

Abstract
A plug-based microfluidic approach was used to perform multiple agglutination assays in parallel without cross-contamination and using only microliter volumes of blood. To perform agglutination assays on-chip, a microfluidic device was designed to combine aqueous streams of antibody, buffer, and red blood cells (RBCs) to form droplets 30–40 nL in volume surrounded by a fluorinated carrier fluid. Using this approach, proof-of-concept ABO and D (Rh) blood typing and group A subtyping were successfully performed by screening against multiple antigens without cross-contamination. On-chip subtyping distinguished common A1 and A2 RBCs by using a lectinbased dilution assay. This flexible platform was extended to differentiate rare, weakly agglutinating RBCs of A subtypes by analyzing agglutination avidity as a function of shear rate. Quantitative analysis of changes in contrast within plugs revealed subtleties in agglutination kinetics and enabled characterization of agglutination of rare blood subtypes. Finally, this platform was used to detect bacteria, demonstrating the potential usefulness of this assay in detecting sepsis and the potential for applications in agglutination-based viral detection. The speed, control, and minimal sample consumption provided by this technology present an advance for point of care applications, blood typing of newborns, and general blood assays in small model organisms.
This paper reports a plug-based microfluidic approach to perform agglutination assays for ABO and D (Rh factor) blood typing and group A subtyping without cross-contamination. This technology also minimizes sample consumption, an important advance for point of care applications, blood typing of newborns, and general blood assays utilizing mice and other model organisms. When antigens on the surface of red blood cells (RBCs) are exposed to antibodies for that antigen, agglutination, or clumping, of RBCs occurs. These clumps are also referred to as agglutinins. Agglutination is utilized to determine a patient’s blood type by indicating the presence of specific antigens on the patient’s RBCs. Solid-phase agglutination gel tests are used for greater than 90% of all ABO and D blood typing in emergency rooms and blood banks.1 The gel tests require microliters of sample, minutes to perform, and give an automated optical readout of agglutination.2 In addition, disposable plastic cards for point of care applications, microtiter plates, laminar microfluidics, and now molecular approaches to analyze expression of blood group antigens are also used in simple ABO and D blood typing.3-6 However, both gel-based and card agglutination assays are performed separately for each blood sample, increasing both labor and sample consumption.
In addition to the common ABO and D blood groups, type A blood is divided into subtypes that have fewer A antigens per RBC. Rare subtypes A3, Ax, Am, and Ael often shown no visible agglutination reaction with the anti-A.1 As a result, A subtypes have a greater potential for being mistyped as O blood and causing a hemolytic transfusion reaction in a recipient upon transfusion.7-10 Currently, subtyping is performed in a separate assay from initial ABO and D typing by using a “test tube” method that requires milliliters of blood and 45 min to complete. A gel-based method of subtyping by using lectins is not yet commercially available.
Here, we describe a plug-based microfluidic method for blood typing that can resolve ABO and D (referred to as + or -) groups as well as subtle agglutination differences between A blood group subtypes. This system utilizes segmented flow microfluidic technology to perform individual agglutination assays in “plugs”, aqueous droplets surrounded by a fluorinated carrier fluid and transported through microfluidic channels. Microfluidics has been used previously to perform kinetic measurements and a number of assays, including determination of glucose level in blood and activated partial thromboplastin time.11-13 Previously, we have developed methods to eliminate cross-contamination between plugs by inserting gas or liquid spacers and perform highthroughput reaction screening by using preformed arrays of reagents.14-21 These advances enable this system to perform serial typing of multiple blood samples in the same experiment. We validate this plug-based microfluidic system by performing agglutination assays for ABO and D typing as proof of principle, A subtyping by using a lectin assay, binding avidity assays to distinguish strong from weak A subtypes, and detecting bacteria. This flexible microfluidic platform could be easily modified to perform continuous blood typing in blood banks or point of care assays by using preformed antibody arrays.
