of m-salicaldehyde allyl ether,xiii was alkylated in high yield with a variety of electrophiles (use of iodobutane is illustrated in Scheme 2). The resulting product was then oxidized using chromic acid to give acid 11.xiv Standard peptide coupling of 11 with glycinamide 12 (EDC, DMAP, CH2Cl2, 83%) provided diamide 13, which was then converted to SF-1 inhibitor analog 14 via ozonolysis of the vinyl group and dehydration of the hemiaminal that is formed in situ. In some cases, the targeted isoquinolinone was obtained directly from standard workup of the ozonolysis reaction. However, in other cases the dehydration of the hemiaminal did not occur spontaneously. Therefore, as a standard procedure, a catalytic amount of I2 iodine was added to a solution of the crude hemiaminal in CH2Cl2 to promote dehydration and aromatization.
In order to probe the SAR of the glycine spacer connected to the isoquinolinone nitrogen, we demonstrated that condensation of hydroxylactone 16 (accessible from ozonolysis of acid 15) and glycine tert-butyl ester 17a provides ester 7 (Scheme 3). Deprotection of 7 provides carboxylic acid 18, which is an intermediate in the sequence summarized in Scheme 1. Alternatively, amino acids such as glycine (17b), alanine and phenylalanine can be coupled directly with 16 to give 18, thereby obviating the need for use of the tert-butyl ester protecting group. Subsequent coupling of carboxylic acid 18 with a range of amines and anilines, using the conditions summarized in Scheme 1, greatly facilitated SAR studies of this amide substituent.
Scheme 3.

(a) ethyl (±)-2-bromopropionate, K2CO3, acetone, 60 °C, 4 h. (b) (i) O3, CH2Cl2, −78 °C (ii) Me2S, 1 h, 23 °C. (c) tert-butyl glycine hydrochloride (17a), 3 equiv. Et3N, AcOH to achieve pH 3 to 5, benzene, 100°C, sealed tube, 12 h (67%). (d) glycine (17b, 3 equiv), benzene, 100°C, 12 h, sealed tube (94%).
Finally, in order to probe the substitution pattern of the isoquinolinone nucleus of 1 and 2, a third synthetic strategy was developed starting from commercially available hydroxyindanones (Scheme 4); the synthesis of 22 starting from 19 is illustrative. Thus, after alkylation of 5-hydroxyindanone with ethyl (±)-2-bromopropionate (K2CO3, acetone, 60 °C, 4 h), indanone 19 was treated with TBS-OTf and Et3Nxv to give the silyl enol ether which, without isolation, was treated with N-methylmorpholine N-oxide (NMO) and catalytic osmium tetroxide.xvi The resulting α-hydroxyindanone 20 was cleaved using NaIO4 to give the hydroxylactone, which was condensed with glycine tert-butyl ester under conditions summarized in Scheme 3. Subsequent coupling of the carboxylic acid derived from deprotection of 21 with aniline 9 provided the regioisomeric isoquinolinone analog 22.
Scheme 4.
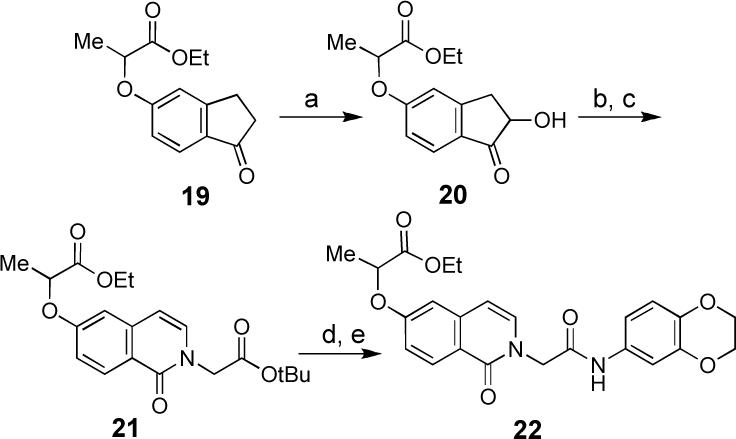
(a) (i). TBSOTf, Et3N, THF (ii) OsO4 (2%), NMO, t-BuOH, acetone. (b) NaIO4, acetone, H2O, THF, t-BuOH (78% from 19). (c) glycine tert-butyl ester hydrochloride, benzene, Et3N and AcOH until pH 3−5, 100°C sealed tube 12 h (21%). (d) 2 M HCl in Et2O and EtOH, (99%) (e) 9, EDC, DMAP, CH2Cl2, (55%).
