

Abstract
Retinol-binding protein (RBP) is secreted out of the cell in its ligand-bound holo-form. The apo-form of RBP is selectively retained within the endoplasmic reticulum (ER) by a mechanism that remains unknown. Using isolated microsomal system, we have recapitulated the biogenesis of RBP involving its oxidative folding and assembly with transthyretin in the ER. In addition to dissecting its pathway of disulfide oxidation, we have analyzed association of its early folding intermediates with ER-chaperones. Our results show that of the three intramolecular disulfides present in RBP (4–160, 70–174, and 120–129) the smallest loop (120–129) was most critical for RBP to fold. Its absence caused RBP to aggregate into an intermolecular disulfide-linked structure. After acquisition of the small loop, formation of one of the two big disulfides (4–160 or 70–174) was sufficient for RBP to acquire a folded state. Using cross-linking in intact microsomes and sedimentation on sucrose gradients, we show that newly synthesized RBP is associated with a complex of chaperones consisting of Grp94, BiP, PDI, and calnexin. The complex was constitutively present in the ER, independent of the presence of folding substrates. RBP dissociated from this complex coincident with the formation of one of the two big disulfide loops, whereas RBP mutant lacking both the large disulfides showed persistent association. While highlighting the matrix-like characteristics of ER in isolated microsomal system our results provide insight into RBP folding and assembly mechanisms that will aid our understanding of its complex secretion properties.
INTRODUCTION
Proteins are secreted outside the cell at different rates (Lodish et al., 1983; Fries et al., 1984). It is widely accepted that transport out of the ER is the rate-limiting step in the biogenesis of secretory and membrane proteins. Processing events occurring inside the lumen of the ER are known to influence the efficiency of protein secretion. In general, proteins are allowed to exit the ER only upon successful completion of their folding itinerary. Partially folded, unfolded, or aggregated proteins are retained within the ER by the quality control machinery made up of ER-chaperones (Helenius et al., 1992; Hammond and Helenius, 1995).
Retention of partially folded proteins is accomplished by binding to ER-resident chaperones. For N-glycosylated cargo, retention is mediated by lectin chaperones calnexin and calreticulin, which depend on the glucosylation states of their substrates as indicators of their unfolded states (Ou et al., 1993; Hebert et al., 1995). In nonglycosylated proteins, chaperone binding seems to depend on exposed hydrophobic patches on unfolded substrates (Flynn et al., 1991; Blond-Elguindi et al., 1993). Thus, chaperones such as BiP and GRP94 bind unfolded substrates by sensing hydrophobic surfaces, which are typically present only when the protein is unfolded. Unlike N-glycosylated proteins, molecular signatures differentiating between folded and unfolded states of nonglycosylated proteins are not well defined.
A large number of secretory and membrane proteins have been shown to associate with chaperones of the ER at one-to-one level. (Roth and Pierce, 1987; Melnick et al., 1992; Lin et al., 1993; Hammond et al., 1994; Kuznetsov et al., 1994; Tatu and Helenius, 1997; Lindquist et al., 1998). These studies have provided important insight into interactions with chaperones such BiP, GRP94, calnexin, calreticulin, PDI, and ERp57. In addition, the concept of ER-matrix has also been discussed in the literature that proposes a collective role for ER-chaperones in the form of a network. Electron microscopic examination of the ER (Palade and Siekevitz, 1956, Ojakian et al., 1977) as well as in vivo cross-linking in intact cells supports presence of such a complex of chaperones in the ER, which specifically interacts with newly synthesized proteins during their early folding (Tatu and Helenius, 1997; Meunier et al., 2002). For disulfide-containing proteins, formation of their disulfides seems to be critical for completion of folding program and transport outside the ER. These proteins are ready to exit the ER upon dissociation from retention mechanisms described above.
Retinol-binding protein (RBP) is an exception to this rule. In RBP, folding and secretion are delinked. RBP is a single domain protein of 21 kDa containing three intramolecular disulfide bonds (Newcomer et al., 1984; Cowan et al., 1990). RBP is designed to function as a carrier of retinol, delivering this important vitamin to various target tissues in the body. Although RBP is primarily synthesized in hepatocytes, its expression has also been detected in many other tissues. RBP circulates in the plasma as a complex with another transport protein, Transthyretin (TTR). This complex has been shown to form in the ER (Melhus et al., 1991; Bellovino et al.; 1996). It is believed that the complex formation prevents the loss of low-molecular-weight RBP and its bound retinol through kidneys. A recent report also points to a role for RBP in diabetes mellitus (Yang et al., 2005). It has been suggested that RBP is an adipocyte derived signal that may contribute to the pathogenesis of type 2 diabetes and that lowering of serum RBP levels could be a new strategy for diabetes treatment.
Like many other secretory proteins, RBP is cotranslationally targeted to the ER, where it undergoes signal peptide cleavage and oxidative folding as the only covalent modifications. On completion of its folding, RBP associates with TTR, which is itself a homotetramer composed of 14-kDa subunits (Melhus et al., 1991; Bellovino et al.; 1996). Despite completion of its maturation program, apo-RBP awaits secretion until it is converted to its ligand-bound holo-form. Secretion of the accumulated pool of apo-RBP is triggered only upon retinol binding (Muto et al., 1972; Smith et al., 1978; Ronne et al., 1983; Melhus et al., 1992). Mechanisms underlying such ligand-dependent secretion are not clearly understood. It is believed that ligand binding may relieve retention of RBP from the quality control machinery in the ER. However, precise interrelationships between RBP folding, ligand binding, and TTR assembly and secretion are not clearly understood.
Kaji and Lodish (1993) have elegantly described folding and secretion properties of RBP in HepG2 cells. Using pulse-chase approach, they reported that RBP completes its oxidative folding within 2 min and acquires dithiothreitol (DTT)-resistant state in the ER within 15 min after synthesis. Apart from a kinetic lag in the rate of acquiring DTT resistance no other difference was found in RBP maturation in the presence or absence of retinol. The study also showed that under optimal conditions of secretion RBP was secreted, with a t1/2 of ∼4 h. The study did not address interaction of newly synthesized RBP with chaperones in the ER. More recently, Bellovino et al. (1996) have reported RBP association with calnexin and TTR in the ER (Bellovino et al., 1996).
We have reconstituted the biogenesis of RBP in isolated microsomes to examine its maturation events and study its interactions with chaperones of the ER. Use of the microsomal system has allowed us to systematically examine early steps in RBP oxidative folding, assembly and provide an account of its sojourn in the ER. We find that newly synthesized RBP transiently associates with a complex of chaperones during early phase of its disulfide oxidation. Of the three intramolecular disulfides present in RBP, the smallest disulfide loop (120–129) was indispensable for RBP to fold. In addition to the formation of the smallest loop, acquisition of any of the two large disulfide loops (4–160 or 70–174) was sufficient for RBP to fold and dissociate from chaperone complexes in the ER. By comparing the folding of RBP mutants lacking specific disulfide loops, we have also determined the possible sequence of its oxidative folding pathway in the ER. Overall, our results show that oxidative folding of RBP occurs in conjunction with a complex of chaperones, including Grp94, BiP, PDI, and CNX. On formation of two of its three intramolecular disulfides (120–129 and 70–174 or 4–160), RBP dissociates from the chaperone complex and assembles with TTR independent of the presence of its ligand.
MATERIALS AND METHODS
The RiboMAX large-scale RNA production system and the cell-free translation system (rabbit reticulocyte lysate, amino acid mixture lacking methionine and RNasin) were purchased from Promega (Madison, WI). Protease inhibitor mix (100×) and Superdex-200 column were purchased from GE Healthcare (Little Chalfont, Buckinghamshire, United Kingdom). Translation grade [35S]methionine was obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Rabbit polyclonal antibodies against human plasma RBP and TTR were obtained from Dako Denmark A/S (Glostrup, Denmark). Antibodies against the ER-chaperones calnexin, calreticulin, BiP PDI, and ERp57 were obtained from Nventa Biopharmaceuticals (San Diego, CA). Restriction enzymes, DNA polymerase, and other molecular biology reagents were from MBI Fermentas (Hanover, MD). All other chemicals, including oxidized glutathione (GSSG) and protein A-Sepharose were from Sigma-Aldrich (St. Louis, MO). Dog pancreas microsomes were prepared by the method of Walter and Blobel (1983), with some modifications.
In Vitro Transcription
The cDNA for human plasma RBP, cloned downstream of the T7 promoter into pET-22b(+) at the NdeI-BamHI restriction sites, was linearized, purified, and transcribed in vitro. The transcription was carried out as described in Promega's technical manual. Briefly, a 20-μl transcription reaction mixture consisted of 4 μl of T7 transcription 5× buffer, 6 μl of rNTP mix (25 mM each), 2 μg of linearized plasmid, and 2 μl of enzyme mix (consisting of RNA polymerase, RNase inhibitor, and yeast inorganic pyrophosphatase). The reaction mix was incubated at 37°C for 2–4 h. At the end of incubation, the products were purified by phenol-chloroform extraction and ethanol precipitation. The transcripts were visually quantified by comparison with appropriate standards on formaldehyde/agarose gels and stored at −80°C.