EXPERIMENTAL SECION
Reagents and Materials
Reagents were used as received unless otherwise specified. Type A+ human RBCs stabilized with citrate were purchased from Innovative Research (Novi, MI), and type A2B+ human RBCs stabilized with citrate-phosphate-dextroseadenine were purchased from Research Blood Components (Brighton, MA). Weakly agglutinating type A human RBCs were donated by the University of Chicago Blood Bank. Antibody IgM serum for anti-A, anti-B, and monoclonal-polyclonal blend anti-D (Rh factor) were obtained from Orthoclinical Diagnostics (Raritan, NJ). The buffer 2-amino-2-(hydroxymethyl)propane-1,3-diol (TRIS) and lyophilized lectin from Dolichous biflorus were purchased from Sigma (St. Louis, MO). Sodium chloride was purchased from Fisher (Hampton, NH). Fluoroinert 3283 was purchased from 3 M (Diegrem, Belgium), and surfactant 1H,1H,2H,2H-perfluoro-1-octanol (PFO) was purchased from Aldrich (Milwaukee, WI). Teflon tubing with an inner diameter (i.d.) of 300 μm used to connect syringes to microfluidics device was purchased from Weico (Edgewood, NY). Telfon tubing (i.d. = 150 μm) used for formation of antibody arrays was purchased from Zeus (Orange-burg, SC). All syringes and 26-gauge needles were from Hamilton (Reno, NV). The Staphylococcus aureus bacteria strain was obtained from American Type Culture Collection 25923 (Manassas, VA). The Staphslide latex test was purchased from Arlington Scientific (Springville, UT). The Mueller Hinton Agar with 5% sheep’s blood was purchased from Fisher (Hampton, NH).
The TRIS buffer was prepared as a 300 OsM solution: 20 mM TRIS and 130 mM sodium chloride and pH adjusted to 7.4 at 25 °C with hydrochloric acid. RBCs were diluted 1:3 v/v with TRIS buffer. In all experiments, the fluorinated carrier fluid consisted of 1:10 v/v of PFO to FC3283.
Fabrication of Microfluidic Devices
All microfluidic devices were fabricated as described previously by a procedure known as rapid prototyping in poly(dimethylsiloxane) (PDMS).22-24 The design for devices used in the assays shown in Figures 1, 3, 4, and 513 and the design for the device for creating preformed arrays in Figure 216 have been previously reported. Microchannels were made hydrophobic by using a previously described silanization procedure.25
Figure 1.
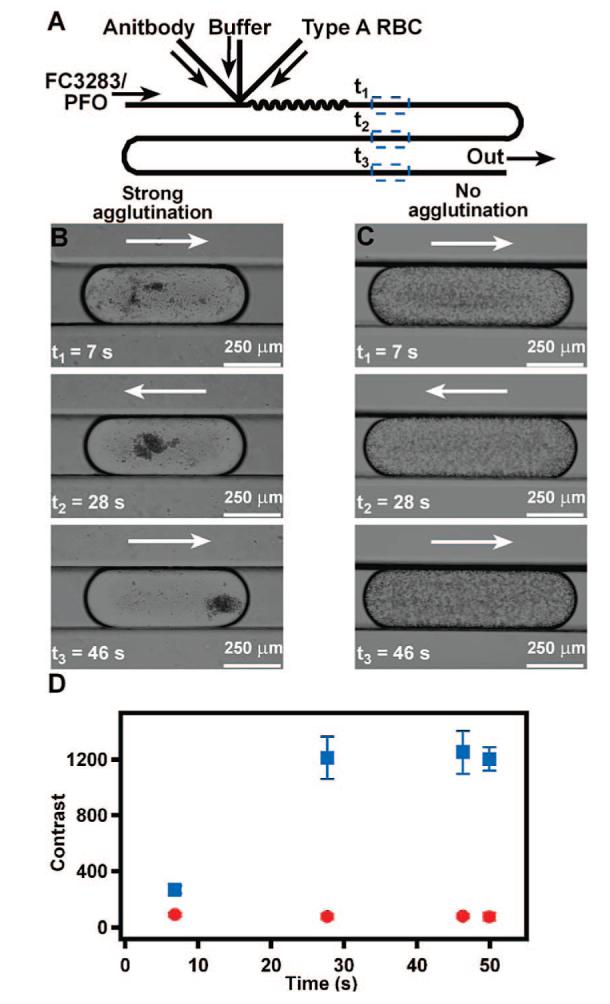