SF-1 inhibition activity of analogs of 1 and 2 are summarized in Table 1, along with comparative inhibition data vs. RORα. All assays were performed as described in ref. 3. Briefly, luciferase-format functional assays were used to measure inhibition of constitutively active SF1(LBD)-Gal4 or RORα (LBD)-Gal4 chimeric constructs. Compounds 1, 2, 22−35 and 40−43 were studied as racemates, or as mixtures of diastereomers (41, 42).
Table 1.
SF-1 Inhibitor Activity of Analogs of 1 and 2 (all IC50 data are in μM).
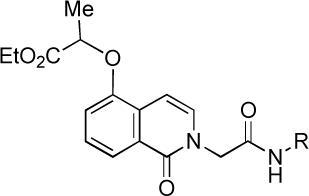 | |||||
|---|---|---|---|---|---|
| IC50 data (μM)a |
|||||
| compound | R | SF-1 | RORα | VP16 | Cell toxb |
| 1 | (see Figure 1) | 0.26 ± 0.06 | >33 | >33 | >33 |
| 2 | (see Figure 1) | 0.76 ± 0.10 | >33 | >33 | inactive |
| 23 | -(CH2)3CH3 | 9.71 ± 5.63 | inactive | inactive | inactive |
| 24 | cyclohexyl | 1.77 ± 0.16 | inactive | inactive | inactive |
| 25 | benzyl | 1.06 ± 0.03 | 3.22 ± 0.34 | inactive | inactive |
| 26 | p-fluorobenzyl | 0.68 ± 0.12 | 1.68 ± 0.43 | inactive | inactive |
| 27 | -CH2-2-furanyl | 4.71 ± 0.86 | >33 | inactive | inactive |
| 28 | phenylethyl | 0.36 ± 0.03 | >33 | inactive | inactive |
| 29 | p-nitrophenyl | 0.53 ± 0.20 | inactive | inactive | inactive |
| 30 | p-methylphenyl | 0.34 ± 0.02 | 36.5 ± 6.0 | >33 | 40.2 ± 3.7 |
| 31 | p-methoxyphenyl | 0.20 ± 0.04 | inactive | inactive | inactive |
| 32 | p-(F3C)Ophenyl | 0.20 ± 0.11 | inactive | inactive | inactive |
| 33 | p-ethoxyphenyl | 0.19 ± 0.01 | 22.6 ± 2.0 | 24.2 ± 5.6 | 37.0 ± 0.7 |
| 34 | p-ethylphenyl | 0.11 ± 0.04 | 25.0 ± 3.9 | >33 | >33 |
| 35 | p-n-butylphenyl | 0.36 ± 0.05 | 30.7 ± 10.7 | inactive | inactive |
 |
||||||
|---|---|---|---|---|---|---|
| compound | R′ | SF-1 | RORα | VP16 | Cell toxb | |
| 14 | -CH2(CH2)2CH3 | 13.7 ± 1.3 | 21.5 ± 3.5 | 24.1 ± 1.9 | >33 | |
| 36 | -CH2CH3CH2CH3 | 7.86 ± 0.22 | 16.5 ± 2.2 | 17.5 ± 1.9 | inactive | |
| 37 | -CH2CH3(CH2)3CH3 | 0.88 ± 0.09 | 23.1 ± 6.1 | 20.7 ± 8.0 | inactive | |
| 38 | -C(CH3)2CO2Et | 0.59 ± 0.12 | inactive | >33 | inactive | |
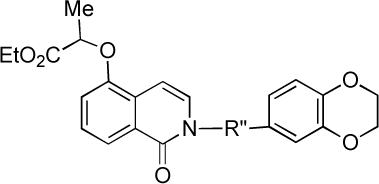 | |||||
|---|---|---|---|---|---|
| compound | R″ | SF-1 | RORα | VP16 | Cell toxb |
| 40 | -CH2CH2CONH- | 8.46 ± 1.67 | inactive | inactive | inactive |
| 41 | -CH(Me)CONH- | inactive | inactive | inactive | inactive |
| 42 | -CH(CH2Ph)CONH- | inactive | inactive | inactive | inactive |
| 43 | -CH2CON(Me)- | 3.57 ± 0.89 | inactive | inactive | inactive |
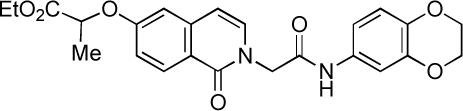 | |||||
|---|---|---|---|---|---|
| compound | SF-1 | RORα | VP16 | Cell toxb | |
| 22 | inactive | inactive | inactive | inactive | |
IC50 value >33 indicates that curve did not fit, and that IC50 was manually assigned to be >33 μM. “Inactive” means <50% inhibition at 100 μM.