In Vitro Translation and Microsomal Translocation of RBP
The [35S]-labeled RBP was translated and translocated into dog pancreas microsomes by using the following lysate mixture: A 20-μl reaction typically consisted of 10.4 μl of nuclease-treated rabbit reticulocyte lysate, 0.4 μl of amino acid mixture without methionine, 3 μl of [35S]methionine, 0.5 μl of 100 mM DTT, 4.0 μl of dog pancreas microsomes, 0.7 μl of nuclease-free water, and 1 μl of RBP mRNA. Protein synthesis was terminated after 1 h at 27°C. The mixture thus contained 4 mM DTT as part of the reticulocyte lysate and 1.5 mM 2-mercaptoethanol of the label.
Protease Protection Assay
RBP was translated in the presence of microsomal membranes in a 25-μl reaction for 1 h at 27°C. The sample was split in three aliquots of 8 μl each. Two microliters of water was added to the first aliquot as mock treatment (control). One microliter of water and 1 μl of proteinase K solution (3 mg/ml) was added to the second aliquot, whereas 1 μl of 10% Triton X-100 and 1 μl of proteinase K solution was added to the third aliquot. The samples were incubated for 10 min at 25°C. Proteolysis was stopped by adding 2 μl of 10 mg/ml phenylmethylsulfonyl fluoride (PMSF) per sample. The samples were then precipitated with 10% trichloroacetic acid (TCA), washed with acetone, and analyzed by 13% SDS-polyacrylamide gel electrophoresis (PAGE).
Posttranslational Disulfide Oxidation of RBP
For posttranslational folding studies, [35S]-labeled RBP was synthesized and translocated under reducing conditions as described above. At the end of 1 h, disulfide oxidation was initiated with the addition of 4 mM oxidized glutathione, and samples were withdrawn at the indicated chase time points. Folding was stopped by alkylation of free sulfhydryls with 20 mM N-ethylmaleimide (NEM).
Immunoprecipitation and SDS-PAGE
The microsomes containing [35S]-labeled RBP were first solubilized in ice-cold 1% Triton X-100 in phosphate-buffered saline (PBS) containing 20 mM NEM and protease inhibitors: 1 mM PMSF, 1 mM EDTA, and 10 μg/ml each of leupeptin, antipain, and pepstatin. The lysate was diluted in 20 volumes of PBS and precleared with protein A-Sepharose for 1 h with end-to-end rotation at 4°C. The precleared lysate was incubated with protein A-Sepharose and anti-RBP rabbit polyclonal antibody at 4°C for 6 h on an end-to-end rotator. The immunoprecipitates were washed three times with radioimmunoprecipitation assay buffer containing 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% SDS, 1% sodium deoxycholate, 1% Triton X-100, 1 mM PMSF, and protease inhibitor cocktail. The final pellets were resuspended in 50 μl of SDS-PAGE sample buffer (200 mM Tris, pH 6.8, 3% SDS, 10% glycerol, 0.004% bromphenol blue, and 1 mM EDTA) and heated to 100°C for 5 min. The samples were divided into two equal parts of which one part was reduced by adding DTT to a final concentration of 100 mM. The supernatants were analyzed by 13% SDS-PAGE followed by fluorography.
Sedimentation Analysis of RBP on Sucrose Gradients
RBP was translated in vitro under reducing conditions at 27°C for 60 min. Disulfide oxidation was initiated by the addition of 4 mM GSSG and further sampling was done at 30 and 90 min of chase. All the samples were alkylated with 20 mM NEM, microsomes were lysed with 1% Triton X-100 containing lysis buffer, and the lysates were loaded on a 12-ml linear sucrose gradient (either 5–25% sucrose in phosphate-buffered saline containing 0.1% Triton X-100). Gradients were centrifuged for 20 h at 40,000 rpm in a SW 41 rotor at 4°C in an Optima ultracentrifuge (Beckman Coulter, Fullerton, CA). Fractions were collected from the top and precipitated with 10% trichloroacetic acid. The precipitates were washed with ice-cold acetone, resuspended in nonreducing SDS-PAGE sample buffer, and heated to 100°C for 5 min. The samples were resolved on a 13% SDS-PAGE and analyzed by fluorography. Sizing of the complex in presence or absence (resting microsomes) RBP translation was carried out in a similar manner with the only exception being that a 10–80% linear sucrose gradient spun at 50,000 rpm for 4 h in TLS 55 at 4°C in a Beckman table top ultracentrifuge.
Gel Filtration Analysis of Chaperone Complex
Superdex-200 column (GE Healthcare) was washed with PBS twice the bed volume before every run. The column was calibrated using a mixture of marker proteins of known molecular weight (Thyroglubulin, 669 kDa; Ferritin, 440 kDa; and bovine serum albumin, 66 kDa). Microsomes treated or untreated with dithiobis(succinimidylpropionate) (DSP) were lysed in 1% Triton X-100, 1× protease inhibitor cocktail in PBS. The sample volume was made up to 0.5 ml with PBS and used for injection. The flow rate was kept at 0.4 ml/min. Twenty fractions of 0.8 ml each were collected after the void volume, precipitated with 10% TCA, and analyzed by 10% SDS-PAGE and Western blotting by using standard procedures.
Cross-linking Analysis in Microsomes
Posttranslational oxidation of RBP was carried out as described above. Samples were withdrawn at the indicated chase times, alkylated with 20 mM NEM, and split in two. One aliquot was treated with 2 mM DSP in dimethyl sulfoxide (DMSO) for 30 min at room temperature, whereas the other was mock treated with just DMSO to serve as control. Excess DSP was quenched with 100 mM glycine, the microsomes were lysed, and RBP was immunoprecipitated and analyzed by 13% SDS-PAGE and fluorography as described above. Cross-linking analysis in microsomes, isolated from dog pancreas not engaged in active translation, was carried out in a similar manner. Cross-linked and control samples were analyzed by 7.5% SDS-PAGE under reducing and nonreducing conditions. The gels were either stained with Coomassie Blue or transferred onto nitrocellulose membrane for Western blot analysis with specific antibodies against the ER-chaperones calnexin, BiP, calreticulin, and PDI.
Generation and Analysis of Disulfide Mutants of RBP
Disulfide mutants of RBP lacking either the 120–129 disulfide bond (120–129 mutant) 4–160 disulfide bond (4–160 mutant), or the 70–174 disulfide bond (70–174 mutant) or both (double mutant) were generated by a PCR based site-directed mutagenesis technique. Cysteines at positions 120, 129, 4, 160, 70, and 174 were mutated to glycine residues in the respective cases. The presence of mutations was confirmed by DNA sequencing. Posttranslational disulfide oxidation, sedimentation assays, and cross-linking analysis of the disulfide mutants were performed in a manner identical to that described for wild-type RBP.
RBP-TTR Cotranslation Assays
Human TTR was cloned from HepG2 cells. Total mRNA was prepared from HepG2 cells by using standard techniques and subjected to reverse transcription-PCR with TTR-specific primers. The resultant TTR cDNA was cloned into pET22b at the NdeI/BamHI sites. In vitro transcription, translation, and microsomal translocation of TTR were performed in a manner similar to that described for RBP earlier. Microsomal translocation was confirmed by the protease protection assay. All other assays including oxidation and cross-linking analyses were carried out exactly as described for RBP.
RESULTS
Oxidative Folding of RBP in Isolated Microsomes
To examine the folding of RBP, we synthesized mRNA for RBP by using a T7 polymerase based in vitro transcription system and carried out in vitro translation and translocation in the reticulocyte lysate system in the presence of microsomes. We labeled newly synthesized RBP with [35S]methionine and at the end of 1 h of translation, we immunoprecipitated RBP from the translation mix. Immunoprecipitations were performed using polyclonal antibodies to RBP as described under experimental procedures. As shown in Figure 1A, in translations performed in the absence of microsomes, we found a single band of ∼23-kDa molecular weight, corresponding to RBP with an intact signal peptide (Figure 1A, lane 1). In translations performed in the presence of microsomes, we found two bands: the top band corresponding to the untranslocated RBP with intact signal, and the bottom band representing the translocated form of RBP smaller in molecular weight by the size of its signal peptide (∼2 kDa; Figure 1A, lane 2). The presence of both translocated as well as untranslocated RBP indicated that RBP synthesized by the reticulocyte lysate was in excess of the microsomal membranes used. As shown in Figure 1A, lane 3, we could titrate out the untranslocated RBP by increasing the amount of microsomal membranes used. As shown in Figure 1B, treatment of intact microsomes with proteinase K resulted in digestion of only the untranslocated form of RBP (Figure 1B, lane 2). Solubilization of microsomes with detergent resulted in digestion of both, untranslocated as well as the translocated forms of RBP (Figure 1B, lane 3).
Figure 1.