Plug-based microfluidic agglutination assay to distinguish type A from type B RBCs. (A) A schematic drawing shows the device design used for the agglutination assay. (B, C) Time-lapse bright-field microphotographs of representative plugs containing antibody, TRIS buffer, and type A RBCs show that type A RBCs agglutinate in the presence of anti-A (B) but not in the presence of anti-B (C). (D) A graph quantifies the change in contrast of plugs, indicative of agglutination, of type A RBCs exposed to anti-A (blue squares) or anti-B (red circles) (n = 3 plugs). Contrast analysis was performed by using ImageJ. Arrows indicate the direction of fluid flow.
Figure 3.
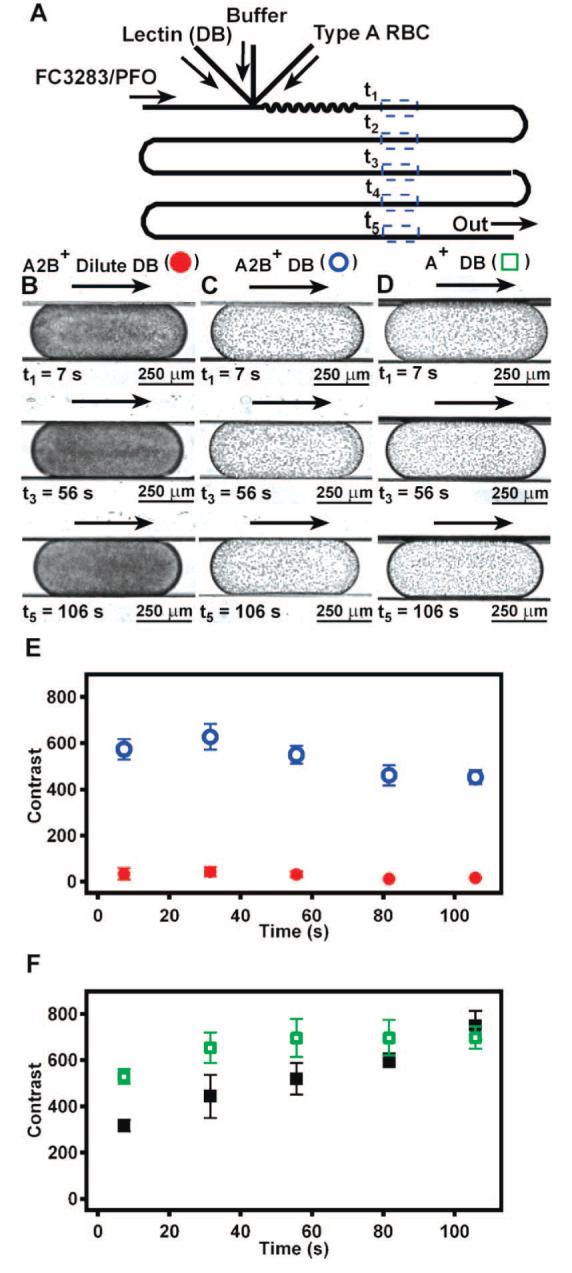
Microfluidic subtyping of type A RBCs by on-chip dilution of lectin from D. biflorus (DB). (A) A schematic drawing shows the microfluidic device used for subtyping. Flow rate of lectin DB and buffer were varied to achieve low and high lectin DB concentrations, while the flow rate of RBCs and the carrier fluid remained constant. (B) Low lectin DB concentration (0.09 mg/mL) did not agglutinate subtype A2B+ RBCs, because plugs were dark and uniform in color, indicating no agglutination. (C) High lectin DB concentration (0.72 mg/mL) resulted in agglutination of subtype A2B+ RBCs, as expected for RBCs of subtype A2. Images in panels B and C correspond to red (solid circles) and blue (hollow circles) data points in panel E, respectively. (D) Type A+ RBCs agglutinate at both low and high lectin DB concentrations (images at low concentration of lectin DB not shown), indicating that these RBCs were subtype A1. Images in panel D correspond to green (hollow) data points in panel F. (E) Contrast variation in plugs containing subtype A2B+ RBCs at low (red solid circles) and high (blue hollow circles) lectin DB concentration quantitatively show the presence of agglutinins over time. (F) Contrast variation in plugs containing type A+ RBCs of unknown subtype at low (black solid squares) and high (green hollow squares) lectin DB concentration quantitatively show the presence of agglutinins over time. In panels E and F, n = 3 plugs. Arrows indicate the direction of flow.
Figure 4.