Cellular toxicity determined by the CellTiterGlo assay (ref. 18)
Initial efforts focused on replacing the oxygenated benzylic amide or anilide units of 1 and 2, but introduction of alkyl amides as in 23 and 24 led to significant loss of SF-1 activity. Replacement of the highly oxygenated benzylic amide unit of 1 with potentially more metabolically stable benzyl (25) or p-fluorobenzyl (26) amides led to 5−50 fold loss of SF-1 activity. However, introduction of a 2-phenylethyl amide (28) yielded a 300 nM SF-1 inhibitor. Still better results were obtained by replacing the potentially metabolically sensitive anilide unit of 2 with other aniline amides, such as the p-methylphenyl (30), p-methoxyphenyl (31), p-trifluoromethoxyphenyl (32), and p-ethoxyethyl (33) amides. These analogs displayed SF-1 inhibitor activity in the 100−200 nM range, a three to six-fold improvement over 2.
All substitutions of the R” unit spanning the isoquinolinone and amide units led to substantial or complete loss of SF-1 activity (analogs 40−43), as did moving the alkoxy substituent from the meta position in 1−2 to the para position in 22. However, clear evidence of a productive SAR for the isoquinolinone alkoxy substituent has been demonstrated by analogs 14 and 36−39. The stereochemistry of the ether substituent in 1 and 2 does not appear to be critical as 38, with a –C(Me2)CO2Et ether at this position, is more potent than 2. In addition, an analog of 2 with a –CH2CO2Et aryl ether (39, not shown), was deemed to be “inactive” in the original HTS campaign.3 These data suggest that hydrophobicity at this position is important for SF-1 inhibitor activity.
All analogs were further assessed in another transactivation assay (Gal4-VP16)xvii to confirm that any SF-1 activity measured was specific to that receptor and not due to transactivation assay artifacts (e.g., promiscuous inhibition of the reporter system); cellular cytotoxicity was also determined (CellTiterGlo assay;xviiisee Table 1).3 The titration curves for compound 32 are shown in Figure 1. This compound showed significant activity only in the SF-1 assay; no activity in the RORα, Gal4-VP16 and cytotoxicity assays were detected at concentrations up to 99 μM.
Figure 1.
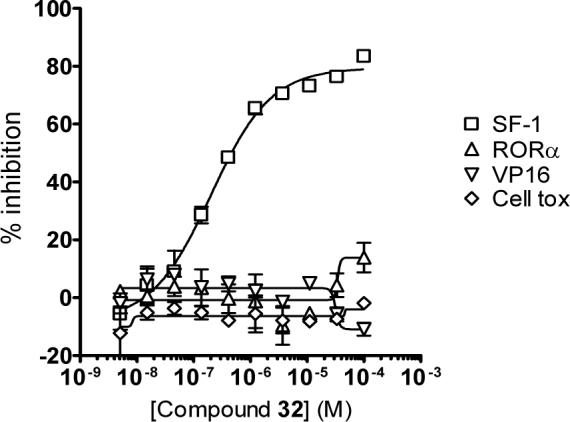
Titration curves for 32 vs. SF-1 (□), RORα(△), VP16 (▽), and the cytotoxicity assays (○). Data represent the mean of three independent experiments with the calculated standard deviation.
In summary, we have developed three routes for the synthesis of substituted isoquinolinones that has enabled us to explore the SAR of isoquinolinone inhibitors of the orphan nuclear receptor steroidogenic factor-1 (SF-1). Of those analogs described herein, 31 and 32 have improved potency and selectivity vs. other nuclear receptor targets compared to the HTS leads 1 and 2.