Oxidative folding of RBP in microsomes. (A) Microsomal translocation of RBP. RBP mRNA was translated in the absence (lane 1) or presence of increasing concentration of microsomal membranes (lanes 2 and 3). RBP translated in the presence of saturating amounts of microsomes showing complete translocation (lane 3). (B) Protease protection assay. RBP was translated in the presence of microsomes and subjected to protease treatment as described under Materials and Methods. Untreated control (lane 1), digestion with proteinase K in the absence of Triton X-100 (lane 2), digestion with proteinase K in the presence of 1% Triton X-100 (lane 3). The positions of the untranslocated and translocated forms of RBP are indicated. (C) Posttranslational oxidation of RBP mRNA was carried out as described under Materials and Methods. Samples were collected at the indicated chase times and subjected to immunoprecipitation with anti-RBP antibody. The immunoprecipitates were analyzed by a 13% nonreducing (left) and reducing (right) SDS-PAGE. The location of reduced (Red), folding intermediate (It) and fully oxidized (Ox) forms of RBP are indicated. (D) Quantification of Red (square), It (circle), and Ox (triangle) as a percentage of totals at the different time points of chase. (E) Posttranslational oxidation of RBP was carried out as described above, and 30- and 90-min samples were subjected to proteinase K treatment, as described above and analyzed by 13% nonreducing SDS-PAGE. The nonreduced panel (lanes 1–4) shows Red, It, and Ox are not sensitive to protease, unlike untranslocated.
The reticulocyte lysate and the microsomal system used to translate RBP were optimized for translocation efficiency by maintaining a reducing environment using DTT, which precludes disulfide oxidation of proteins. To facilitate the formation of the three intramolecular disulfides in RBP, we adjusted the redox conditions of the microsomal system with oxidized glutathione posttranslationally, and we examined the status of RBP at different time intervals. Aliquots from the translation mix were withdrawn at 0, 15, 30, 60, and 90 min after addition of GSSG, and NEM was added to quench any free –SH on proteins. RBP was immunoprecipitated as described under Materials and Methods, and the immunoprecipitates were analyzed by nonreducing as well as reducing SDS-PAGE. As shown in Figure 1C, in nonreducing SDS-PAGE before the addition of GSSG (0 min), we could see a single band corresponding to reduced RBP (Figure 1C, lane 1, Red). After 15 min of incubation in the presence of GSSG, in addition to reduced RBP, we saw two closely spaced bands of increasing mobility (Figure 1C, lane 2). Faster mobilities of these bands compared with reduced RBP indicated that the band with an intermediate mobility corresponded to the partially disulfide bonded intermediate (labeled as It), whereas the fastest moving band corresponded to the fully oxidized form (labeled as Ox). The pattern was in agreement with that described by Kaji and Lodish (1993) in their study on posttranslational folding of RBP in HepG2 cells. At 30 min of oxidation, the band corresponding to reduced RBP had decreased in intensity, and there was a corresponding increase in the signals for the intermediate form as well as the fully oxidized form of RBP (Figure 1C, lane 3). At 90 min, all the reduced and partially oxidized intermediate of RBP had disappeared, and only the fully oxidized form was seen (Figure 1C, lane 5). In contrast to multiple bands seen in nonreduced samples after initiation of folding, the reduced samples showed only one band corresponding to reduced RBP (Red) at all the time points before and after addition of GSSG (Figure 1C, right). Figure 1D shows the quantitation of bands corresponding to Red, It, and Ox at different times of folding. A similar pattern of bands was seen when we analyzed the oxidative folding of RBP in microsomes cotranslationally (data not shown). Based on these results, RBP showed a t1/2 of ∼30 min for its oxidative folding in isolated microsomes.
To confirm that RBP bands of faster mobility appearing upon GSSG addition were indeed oxidized forms of RBP in the ER lumen, we incubated 30- and 90-min folding samples of RBP with or without proteinase K, and then we examined the samples in nonreduced as well as reduced forms. As shown in Figure 1E in control samples at the 30-min time point (lane 1), we could see an untranslocated form of RBP, a band corresponding to the translocated form of reduced RBP (Red), and also faint bands corresponding to the intermediate (It) as well as the oxidized form (Ox). At 90 min, as expected, the reduced and intermediate forms of RBP had disappeared and given rise to the oxidized form of RBP (lane 2). On proteinase K digestion, the untranslocated form of RBP was fully digested, but the translocated forms (Red, It, and Ox) were protected (lane 3 and 4), suggesting that these bands were inside the microsomal lumen and did not arise from the untranslocated form of RBP. Samples analyzed in their reduced forms confirmed that translocated forms of RBP were proteinase K resistant, whereas the untranslocated form could be readily digested with the protease. (Figure 1E, lanes 5–8).
Oligomeric State of Folding RBP
To study association of newly synthesized RBP with resident proteins of the ER, we examined its oligomeric state on sucrose gradients. Translation and translocation of RBP were performed in the presence of [35S]methionine as described above. Oxidation of disulfides was initiated by adding GSSG and at 0, 30, and 90 min, samples were withdrawn and quenched with NEM. Samples were layered on a linear gradient of 5–25% sucrose and centrifuged at 40,000 rpm for 20 h. At the end of the centrifugation, fractions were collected from the top, precipitated with 10% TCA, and the precipitates were analyzed by SDS-PAGE and fluorography. As shown in Figure 2A (top), at 0 min of folding, RBP was predominantly found in the pellet of the gradient, indicating its presence in a large complex. From the sedimentation of the markers analyzed independently on the same gradient we estimated the size of the complex to be ∼20 S20, w. At 30 min of folding, we found a significant amount of oxidized RBP (>50%) present in fractions 2 and 3, indicating its monomeric nature (Figure 2, middle). At 90 min of folding (Figure 2, bottom), the majority of RBP (∼80%) had moved out of the pellet fraction into the monomeric form in fractions 2 and 3. The result indicated that the reduced form of RBP was present in a dynamic complex, probably containing with ER-chaperones. On addition of oxidized GSSG, the complex readily gave rise to disulfide bonded, mature form of RBP.
Figure 2.

Oligomeric state of newly synthesized RBP. (A) RBP mRNA was translated in a rabbit reticulocyte lysate system supplemented with dog pancreas microsomes under reducing conditions for 60 min at 27°C. Disulfide oxidation was initiated by the addition of 4 mM GSSG and chased for a further period of 30 and 90 min. At the end of the chase, the microsomes were lysed and the lysate subjected to sedimentation analysis on a 5–25% sucrose gradient. Fractions were collected from the top and precipitated with 10% TCA. The precipitates were analyzed on a 13% nonreducing SDS-PAGE followed by autoradiography. The positions of reduced (Red), intermediate (It), and fully oxidized (Ox) forms are indicated. The pellet fractions are shown at a lighter exposure. (B) To estimate the size of the complex, RBP was subjected to posttranslational folding as described above, and samples taken at 0 and 90 min were sedimented on a tabletop ultracentrifuge using 10–80% glycerol gradient at 50,000 rpm for 4 h. Fractions were collected from the top, TCA precipitated, and analyzed on a 10 or 13% nonreducing SDS-PAGE. Gels (13%) were followed by autoradiography. The positions of the reduced (Red) and fully oxidized (Ox) forms are indicated. Gels (10%) were analyzed by Western blotting using antibodies against ER-chaperones (BiP, PDI, and CNX). (C) Cotranslational oxidation of RBP was performed by translating in presence of GSSG. After 30 and 60 min of initiation of translation, the samples were taken and analyzed on 10–80% glycerol gradient as described above.
To estimate the size of the folding complex more accurately, we analyzed samples for 0- and 90-min folding times on a 10–80% linear glycerol gradient spun at 50,000 rpm for 4 h in a tabletop ultracentrifuge (see Materials and Methods). As shown in Figure 2B (top), at 0 min of folding, the reduced form of RBP was predominantly present in fractions corresponding in S value above 18S20, w in size. A population of reduced RBP was also seen in fractions ranging in S value of 11–18 S20, w. The latter was likely to arise from partial disruption of the larger complex seen in fraction 8. At 90 min of folding time (Figure 2B, bottom), RBP was predominantly found at the top of the gradient, corresponding to the folded, monomeric form. Importantly, Western blot analysis of fractions from 0-min sample containing reduced RBP showed presence of BiP, PDI, and CNX in fractions overlapping with that of reduced RBP (see bottom panels in Figure 2B). The results suggest that reduced RBP sedimenting in large complexes was associated with chaperones in the ER. Experiments shown subsequently (see Figure 4C) confirmed that newly formed RBP is in fact associated with pre-existing complex of ER-chaperones, which was present even in the absence of RBP translation.
Figure 4.

Analysis of chaperone complex in resting microsomes. (A) Microsomes isolated from dog pancreatic tissue were subjected to cross-linking with DSP as described above under steady-state conditions. Reduced (lanes 2 and 4) and nonreduced (lanes 1 and 3) samples were analyzed by 7.5% SDS-PAGE. The positions of the chaperone proteins are indicated. (B) Western blot analysis of cross-linked microsomal samples from dog pancreas using antibodies against BiP, calreticulin, calnexin, and PDI. Nonreduced (lane 1) and reduced (lane 2). (C) The microsomes treated with DSP or DMSO alone, as described above, were analyzed by 10–80% glycerol gradient as described under Materials and Methods. The fractions collected were TCA precipitated and analyzed by 10% SDS-PAGE and Western blotting using antibodies against BiP, PDI, and CNX. Bands were quantitated as described under Materials and Methods. Open circles indicate uncross-linked samples (−DSP), and closed circles indicate cross-linked samples (+DSP). (D) Microsomes treated with DSP (as above) were analyzed by Superdex-200 column, and fractions were processed similarly.