Binding avidity of type A RBCs distinguished by using shear in plug-based microfluidics. (A) A schematic drawing shows the device used to characterize the binding avidity of type A RBCs by using shear, and the enlarged inset shows the winding, bumpy channels included to promote mixing. (B, C) Type A RBCs with strong binding avidity agglutinate at a shear rate of 13 s-1 (B), while type A RBCs with weak binding avidity do not agglutinate (C). (D, E) When the shear rate was reduced from 13 to 0 s-1, RBCs with strong binding avidity remained agglutinated (D), and those with weak binding avidity agglutinated (F). (E, G) When the shear rate was increased to 530 s-1, RBCs with strong binding avidity remained agglutinated (E), while agglutinins from RBCs with weak binding avidity broke up (G). (H, I) Contrast analysis of plugs at shear rates of 0 (blue squares), 13, (red circles), and 530 s-1 (black triangles) indicates that agglutination of type A RBCs with strong binding avidity is independent of shear rate (H), while agglutination of type A RBCs with weak binding avidity is affected by shear rate (I). A decrease in contrast within plugs correlates with the breakup of agglutinins (n = 2 plugs in all data points). Symbols in panels B—G correspond to symbols in panels H and I. Note the difference in scale of the Y-axis in graphs H and I.
Figure 5.

Plug-based microfluidic agglutination assays for the detection of S. aureus. (A) A schematic drawing shows the device used in the agglutination assay to detect bacteria. (B) Time-lapse microphotographs show that agglutination in the presence of bacteria begins in as little as 20 s. (C) Time-lapse microphotographs of a control plug with no bacteria shows that latex beads alone do not cause agglutination. (D) Contrast analysis of plugs with (red squares) and without (blue circles) bacteria quantitatively shows agglutination over time (n = 3 plugs). Arrows indicate the direction of flow.
Figure 2.
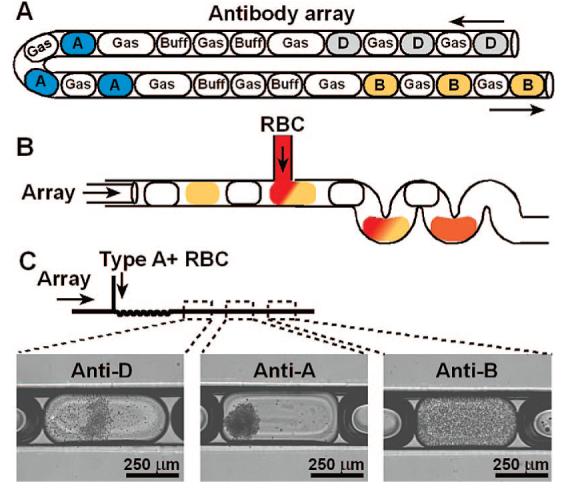
ABO and D blood typing in a single experiment by using a preformed antibody array coupled to a microfluidic device. (A) A schematic drawing illustrates the antibody array that was used for ABO and D blood typing (gas = air, buff = TRIS buffer; D = anti-D; A = anti-A; B = anti-B). (B) A schematic drawing illustrates merging of plugs in the antibody array with a solution of RBCs. (C) A schematic drawing and bright-field microphotographs show that merging a 3% solution of type A+ RBCs with the antibody array resulted in agglutination in plugs of anti-D or anti-A merged with RBCs, but not in plugs of anti-B merged with RBCs. Arrows indicate the direction of fluid flow.