Supplementary Material
Acknowledgements
This research supported by the NIH Molecular Library Screening Center Network (MLSCN) grant U54MH074404 (Dr. Hugh Rosen, Principal Investigator). Lina DeLuca and Pierre Baillargeon are thanked for compound management. Dr. Louis Scampavia is thanked for quality control of SF-1 inhibitor analogs by LC-MS. The construct for the Gal4-VP16 assays was generously provided by Prof. Michael Conkright. Prof. Pat Griffin is thanked for numerous helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- i.Giguere V. Endocr. Rev. 1999;20:689. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- ii.Moore JT, Collins JL, Pearce KH. ChemMedChem. 2006;1:504. doi: 10.1002/cmdc.200600006. [DOI] [PubMed] [Google Scholar]
- iii.Madoux F, Li X, Chase P, Zastrow G, Cameron MD, Conkright JJ, Griffin PR, Thacher S, Hodder P. Mol. Pharmacol. 2008 doi: 10.1124/mol.108.045963. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- iv.a Morohashi K, Honda S, Inomata Y, Handa H, Omura T. J. Biol. Chem. 1992;267:17913. [PubMed] [Google Scholar]; b Bakke M, Zhao L, Hanley NA, Parker KL. Mol. Cell. Endocrinol. 2001;171:5. doi: 10.1016/s0303-7207(00)00384-1. [DOI] [PubMed] [Google Scholar]; c Val P, Lefrancois-Marinez AM, Veyssiere G, Martinez A. Nucl. Recept. 2003;1:8. doi: 10.1186/1478-1336-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- v.a Ikeda Y, Luo X, Abbud R, Nilson JH, Parker KL. Mol. Endocrinol. 1995;9:478. doi: 10.1210/mend.9.4.7659091. [DOI] [PubMed] [Google Scholar]; b Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, Bakke M, Zhao L, Frigeri C, Hanley NA, Stallings N, Schimmer BP. Recent. Prog. Horm. Res. 2002;57:19. doi: 10.1210/rp.57.1.19. [DOI] [PubMed] [Google Scholar]; c Val P, Lefrancois-Marinez AM, Veyssiere G, Martinez A. Nucl. Recept. 2003;1:8. doi: 10.1186/1478-1336-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.Luo X, Ikeda Y, Parker KL. Cell. 1994;77:481. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- vii.Doghman M, Karpova T, Rodrigues GA, Arhatte M, De Moura J, Cavalli LR, Virolle V, Barbry F, Zambetti GP, Figueiredo BC, Heckert LL, Lalli E. Mol. Endocrinol. 2007;21:2968. doi: 10.1210/me.2007-0120. [DOI] [PubMed] [Google Scholar]
- viii.a Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD, Parker KL. Endocrinology. 2002;143:607. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]; b Zhao L, Bakke M, Hanley NA, Majdic G, Stallings NR, Jeyasuria P, Parker KL. Mol. Cell. Endocrinol. 2004;215:89. doi: 10.1016/j.mce.2003.11.009. [DOI] [PubMed] [Google Scholar]
- ix.a Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thorton JW, Fletterick RJ, Wilson TM, Ingraham HA. Cell. 2005;120:343. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]; b Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, Bingham NC, Kliewer SA, Xu HE. Mol. Cell. 2005;17:491. doi: 10.1016/j.molcel.2005.02.002. [DOI] [PubMed] [Google Scholar]; c Wang W, Zhang C, Marimuthu A, Krupka HI, Tabrizizad M, Shelloe R, Mehra U, Eng K, Nguyen H, Settachatgul C, Powell B, Milburn MV, West BL. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7505. doi: 10.1073/pnas.0409482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- x.a Whitby RJ, Dixon S, Maloney PR, Delerive P, Goodwin BJ, Parks DJ, Willson TM. J. Med. Chem. 2006;49:6652. doi: 10.1021/jm060990k. [DOI] [PubMed] [Google Scholar]; b Del Tredici AL, Andersen CB, Currier EA, Ohrmund SR, Fairbain LC, Lund BW, Olsson R, Piu F. Mol. Pharm. 2008;73:900. doi: 10.1124/mol.107.040089. [DOI] [PubMed] [Google Scholar]
- xi. HTS reports are published in PubChem AID 525, 599 and 600 ( http://pubchem.ncbi.nlm.nih.gov/)
- xii.Robison MM, Robison BL. J. Org. Chem. 1957;21:1337. [Google Scholar]
- xiii.Danishefsky SJ, Harrison PJ, Webb RR, II, O'Neill BT. J. Am. Chem. Soc. 1985;107:1421. [Google Scholar]
- xiv.Bowden K, Heilbron IM, Jones ERH, Weedon BCL. J. Chem. Soc. 1946:39. [Google Scholar]
- xv.Emde H, Gotz A, Hofmann K, Simchen G. Liebigs Ann. Chem. 1981:1643. [Google Scholar]
- xvi.McCormick JP, Tomasik W, Johnson MW. Tetrahedron Lett. 1981;22:607. [Google Scholar]
- xvii.Sadowski I, Ma J, Triezenberg S, Ptashne M. Nature. 1988;335:563. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- xviii.Crouch SP, Kozlowski R, Slater KJ, Fletcher J. J. Immunol. Methods. 1993;160:81. doi: 10.1016/0022-1759(93)90011-u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.