We also analyzed translation mixtures in which RBP was folded cotranslationally. The translations were performed as above except that GSSG was included in the reaction mixture at the time of translations. Analysis of samples at 30 min of translation by sucrose gradients revealed a population of RBP present in complexes ranging from 11S to 18S (∼40%). The remaining RBP pool was found in the region corresponding to the monomeric form. At 60 min of synthesis, the total amount of labeled RBP increased in intensity but was predominantly found in monomeric fractions (Figure 2C). The result indicated that during cotranslational folding as well a population of reduced RBP seemed in large complexes of size ranging from 11S to 18S. This population was short lived and hence did not accumulate significantly during cotranslational folding.
Folding RBP Can Be Transiently Cross-linked to the Chaperone Complex
The above-mentioned results suggested that newly synthesized, reduced form of RBP was present in a large complex. On completion of its oxidative folding, RBP monomers are released from this complex. To examine whether newly synthesized RBP can be cross-linked into a complex without breaking open the microsomes, we made use of a membrane-permeable, homobifunctional, and thiol-cleavable cross-linking agent, DSP.
We carried out in vitro translation and translocation of RBP in the presence of [35S]methionine as noted above, and disulfide oxidation was initiated with addition of GSSG. At 0, 30, and 90 min of oxidation, we divided the samples into two aliquots, and we added DSP in carrier DMSO to one aliquot and further continued incubation at room temperature for 30 min. DMSO alone was added to the control. After cross-linking, excess cross-linker was quenched using 100 mM glycine. The microsome samples were then solubilized in detergent containing buffer, and RBP was immunoprecipitated as described above. The immunoprecipitates were analyzed in the nonreduced as well as reduced form by SDS-PAGE and fluorography. As shown in Figure 3A (left, lanes 1–3), in addition to the untranslocated form of RBP (Ut) the noncross-linked samples in their nonreduced forms showed the presence of reduced RBP (Figure 3A, left, lane 1, Red) at 0 min, some partially oxidized (It) as well as fully oxidized RBP (Ox) at 30 min (Figure 3A, left, lane 2) and predominantly the fully oxidized form (Ox) at 90 min of folding (Figure 3A, left, lane 3). A single band corresponding to Red was visible at all three time points in reduced samples (Figure 3A, right).
Figure 3.

Association of newly synthesized RBP with ER-chaperones. (A) Posttranslational disulfide oxidation of RBP was carried out as described previously. At chase times of 0, 30, and 90 min, samples were alkylated with 20 mM NEM and split in two. One was treated with 2 mM DSP in DMSO at room temperature for 30 min, whereas the other was mock treated with just DMSO to serve as control. The excess DSP was quenched with 100 mM glycine, microsomes were lysed and subjected to immunoprecipitation with anti-RBP antibody. The immunoprecipitates were analyzed by 13% nonreducing and reducing SDS-PAGE followed by autoradiography. (Left) Nonreduced SDS-PAGE of control and cross-linked samples (lanes 1–6). (Right) Reducing SDS-PAGE of the same samples (lanes 7–12). The positions of the untranslocated (Ut), reduced (Red), folding intermediate (It), and the fully oxidized (Ox) forms are indicated. (B) HepG2 cells were subjected to metabolically labeled, cross-linked and immunoprecipitated with antibodies to RBP, Grp94, BiP, and PDI. The samples were analyzed on reducing, 8–15% gradient SDS-PAGE. (C) Posttranslational oxidation of RBP was carried out as described under Materials and Methods. The samples were lysed with 2% CHAPS, 1× protease inhibitor cocktail, in phosphate-buffered saline, pH 7.2. The lysate was divided into three aliquots and subjected to immunoprecipitation using anti-RBP, anti-BiP, and anti-calnexin antibodies. The immunoprecipitates were analyzed by 13% SDS-PAGE. (D) Quantitation of RBP bands is shown for RBP immunoprecipitated by anti-calnexin (closed squares) and anti-BiP (open squares). The RBP immunoprecipitated by anti-RBP antibody was taken as 100% for respective chase point. (E) RBP was translated in presence of microsomes and cross-linked as described for Figure 3A. Sample corresponding to 0 min was lysed and equal aliquots were immunoprecipitated with antibodies to RBP, Grp94, and BiP. Samples were analyzed by reduced SDS-PAGE.
In cross-linked samples shown in Figure 3A (left panel, lanes 4–6), at 0 min we found all the reduced RBP (Red) in large cross-links that did not enter the resolving gel (Figure 3A, left, lane 4). At 30 min of folding time, although a part of RBP continued to occur in the large cross-links, a small amount corresponding to non–cross-linkable RBP (Ox) also occurred within the resolving gel (Figure 3A, left, lane 5). At 90 min of folding time, all the labeled RBP was present as non–cross-linkable, mature form (Figure 3A, left, lane 6). Because the cross-linker used in this experiment was reversible, when analyzed in reduced state (Figure 3, right), the cross-links dissociated and gave rise to a labeled band corresponding to reduced RBP (Red). The experiment confirmed results of our sucrose gradient experiment, showing that in its reduced form, the folding RBP precursor is associated with a large complex, which can be readily cross-linked. On completion of its oxidative folding, the mature RBP is no longer cross-linked.
To further examine the nature of the folding complex, we resorted to the use of human hepatoma cell line HepG2, wherein individual ER-chaperones could be examined by prelabeling and immunoprecipitation analysis. HepG2 cells were labeled for 24 h with [35S]cysteine and methionine to label cellular protein pool, including chaperones of the ER. At the end of this labeling period, cells were further labeled for 10 min to label newly synthesized proteins in presence of 5 mM DTT, including secretory proteins such as RBP. At the end of this labeling, cells were treated with ice cold 20 mM NEM in PBS and reacted with DSP to cross-link interacting proteins. Samples were lysed in detergent-containing buffer, and equal aliquots were immunoprecipitated with antibodies specific to Grp94, BiP, PDI, and RBP. An aliquot of the cell lysate was also incubated with protein A-linked beads alone to visualize any nonspecific binding. The immunoprecipitates were analyzed in the reduced form.
As shown in Figure 3B, in samples incubated with protein A beads alone, we found a nonspecifically interacting protein band of ∼45-kDa from HepG2 cells (lane 1). In lysates precipitated using antibodies to RBP (lane 2), we found a band corresponding in mobility to RBP as well as several higher molecular weight bands corresponding in size to specific ER-chaperones. Comparison of these bands with immunoprecipitates using antibodies to BiP (lane 3), Grp94 (lane 4), and PDI (lane 5) showed that these chaperone bands were indeed coprecipitated with antibodies to RBP (see bands marked with an asterisk [*]). In addition, there were other additional bands seen in RBP immunoprecipitates that may correspond to other ER-chaperones. Importantly, in Grp94 immunoprecipitates we were able to detect a faint band corresponding to RBP as well. The low recovery of RBP in Grp94 immunoprecipitates was due to Grp94 itself associating with large number of newly synthesized proteins in the ER of HepG2 cells and immunoprecipitation with antibodies to Grp94 pulls down a fraction of these complexes, among which RBP–GRP94 complex is a minor population. The results indicated that newly synthesized RBP was associated with a complex of chaperones containing BiP, PDI, and Grp94 as well as additional unidentified ER proteins.
We also used a coimmunoprecipitation approach to demonstrate interactions between newly synthesized RBP and ER-chaperones more directly. We performed a posttranslational folding experiment as described in Figure 1, and samples corresponding to 0, 30, and 60 min of folding were divided into three aliquots and immunoprecipitated with antibodies specific to RBP, BiP, or CNX. As shown in Figure 3C, the immunoprecipitates were analyzed in nonreduced and reduced forms. In nonreduced forms of RBP immunoprecipitates, as expected, we observed a reduced form of RBP at 0-min time interval and then the oxidized forms at 30 and 60 min of folding (Figure 3C, lanes 1–3). RBP immunoprecipitates also showed small amount of signal corresponding to labeled RBP at the top of the resolving gel (Figure 3C, see arrows in lanes 2 and 3). Absence of these bands in the reduced samples (Figure 3C, lanes 11 and 12) indicated that these were disulfide bonded aggregates. In BiP as well as CNX immunoprecipitates, we found RBP band coprecipitating mainly at 0-min time point when it had not acquired its disulfide bonds (Figure 3C, lanes 4 and 7). The oxidized forms of RBP seen in lanes 2 and 3 did not significantly coprecipitate with antibodies to BiP and CNX (Figure 3C, lanes 5 and 6 and 8 and 9). The disulfide bonded aggregates seen in lanes 2 and 3 were faintly visible in CNX immunoprecipitates (Figure 3C, lanes 8 and 9).
As noted above, in the reduced samples also (Figure 3C, right) we could see a bands corresponding to RBP in immunoprecipitates corresponding to RBP, CNX as well as BiP mainly at 0-min time point (Figure 3C, lanes 10, 13, and 16). At folding time points 30 and 60 min, very faint bands corresponding to RBP were visible in BiP and CNX immunoprecipitates (Figure 3C, lanes 14 and 15 and 17 and 18).