Imaging and Quantification of Agglutination Results
We evaluated the presence of agglutination by monitoring individual plugs for an increase in contrast over time. To monitor the agglutination reaction within each plug, plugs were manually imaged with Metamorph imaging software on a Leica DMI 6000 epifluorescence microscope (Wetzler, Germany) at approximately 2, 6, and 12 cm from the junction of the microfluidic device (x = 0 cm), where plugs containing reagents are formed and the reaction is initiated. The reaction time, t, was obtained from total volumetric flow rate by using t = v/d, where v is linear velocity of fluid flow from volumetric flow rate and channel cross-sectional area and d is distance from the merging junction as described previously.20 Images of different plugs were manually captured with Metamorph, and these images were manually analyzed by mean and variance filters in ImageJ to quantify the presence and extent of agglutinins by contrast. Briefly, images were assembled into a stack that contained all of the images taken at each time point during a single assay. In all figures, N = 3 plugs, except Figure 4, where N = 2 plugs. The value of N = 3 plugs was chosen to maintain consistency with Figure 2, where technical limitations dictated the value of N. In Figure 4, the microscope viewing area is limited to N = 2 plugs. A threshold for minimal agglutinin pixel size was measured with a line scan to determine the difference in resolution between agglutinins and nonagglutinated cells. Contrast values below the threshold represent nonagglutinated RBCs and are not counted by ImageJ. Individual plugs in the stack were selected and analyzed with a variance filter to measure the contrast in brightness in each plug. This contrast analysis method examined changes in intensity between pixels in a given plug over time, eliminating the need to consider changes in intensity from plug to plug or from day to day. An increase in contrast within a plug over time indicated the presence of agglutination, while a plug that remained at a constant contrast was not agglutinated.
Experiments To Distinguish Blood Type A from Type B
The carrier fluid flow rate was 5.5 μL/min in the device. Streams of the three aqueous solutions were merged to form plugs. The first aqueous stream consisted of undiluted antibody serum at a flow rate of 0.75 μL/min in the device. In separate experiments, this antibody serum was either anti-A or anti-B. The flow rate of the antibody serum was kept low to minimize reaction between the antibody and RBCs before plug formation. The second aqueous stream consisted of a solution of TRIS buffer at a flow rate of 4 μL/min in the device. This aqueous stream was used to minimize the reaction between antibody and RBCs before plug formation. The third aqueous stream consisted of RBCs diluted from 100% as received to 33% v/v with TRIS buffer at a flow rate of 0.25 μL/min in the device. RBCs were further diluted to ∼2% v/v on-chip with other aqueous components. The total flow rate in the device was 10.5 μL/min (aqueous and carrier fluid). Plugs of 30–40 nL in volume with anti-A or anti-B, respectively, were imaged by using Metamorph at four locations in the device, with the first three locations denoted as t1, t2, and t3 in Figure 1A for type A RBCs. Because the antibody stream wets the walls of the inlet channel in this device, the agglutination assays using anti-A and anti-B were performed by using two different devices to avoid mixing of antibodies.