The interaction of newly synthesized RBP with ER chaperones was further stabilized by cross-linking microsomal samples. RBP was translated and cross-linked as described for Figure 3A. The cross-linked, 0-min sample was lysed, and equal aliquots were immunoprecipitated with antibodies specific to RBP, Grp94, and BiP. As shown in Figure 3E a convincing signal corresponding to RBP was seen in samples immunoprecipitated with antibodies to BiP and Grp94. In all, the results indicated that newly synthesized, labeled RBP transiently associated with Grp94, BiP as well as CNX at early times of folding and dissociated coincident with its acquisition of intramolecular disulfides.
Preexisting Complex of Chaperones in ER-derived Microsomes
To investigate the nature of the complex described above, we examined the status of ER-chaperones in microsomes not engaged in translation using a similar approach. Microsomes, isolated from either dog pancreas or rat liver, were subjected to cross-linking with DSP or DMSO carrier alone as described under Materials and Methods and examined under nonreduced as well as reduced conditions by SDS-PAGE and Coomassie Blue staining to visualize the status of its proteins. As shown in Figure 4A, microsomes treated with DMSO alone (Figure 4A, lane 1) showed the presence of several protein bands corresponding to abundant chaperone proteins in ER lumen. On treatment with DSP (Figure 4A, lane 3), a majority of these proteins were seen cross-linked in a large complex that did not enter the resolving gel. Examination of these samples in the reduced form showed individual chaperones released out of this complex (Figure 4A, lane 4). Analysis of these samples by Western blotting using antibodies to calnexin, BiP, calreticulin, and PDI confirmed that Coomassie-stained bands seen in microsomal samples corresponded to these ER-chaperones (Figure 4B). The results indicated that chaperones of the ER-derived microsomes could be cross-linked into a large complex under steady-state conditions, even in the absence of active translation.
To further investigate the complexes of chaperones we used chemical cross-linking approach in combination with velocity sedimentation as well as size exclusion chromatography. In an experiment similar to that described above, we treated intact microsomes with DSP or the carrier DMSO alone and divided the lysates into two equal aliquots. One aliquot of each lysate was layered on glycerol gradients and spun on table top ultracentrifuge at 50,000 rpm for 4 h. Fractions were collected from the top, precipitated with TCA and analyzed for the presence of BiP, PDI, and CNX in various fractions by SDS-PAGE and Western blotting (Figure 4C). The other aliquot of lysates was analyzed on size exclusion chromatography using Superdex 200 gel filtration column. Chromatographic fractions were TCA precipitated and analyzed for the presence of BiP, PDI, and CNX by SDS-PAGE and Western blotting approach (Figure 4D).
In velocity sedimentation experiment shown in Figure 4C, we observed BiP and PDI sedimenting in fractions 1–3 corresponding in size from 4S to 11S. The sizes were somewhat bigger than those expected of BiP and PDI in their monomeric forms. On cross-linking, we found almost all of signal corresponding to BiP in fractions 6–8 indicative of a complex of size about 18S. In PDI and CNX, we also could see a shift in sedimentation to fractions 6–8 upon cross-linking. The results suggested that it was possible to stabilize complexes of BiP, PDI, and CNX corresponding in size up to 18S upon cross-linking. Similarly in size exclusion chromatography experiment shown in Figure 4D we found stabilization of complexes containing chaperones between 440 and 660 kDa upon chemical cross-linking. It is important to note that, even in the absence of chemical cross-linker, faint but detectable signal corresponding to ER-chaperones was visible in complexes of size up to 650 kDa (fractions 1–3). The observation supports the possibility that chemical cross-linking merely stabilized pre-existing complexes that would otherwise disrupt due to detergent treatments used in the lysis protocols.
Overall, the results described in the sections above suggest that newly synthesized proteins, such as RBP, transiently associated with a pre-existing complex of ER-chaperones and dissociated at late stages of their maturation process. It was possible that substrate association further stabilized the complex of chaperones.
Deletion of Two Large Disulfides Halts Oxidative Folding of RBP, whereas Removal of the Small Loop Diverts RBP into a Disulfide-linked Aggregate
To examine the role of the two major disulfides, namely, 4–160 and 70–174 in the folding of RBP, we created three mutant forms of RBP lacking one of these disulfide pairs. We replaced 1) cysteine residues at position 4 and 160 to glycines to eliminate 4–160 disulfide pair (mutant 4–160); 2) cysteines at positions 70 and 174 to glycines to remove 70–174 disulfide pair (mutant 70–174); 3) cysteines 4, 160, 70, and 174 to glycines to create the double mutant lacking both the major disulfide bonds (double disulfide mutant); and 4) cysteines at 120 and 129 to glycines to 120–129 disulfide pair (mutant 120–129). The wild-type and the four mutant forms of RBP described were then analyzed for their ability to fold in isolated microsomes as mentioned above.
As shown in Figure 5, although wild-type RBP (Figure 5, lanes 1–3, top) containing all three disulfides oxidized to the compactly folded state by 120 min of folding, 4–160 mutant (Figure 5, lanes 4–6, top) and 70–174 mutant (Figure 5, lanes 7–9, top) lacking one of the two major disulfides showed a decreased mobility on nonreducing SDS-PAGE. The fully oxidized form in both these cases resembled the partially folded form of WT RBP (It). The double mutant lacking both the major disulfides failed to show any faster mobility and continued to migrate at the position of reduced RBP (Figure 5, lanes 11–13, top). The results indicated that formation of one of the two major disulfides, namely, 4–160 or 70–174 was sufficient to form the intermediate (It) and formation of the remaining second big loop completed the folding to the compact state of RBP.
Figure 5.

Folding of RBP-disulfide mutants. WT and the disulfide mutants of RBP, generated as described under Materials and Methods, were translated in a rabbit reticulocyte lysate system supplemented with dog pancreas microsomes under reducing conditions for 60 min at 27°C. Disulfide oxidation was initiated by the addition of 4 mM GSSG, and samples were collected at the indicated chase times. The samples were alkylated with 20 mM NEM and subjected to immunoprecipitation with anti-RBP antibody. The immunoprecipitates were analyzed by 13% nonreducing SDS-PAGE (top) or reducing SDS-PAGE (bottom). WT (lanes 1–3), 4–160 mutant (lanes 4–6), 70–174 mutant (lanes 7–9), and the double mutant (lanes 11–13) are shown. The positions of untranslocated (Ut), reduced (Red), folding intermediate (It), and fully oxidized (Ox) forms are indicated. (Bottom) Analysis of the same samples in reduced form.
To assess the role of the smallest of the three intramolecular disulfide loops, we mutated cysteine 120 as well as 129 to glycine, maintaining cysteines involved in the other two disulfides, namely, 4–160 and 70–174 intact. The wild-type and 120–129 mutant of RBP were analyzed for their ability to fold in isolated microsomes as described under Materials and Methods. As shown in Figure 6, although the wild-type RBP progressed from reduced (r) to its oxidized form (lanes 1–3), the mutant form failed to fold and got irreversibly trapped in an aggregate possibly, visible at the beginning of the resolving gel (Figure 6, see asterisk [*] in lanes 5 and 6). Analysis of samples in their reduced form showed reappearance of bands corresponding to reduced RBP (Figure 6, lanes 10–12), suggesting that the aggregates were indeed due to intermolecular disulfide bonds.
Figure 6.

Deletion of the small disulfide loop abrogates RBP folding. The 120–129 mutant was generated as described under Materials and Methods. Posttranslational oxidation was performed as described above, and samples were taken at indicated time points, alkylated with NEM, and immunoprecipitated with anti-RBP antibody. The immunoprecipitates were analyzed by 13% nonreducing and reducing SDS-PAGE.
Substrate Association with the Chaperone Complex Is Sensitive to Its Folded State
To examine whether perturbation of RBP folding would influence its association with the chaperone complex, we examined its association with RBP mutants lacking one or two of its disulfide bonds. We translated wild-type as well as the mutant forms of RBP, and we performed cross-linking as described above. Cross-linked samples were analyzed by nonreducing SDS-PAGE. As shown in Figure 7A, the wild-type RBP could be cross-linked into a big complex at early times of folding when it was still in the reduced form (Figure 7A, lane 1, compare top and bottom panels). As RBP progressed in its oxidative folding, it became progressively less cross-linkable (Figure 7A, lane 3). Interestingly, in mutant forms of RBP lacking one of the two big loops, both were released from the cross-link by 120 min of oxidation (Figure 7A, lanes 6 and 9). In the double mutant lacking both the major loops, the reduced form of RBP remained cross-linked to ER-chaperones even at 120 min of oxidation (Figure 7A, lane 12). The results are presented in the form of a bar graph in Figure 6B. The results suggested that despite the absence of one of the two big loops, RBP mutants were considered “folded” by the chaperones of the ER. This was consistent with sedimentation analysis of RBP mutants lacking either one or both the big disulfide loops. As shown in Figure 7C, similar to the wild-type form, mutant RBP lacking either 4–160 or 70–174 disulfides were released from the fast sedimenting complexes upon oxidation in the presence of GSSG. However RBP mutant form lacking both the above disulfides (4–160 and 70–174) persisted in large complexes on the sucrose gradient even after 2 h of incubation with GSSG. The aggregated nature of 120–129 mutant even at early times of folding (shown in Figure 6) precluded its cross-linking analysis.