Experiments To Perform ABO and D Blood Typing
Plugs were aspirated manually into Teflon tubing (i.d. = 150 μm) in the following sequence: three plugs of anti-A, two plugs of TRIS buffer, three plugs of anti-B, two plugs of TRIS buffer, three plugs of anti-D, and two plugs of TRIS buffer. Air spacers were included between aqueous plugs as illustrated in Figure 2A. The approximate volume of each plug in the array was 15–20 nL. The array was then connected to the device illustrated in Figure 2B with capillary wax. RBCs at a flow rate of 0.5 μL/min through a hydrophilic capillary were injected into aqueous droplets also at a flow rate of 0.5 μL/min, diluting type A+ RBCs on-chip to a final concentration of 1.5% v/v.
Experiments To Perform Subtyping of Type A Blood
Using the device illustrated in Figure 3A, aqueous streams of lectin, TRIS buffer, and RBCs were merged to form plugs, and the resulting ∼40-nL plugs were monitored at five different time points, t1—t5 illustrated in Figure 4A. We performed this assay with both A2B+ RBCs and A+ RBCs at both high and low concentrations of lectin. In all trials, the flow rate of RBCs was kept constant at 0.5 μL/min, and the final on-chip concentration of RBCs was 6% v/v. For assays at low lectin concentration, flow rates of 0.25 μL/min for lectin and 2 μL/min for buffer were used to dilute lectin from 1 to 0.09 mg/mL. Lower overall flow rates resulted in longer assay times of ∼100 s. For assays at high lectin concentration, flow rates were 2 μL/min for lectin and 0.25 μL/min for buffer to dilute lectin from 1 to 0.72 mg/mL. We also performed a control experiment with two different type B RBCs (gift from University of Chicago Blood Bank). Conditions for the control experiment were the same as those experiments using type A RBCs.
Experiments To Resolve Weak Subtypes of Type A Blood by Using Shear
For these experiments, the device design illustrated in Figure 3A was modified to include bumps on the walls of the winding channel to facilitate mixing and more uniform shear rate within plugs.26 Plugs were formed by merging aqueous streams of anti-A, TRIS buffer, and type A RBCs. Flow rates were 0.375 μL/min for anti-A, 1.0 or 1.5 μL/min for TRIS buffer, 0.25 μL/min for type A RBCs, and 4 μL/min for carrier fluid. When using strongly agglutinating type A RBCs, the flow rate of the buffer was adjusted to 1.0 or 1.5 μL/min to minimize agglutination at the merging junction. Shear rate was determined by dividing the total volumetric flow rate by the product of the cross-sectional area and the diameter of the microfluidic channel. Cross sections were measured with a Leica MZ 125 stereoscope at 6.3× magnification. Shear rates given here are an approximation of the average shear experienced by a plug. After plugs filled ∼32 mm of the device, the flow of all streams was stopped to reduce the shear rate to 0 s-1, and plugs were observed for agglutination. Next, the flow rate of the carrier fluid was increased to 250 μL/min to test the influence of shear rate on agglutinated RBCs, and images were captured at 5× magnification. To distinguish agglutination of type O RBCs from that of weakly agglutinating type A RBCs, a control experiment using type O+ RBCs (Innovative Research) was performed by using the same procedure as that described above with the following exceptions. For type O+ RBCs, fluid flow rates for all reagents, including carrier fluid, were resumed at initial conditions after the ∼10-min observation period to show that agglutinins possess no binding avidity. This control experiment for type O RBCs was compared to a control experiment with the weakly agglutinating type A RBCs where flow rate of the carrier fluid was resumed after ∼10 min at a flow rate of 25 μL/min.