Figure 7.

Cross-linking analysis of RBP disulfide mutants. (A) Posttranslational disulfide oxidation of WT and disulfide mutants of RBP was carried out as described previously. At chase times of 0, 30, and 120 min, samples were alkylated with 20 mM NEM and split in two. One sample was treated with 2 mM DSP in DMSO at room temperature for 30 min, whereas the other sample served as control. The excess DSP was quenched with 100 mM glycine, and microsomes were lysed and subjected to immunoprecipitation with anti-RBP antibody. The immunoprecipitates were analyzed by 13% nonreducing SDS-PAGE. (Top) Noncross-linked controls. (Bottom) Cross-linked samples. WT (lanes 1–3), 4–160 mutant (lanes 4–6), 70–174 mutant (lanes 7–9), and the double mutant (lanes 10–12) are shown. The positions of the reduced (Red), folding intermediate (It) and fully oxidized (Ox) forms are indicated. (B) Bar diagram showing the percentage of cross-linked RBP at different time points of chase. (C) Sedimentation profiles of WT and disulfide mutants of RBP shown. Open circles indicate 0 min, and closed circles indicate 120 min posttranslation oxidation time point.
These results suggested that the chaperone complex was able to sense the folded state of its substrate. Unlike the wild-type RBP, which associated with the complex only transiently the mutants exhibited binding time-scale proportionate to their respective folding defects. Thus, the mutant lacking one of the two large disulfides showed prolonged association with the complex, whereas the double mutant remains bound persistently.
RBP Assembly with TTR in Isolated Microsomes
In the above-mentioned scenario, newly synthesized RBP underwent disulfide oxidation in association with a complex of chaperones and dissociated upon completion of its oxidative folding. To examine whether RBP association with TTR coincided or followed its oxidative folding, we cotranslated RBP and TTR together and performed cross-linking at early as well as late chase times. As described above, RBP and TTR were translated and translocated together in isolated microsomes in the reduced state, and cross-linking was performed either at early stage when it was still reduced or 2 h after addition of oxidized GSSG to allow disulfide oxidation. Microsomes were lysed with detergent containing buffer and immunoprecipitated with antibodies to RBP or TTR. An aliquot of the lysate was incubated with protein-A agarose alone to serve as a control. Immunoprecipitates were analyzed in their reduced or nonreduced forms by SDS-PAGE and PhosphorImage analysis. As shown in Figure 8A, in samples precipitated with antibodies to RBP, at 0-h time point we saw bands corresponding to reduced RBP alone (Figure 8, lanes 1 and 5). In TTR immunoprecipitates of the same samples (Figure 8A, lanes 2 and 6), we saw bands corresponding in size to TTR (14 kDa) and in addition a band probably corresponding to a dimer of TTR. At 2 h of chase with GSSG, we could see the oxidized form of RBP (Figure 8A, lanes 3 and 7) migrating faster than the reduced form seen at 0 h. In addition, in cross-linked samples two additional bands could be seen corresponding in size to ∼40 kDa (CI) and 77 kDa (CII). In TTR immunoprecipitates of the same samples, we saw both the above-mentioned bands in addition to the band of TTR-dimer discussed above. Presence of these bands in the immunoprecipitates of both RBP as well as TTR indicated that they were RBP–TTR complexes. Based on the size, the 77-kDa band (CII) is likely to be a cross-linked complex containing RBP and TTR in 1:4 ratio. The intense 40 kDa (CI) cross-linked band is likely to correspond to an intermediate in the assembly containing RBP and TTR in 1:1 ratio. The 77-kDa complex of RBP-TTR is in agreement with the previously proposed stoichiometry of 1:4 for RBP–TTR interactions. The presence of RBP-TTR cross-link only at 2 h of chase showed that RBP forms a complex with TTR only upon completion of its disulfide oxidation in the ER when it is no longer associated with chaperones.
Figure 8.

RBP-TTR assembly in microsomes. (A) RBP and TTR were cotranslated in the presence of microsomes under reducing conditions and disulfide oxidation was carried out as described in text. Samples were withdrawn at 0 h (lanes 1, 2, 5, and 6) and 2 h (lanes 3, 4, 7, and 8) of chase and subjected to cross-linking with DSP. Noncross-linked (left) and cross-linked (right) samples were immunoprecipitated with antibodies against RBP (lanes 1, 3, 5, and 7) and TTR (lanes 2, 4, 6, and 8) and analyzed by 13% nonreducing SDS-PAGE and fluorography. The positions of reduced and oxidized RBP and TTR are indicated. Bands corresponding to TTR dimer (28 kDa), 1:1 RBP-TTR complex (CI, 35 kDa) and 1:4 RBP-TTR complex (CII, 77 kDa) are indicated by arrows in the gel. (B) Similar procedure was adopted as described above, where WT or mutant RBP mRNA was used for translation. The 2-h chase samples subjected to cross-linking with DSP and immunoprecipitated using antibody against RBP (lanes 1–3) or TTR (lanes 4–6). Immunoprecipitates were analyzed by nonreducing or reducing SDS-PAGE (top and bottom, respectively). CI and CII correspond to RBP–TTR complexes described above.
We also examined whether TTR was able to assemble with RBP mutants lacking one or two disulfides described above. As described above, we cotranslated TTR mRNA with wild-type RBP or 70–174 mutant or 4–160 mutant or the double mutant lacking both 4–160 and 70–174 disulfide bonds. RBP was allowed oxidize for 2 h after addition of GSSG, and the reaction mixture was divided into two aliquots. One aliquot of sample was cross-linked with DSP, whereas the other aliquot was treated with carrier DMSO alone. The samples were lysed and immunoprecipitated with antibodies to RBP or TTR as described under Materials and Methods. The immunoprecipitates were analyzed in nonreduced and reduced form by SDS-PAGE and PhophorImaging. As shown in Figure 8B (top), we found wild-type RBP to form cross-links corresponding in size to RBP–TTR interactions as shown above (Figure 8B, see arrows). Both CI and CII complexes were visible in immunoprecipitations carried out with antibodies to RBP (Figure 8B, lane 1, left) as well as antibodies to TTR (Figure 8B, lane 4, right). Single mutants 4–160 and 70–174 also showed presence of these cross-links (Figure 8B, lanes 2 and 3 and lanes 5 and 6). Analysis of the TTR and RBP immunoprecipitates of cross-linked samples in reduced form (wherein the cross-linked partners would dissociate) revealed bands corresponding to RBP and TTR, respectively, confirming that the cross-links seen in the nonreduced form indeed corresponded to RBP-TTR complexes (Figure 7B, bottom). Analysis of the double mutants in a similar way failed to show presence of RBP-TTR cross-links (data not shown). The result indicated that RBP mutants lacking single disulfides (4–160 or 70–174) were able to associate with TTR while the mutant lacking both the disulfides remained unassembled.
DISCUSSION
It is well known that chaperones in the ER assist newly synthesized proteins to attain their natively folded states. Only correctly folded proteins are allowed exit out of the ER, whereas partially folded, misfolded, and aggregated proteins are effectively retained. This quality control system of the ER is the main checkpoint to control export of secretory proteins outside the cell. Many secretory and membrane proteins have been shown to conform to this quality control rule. Mechanisms underlying this conformation based sorting within the ER are only partly understood. Selective association of chaperones with partially folded and unfolded proteins is thought to govern their retention in the ER. However molecular signatures defining the unfolded or partially folded states of proteins in the ER are not fully deciphered.
In proteins containing N-linked glycans, the glucosylation state of their N-glycan is known to dictate their binding to chaperones such as calnexin and calreticulin (Sousa et al., 1992; Hammond et al., 1994). Glucosylation state of substrates in turn depends on how well the protein is folded. Rules that govern binding of nonglycosylated proteins to other ER-chaperones are less well defined. It is believed that hydrophobic patches on partially folded and unfolded proteins form effective binding sites for chaperones such as BiP and GRP94 (Flynn et al., 1991; Blond-Elguindi et al., 1993). Although differentiation between unfolded, partially folded, and folded proteins is obvious for many well studied examples, this distinction is somewhat subtle in other cases. MHC class I assembly is one such example where, in addition to the folding requirements peptide loading is essential for acquisition of transport competent state (Ortmann et al., 1994; Suh et al., 1994). RBP is another example of a protein whose secretion does not conform to the general rules of quality control in the ER (Ronne et al., 1983).
Among secretory proteins, RBP is unique in being secreted only in response to availability of its ligand retinol. In the absence of retinol, RBP synthesis continues but the apo-form of RBP is retained within the ER. It is believed that apo-form of RBP may not be fully folded and may therefore be retained by the quality control system composed of chaperones in the ER. Previous studies by Kaji and Lodish (1993) have addressed conformational differences between apo- and holo-forms of RBP in the ER of human hepatoma cells. Using DTT resistance as a marker for RBP maturation, they showed that RBP completes its oxidative folding and attains a DTT-resistant, mature state both in the presence or the absence of its ligand. Apart from a small difference in their kinetics of turning DTT resistant, there were no obvious differences in the folded states of RBP in its apo or the holo-form. These studies, however, did not address association of RBP with the quality control machinery. It was not clear whether RBP dissociated from chaperone proteins of the ER in its apo form. Secretion of RBP may also depend on energetic considerations similar to those proposed for TTR secretion (Sekijima et al., 2005). It is possible that ligand unbound, apo form of RBP is conformationally flexible and energetically less stable compared with its rigorously folded and stable holo-form. This possibility is consistent with the previous observation that ligand bound form of RBP is DTT resistant.
Despite its simple globular shape and lack of complex modifications, RBP is extremely sluggish in terms of its secretion. Reasons for its prolonged stay in the ER remain unclear. Moreover, the chronology of RBP maturation events in the ER is still unknown. There is also very little known about association of RBP with chaperones of the ER. Bellovino et al. (1996) have found newly synthesized RBP to be associated with calnexin in the ER. However, this is not consistent with the prevailing notion that interaction with lectin-like chaperones is restricted to N-glycosylated proteins. Apart from this report, there are no other studies on the interaction status of newly synthesized RBP in the ER.
In addition to dissecting the oxidative folding events of newly synthesized RBP, our studies in isolated microsomes also address its interactions with the quality control machinery in the ER. Oxidative folding steps of RBP earlier described by Kaji and Lodish (1993) could be faithfully reproduced in isolated microsomes. Consistent with the slower rates of folding reported for many other proteins in isolated microsomes, we also found RBP folding in microsomes to have slowed down considerably. Although the posttranslational folding of RBP was complete within 2 min in vivo, the t1/2 of RBP folding in microsomes was ∼30 min. Acquisition of DTT-resistant state by RBP translated in microsomes indicated that the delay in protein folding was only kinetic and the end result of the folding process was similar to the mature RBP seen in vivo.
Analysis of RBP mutants lacking one of the two major disulfides (4–160 or 70–174) revealed that the folding intermediate of RBP possessed one of the two large disulfide loops. RBP folding was complete as soon as the second disulfide loop was acquired. Although it was not possible to determine the order in which the two disulfides formed, it was apparent that formation of both the above-mentioned disulfides was necessary to complete the folding process. Mutants lacking one of the two major loops showed decreased mobility on nonreducing gels and resembled the It form of wild-type RBP. Mutating both the above-mentioned disulfide loops abolished its mobility and the double mutant was found to migrate at the position of reduced RBP. Remarkably the smallest disulfide, 120–129, seemed to play the most critical role in committing RBP to a productive folding pathway. In the absence of this small disulfide, RBP failed to acquire the other two disulfides, unproductively deviating into an intermolecular disulfide-bonded aggregate. The result suggested that formation of this small loop possibly provided a nucleation center for a structure that directs RBP to a productive folding pathway at an early stage of its folding. The results suggested a sequence from [Red to (120–129) to (4–160) or (70–174) to (70–174) or (4–160) to Ox] for RBP folding in the ER (Figure 9).
Figure 9.

A model depicting biogenesis of RBP in the ER. Newly synthesized RBP undergoes oxidative folding bound to a pre-existing complex of chaperones. It remains associated with chaperones in the ER until two of its three intramolecular disulfides are formed. On formation of disulfide bonds, RBP dissociates from the chaperones and assembles with a tetramer of TTR in the ER. Cysteines ( ), disulfide bond (
), disulfide bond (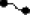 ), and transthyretin monomer (
), and transthyretin monomer (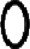 ).
).
Sedimentation analysis of RBP during its oxidative folding indicated that early forms of RBP, namely, the reduced form (Red) and the folding intermediate (It), were organized in a large complex. In the presence of oxidizing conditions these complexes readily gave rise to monomeric RBP (Ox). Both cotranslational as well as post-translational forms of unfolded and partially folded RBP were seen in similar complexes of size >18 S20, w. Dynamic nature of these complexes suggested them to be folding complexes containing chaperone machinery of the ER. These results also were supported by cross-linking analysis performed with homo-bifunctional cross-linker DSP. We found the reduced (Red) and the folding intermediate (It) forms of RBP to be transiently cross-linked into a large complex consisting of ER-chaperones BiP, PDI, and CNX in addition to RBP itself. Association of RBP with CNX in the cross-linked complex may be indirect as RBP is devoid of N-glycans, known to be required for binding to these lectin chaperones in the ER. The oxidized form of RBP, however, could not be cross-linked into this complex, indicating that it had dissociated from the chaperone machinery in the ER. Our observations show that RBP associated with a complex of chaperones in a transient manner. It could be trapped in association with the complex soon after synthesis and was dissociated upon completion of it oxidative folding.
Cross-linking analysis in microsomes, isolated from both dog pancreas and rat liver, not engaged in active protein translation, suggested that chaperones of the ER were constitutively organized in a complex. Its assembly was not dependent on the translocating polypeptide substrate or early folding intermediates of newly synthesized proteins. The result was in agreement with organization of lumenal proteins in the form of a matrix consisting of ER-chaperones and accessory factors. In this scenario, newly synthesized protein substrates would transiently associate with a pre-existing complex of chaperones and dissociate during late stages of their maturation. Thus, regardless of the folding state of RBP, chaperones themselves continued to exist in a complex under steady-state conditions.
Presence of a complex of ER chaperones and organization of ER-resident proteins in the form of a matrix has been suggested by three independent studies previously (Reddy et al., 1996; Tatu and Helenius, 1997; Meunier et al., 2002). In these studies, a model viral glycoprotein and an endogenous immunoglobulin heavy chain were shown to transiently interact with a complex of chaperones in vivo. This is the first report of complexes containing folding intermediates of a small, globular, secretory protein devoid of glycosylation in isolated microsomes. The presence of a complex of ER chaperones shown here highlights yet another facet of the ER microenvironment that is preserved in the isolated microsomal system.
It must be mentioned here that fluorescence measurements of GFP fusion constructs of calreticulin were not able to capture the matrix-like essence of ER microenvironment (Snapp et al.,2006). It is possible that ectopic expression of fusion constructs may not fully recapitulate the native organization of the ER. A similar contradiction was reported previously between live-cell experiments performed with GFP fusion of a temperature-sensitive form of vesicular stomatitis virus G-protein and biochemical studies explaining retention mechanisms in the ER (Nehls et al., 2000). It is possible that different experimental approaches highlight different aspects of ER organization and further detailed experiments are required to better appreciate this facet of the ER.
What determines the time span of substrate association with the chaperone complex? Sedimentation and cross-linking analysis of RBP mutants indicated that although the double mutant remained irreversibly associated with the large complex, the single disulfide mutants (lacking either 4–160 or 70–174 disulfide) could be released out of the large complex with delayed kinetics. The observation was consistent with the acquisition of DTT-resistant state by these mutants and emphasized that association with the complexes was sensitive to the folded state of the substrate. Release of the single disulfide mutants out of the large complex suggested that despite the lack of one of the two major disulfides, these mutants were considered “folded” by the quality control machinery in the ER. Persistent cross-linking of RBP mutant lacking two major disulfides suggested that in the absence of both of these two disulfides RBP may resemble a molten globule state and classified “unfolded” by the quality control.
Although RBP assembly with TTR has been shown to occur in the ER (Bellovino et al., 1996), the stoichiometry of this assembly has not been studied systematically. Estimates of 1:4 M ratio of RBP to TTR have been made based on their relative concentrations in the serum (Newcomer and Ong, 2000). Our cross-linking results for the first time provide evidence for RBP–TTR complexes, suggesting both 1:1 as well as 1:4 stoichiometries. It is possible that the former is an intermediate in the assembly leading to a mature 1:4 complex. Analysis of TTR assembly with RBP disulfide mutants indicated that dissociation from the chaperone complex was essential for RBP to assemble with TTR. RBP mutants that dissociated from the folding complex formed RBP-TTR cross-links qualitatively similar to those for wild-type RBP. The double mutant lacking both 4–160 and 70–174 disulfides, showing persistent association with the folding complex, failed to assemble with TTR.
In all, our results suggest a two-step maturation of RBP in the ER wherein newly synthesized RBP undergoes disulfide oxidation bound to a complex of chaperones in the ER and assembles with TTR only upon completion of its early folding and dissociation from the chaperone complex (Figure 9). Dissociation from the quality control is an early event in the maturation of RBP, not related to its ligand binding and the trigger for its ligand dependent secretion may operate in the postfolding phase independent of the mechanisms that support its folding. It is possible that ligand binding and/or TTR association may provide a positive signal for the egress of RBP out of the ER. This possibility is consistent with the emerging concept of selective export dependent on positive signals in the form of sequence motifs or glycosylation state of proteins in the ER. Finally, preservation of a matrix like network of chaperones in isolated microsomes, described here, opens new possibilities to analyze mechanisms used by the ER to negotiate complex decisions related to folding, retention and transport of secretory proteins.
ACKNOWLEDGMENTS
We acknowledge kind gift of antibodies to Grp94 from Prof. Christopher Nicchita (Duke University, NC) and BiP from Prof. R. Zimmerman (University of Saarlandes, Germany). This work was supported by a grant from the Department of Atomic Energy, Department of Biotechnology, Council of Science and Industrial Research, India.
Abbreviations used:
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- GSSG
oxidized glutathione
- NEM
N-ethylmaleimide
- PMSF
phenylmethylsulfonyl fluoride
- RBP
retinol-binding protein
- TTR
Transthyretin.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E08-01-0026) on September 24, 2008.
REFERENCES
- Bellovino D., Morimoto T., Tosetti F., Gaetani S. Retinol binding protein and transthyretin are secreted as a complex formed in the endoplasmic reticulum in HepG2 human hepatocarcinoma cells. Exp. Cell. Res. 1996;222:77–83. doi: 10.1006/excr.1996.0010. [DOI] [PubMed] [Google Scholar]
- Blond-Elguindi S., Cwirla S. E., Dower W. J., Lipshutz R. J., Sprang S. R., Sambrook J. F., Gething M. J. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717–728. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- Cowan S. W., Newcomer M. E., Jones T. A. Crystallographic refinement of human serum retinol binding protein at 2A resolution. Proteins. 1990;8:44–61. doi: 10.1002/prot.340080108. [DOI] [PubMed] [Google Scholar]
- Flynn G. C., Pohl J., Flocco M. T., Rothman J. E. Peptide-binding specificity of the molecular chaperone BiP. Nature. 1991;353:726–730. doi: 10.1038/353726a0. [DOI] [PubMed] [Google Scholar]
- Fries E., Gustaffson L., Peterson P. A. Four secretory proteins synthesized by hepatocytes are transported from endoplasmic reticulum to Golgi complex at different rates. EMBO J. 1984;3:147–152. doi: 10.1002/j.1460-2075.1984.tb01775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C., Braakman I., Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. USA. 1994;91:913–917. doi: 10.1073/pnas.91.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C., Helenius A. Quality control in the secretory pathway. Curr. Opin. Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Hebert D. N., Foellmer B., Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81:425–433. doi: 10.1016/0092-8674(95)90395-x. [DOI] [PubMed] [Google Scholar]
- Helenius A., Marquardt T., Braakman I. The endoplasmic reticulum as a protein-folding compartment. Trends Cell Biol. 1992;2:227–231. doi: 10.1016/0962-8924(92)90309-b. [DOI] [PubMed] [Google Scholar]
- Kaji E. H., Lodish H. F. Unfolding of newly made retinol-binding protein by dithiothreitol. Sensitivity to retinoids. J. Biol. Chem. 1993;268:22188–22194. [PubMed] [Google Scholar]
- Kuznetsov G., Chen L. B., Nigam S. K. Several endoplasmic reticulum stress proteins, including ERp72, interact with thyroglobulin during its maturation. J. Biol. Chem. 1994;269:22990–22995. [PubMed] [Google Scholar]
- Lin H. Y., Masso-Welch P., Di Y. P., Cai J. W., Shen J. W., Subjeck J. R. The 170-kDa glucose regulated stress protein is an endoplasmic reticulum protein that binds immunoglobulin. Mol. Biol. Cell. 1993;4:1109–1119. doi: 10.1091/mbc.4.11.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist J. A., Jensen O. N., Mann M., Hammerling G. J. ER-60, a chaperone with thiol-dependent reductase activity involved in MHC class I assembly. EMBO J. 1998;17:2186–2195. doi: 10.1093/emboj/17.8.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodish H. F., Kong N., Snider M., Strous G. J. Hepatoma secretory proteins migrate from rough endoplasmic reticulum to Golgi at characteristic rates. Nature. 1983;304:80–83. doi: 10.1038/304080a0. [DOI] [PubMed] [Google Scholar]
- Melhus H., Laurent B., Rask L., Peterson P. A. Ligand-dependent secretion of rat retinol-binding protein expressed in HeLa cells. J. Biol. Chem. 1992;267:12036–12041. [PubMed] [Google Scholar]
- Melhus H., Nilsson T., Peterson P. A., Rask L. Retinol-binding protein and transthyretin expressed in HeLa cells form a complex in the endoplasmic reticulum in both the absence and the presence of retinol. Exp. Cell. Res. 1991;197:119–124. doi: 10.1016/0014-4827(91)90488-g. [DOI] [PubMed] [Google Scholar]
- Melnick J., Aviel S., Argon Y. The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J. Biol. Chem. 1992;267:21303–21306. [PubMed] [Google Scholar]
- Meunier L., Usherwood Y. K., Chung K. T., Hendershot L. M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell. 2002;13:4456–4469. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto Y., Smith J. E., Milch P. O., Goodman D. S. Regulation of retinol-binding protein metabolism by vitamin A status in the rat. J. Biol. Chem. 1972;247:2542–2550. [PubMed] [Google Scholar]
- Nehls S., Snapp E. L., Cole N. B., Zaal K. J., Kenworthy A. K., Roberts T. H., Ellenberg J., Presley J. F., Siggia E., Lippincott-Schwartz J. Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol. 2000;2:288–295. doi: 10.1038/35010558. [DOI] [PubMed] [Google Scholar]
- Newcomer M. E., Ong D. E. Plasma retinol binding protein: structure and function of the prototypic lipocalin. Biochim. Biophys. Acta. 2000;1482:57–64. doi: 10.1016/s0167-4838(00)00150-3. [DOI] [PubMed] [Google Scholar]
- Newcomer M. E., Jones T. A., Aqvist J., Sundelin J., Eriksson U., Rask L., Peterson P. A. The three-dimensional structure of retinol-binding protein. EMBO J. 1984;3:1451–1454. doi: 10.1002/j.1460-2075.1984.tb01995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojakian G. K, Kreibich G., Sabatini D. D. Mobility of ribosomes bound to microsomal membranes. A freeze-etch and thin-section electron microscope study of the structure and fluidity of the rough endoplasmic reticulum. J. Cell Sci. 1977;72:530–551. doi: 10.1083/jcb.72.3.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortmann B., Androlewicz M. J., Cresswell P. MHC class I/β2-microglobulin complexes associate with TAP transporters before peptide binding of outstanding interest. Nature. 1994;368:864–867. doi: 10.1038/368864a0. [DOI] [PubMed] [Google Scholar]
- Ou W. J., Cameron P. H., Thomas D. Y., Bergeron J. J. Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature. 1993;364:771–776. doi: 10.1038/364771a0. [DOI] [PubMed] [Google Scholar]
- Palade G. E., Siekevitz P. Liver microsomes; an integrated morphological and biochemical study. J. Biophys. Biochem. Cytol. 1956;2:171–200. doi: 10.1083/jcb.2.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P., Sparvoli A., Fagioli C., Fassina G., Sitia R. Formation of reversible disulfide bonds with the protein matrix of the endoplasmic reticulum correlates with the retention of unassembled Ig light chains. EMBO J. 1996;15:2077–2085. [PMC free article] [PubMed] [Google Scholar]
- Ronne H., Ocklind C., Wiman K., Rask L., Obrink B., Peterson P. A. Ligand-dependent regulation of intracellular protein transport: effect of vitamin A on the secretion of the retinol-binding protein. J. Cell Biol. 1983;96:907–910. doi: 10.1083/jcb.96.3.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth R. A., Pierce S. B. In vivo cross-linking of protein disulfide isomerase to immunoglobulins. Biochemistry. 1987;26:4179–4182. doi: 10.1021/bi00388a001. [DOI] [PubMed] [Google Scholar]
- Sekijima Y., Wiseman R. L., Matteson J., Hammarstorm P., Miller S. R., Sawkar A. R., Balch W. E., Kelly J. W. The biological and chemical basis for Tissue Selective Amyloid Disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Smith J. E., Borek C., Goodman D. S. Regulation of retinol-binding protein metabolism in cultured rat liver cell lines. Cell. 1978;15:865–873. doi: 10.1016/0092-8674(78)90271-4. [DOI] [PubMed] [Google Scholar]
- Snapp E. L., Sharma A., Lippincott-Schwartz J., Hegde R. S. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. USA. 2006;103:6536–6541. doi: 10.1073/pnas.0510657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa M. C., Ferrero-Garcia M. A., Parodi A. J. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry. 1992;31:97–105. doi: 10.1021/bi00116a015. [DOI] [PubMed] [Google Scholar]
- Suh W.-K, Cohen-Doyle M. F., Fruh K., Wang K, Peterson P., Williams D. B. Interaction of MHC class I molecules with the transporter associated with antigen processing of outstanding interest. Science. 1994;264:1322–1326. doi: 10.1126/science.8191286. [DOI] [PubMed] [Google Scholar]
- Tatu U., Helenius A. Interactions between newly synthesized glycoproteins, calnexin, and a network of resident chaperones in the endoplasmic reticulum. J. Cell Biol. 1997;136:555–565. doi: 10.1083/jcb.136.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Blobel G. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol. 1983;96:84–93. doi: 10.1016/s0076-6879(83)96010-x. [DOI] [PubMed] [Google Scholar]
- Yang Q., Graham T. E., Mody N., Preitner F., Peroni O. D., Zabolotny J. M., Kotani K., Quadro L., Kahn B. B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]