Experiments To Detect Bacteria
In these experiments, the same device design was used as that illustrated in Figure 1A, shown again in Figure 5A. Plugs were formed by merging aqueous streams of antibody-coated latex beads at a flow rate of 0.25 μL/min, TRIS buffer at a flow rate of 0.25 μL/min, and bacteria at a flow rate of 0.5 μL/min. The bacteria used in these experiments was S. aureus, a common pathogenic bacteria, which was suspended in TRIS buffer at a concentration of ∼ 1 × 108 colony forming units (cfu)/mL. The concentration of bacteria used in these proof-of-concept experiments may not be applicable in a clinical setting. Bacteria were grown overnight on 5% sheep’s blood agar plates. Plug size was ∼45 nL for bacteria assays. Plugs were monitored for agglutination at two time points, t1 and t2, as illustrated in Figure 5A.
RESULTS AND DISCUSSION
To perform agglutination assays on-chip, we designed a microfluidic device that combines aqueous streams of antibody, buffer, and RBCs to form plugs of ∼40 nL in volume surrounded by a fluorinated carrier fluid. All devices were fabricated from PDMS as previously described.22,23 The ratio of aqueous solutions in the plugs was controlled by adjusting the flow rates of the three aqueous streams. By controlling the concentration of each reagent in a plug, reagents can be diluted on-chip, and multiple reaction conditions can be screened in the same experiment.14,19 To achieve assay times comparable to gel-based agglutination assays, a winding region was included immediately downstream of the merging junction to promote rapid mixing of reagents by chaotic advection.19,20 Because the effectiveness of chaotic mixing is only weakly dependent on the diffusivity of the species being mixed, this method will be applicable to a wide range of species.19,20 The rate of mixing was controlled by flow rate of the fluorinated carrier fluid.27 After mixing, plugs of antibody and RBCs were monitored for agglutination at points downstream of the winding region.
A simplified agglutination assay demonstrated that this plug-based microfluidic system can distinguish type A RBCs from type B RBCs. Solutions were delivered to the device as illustrated in Figure 1A with pressure-driven flow, and plugs were imaged at approximately 2, 6, 10, and 12 cm from the merging junction. The first three time points, t1-t3, are shown in Figure 1B and 1C. Assays utilizing anti-A and anti-B were performed in two separate experiments with two different devices to avoid mixing of antibodies. When type A RBCs were exposed to anti-A (Figure 1B; Figure 1D, blue squares), agglutination occurred, indicated by an increase in contrast in plugs over time. Visually, agglutination of type A blood appeared complete after 46 s (Figure 1B), and quantitative image analysis confirmed that agglutination was complete in 28 s (Figure 1D). On the other hand, when type A RBCs were exposed to anti-B, no agglutination was observed, and the contrast within plugs did not change (Figure 1C; Figure 1D, red circles). Differences in the size of plugs between Figure 1B (30 nL) and C (40 nL) did not affect analysis of agglutination by the imaging software.
To enable screening of multiple antigens against a single blood sample without cross-contamination, we used three-phase liquid—liquid—gas flow. Preformed arrays of antibody plugs (Figure 2A) were generated by alternately aspirating plugs of antibody, air, and buffer by using previously described methods.14-17 In the preformed arrays, buffer and air plugs were placed between aqueous plugs16 to avoid cross-contamination of RBCs, agglutinins, or antibodies between plugs. To generate a preformed array of antibodies on-chip, reagent plugs could be separated by a third immiscible liquid as previously described.21 Preformed antibody arrays can be generated in advance of the agglutination assay, providing reduced assay time and the potential for point of care testing. The antibody plugs in a preformed array were merged with RBCs (Figure 2B), and the resulting plugs were flowed through a winding region to facilitate mixing. Plugs were monitored as described above. We confirmed consistent loading of RBCs into plugs by comparing the size of plugs before and after merging with RBCs. When type A+ RBCs were merged with plugs of the preformed antibody array, agglutination only occurred in plugs containing anti-A or anti-D, but not in plugs containing anti-B (Figure 2C). Therefore, this assay correctly typed the sample RBCs as type A+, providing proof of principle for this plug-based microfluidic method of blood typing. Of the plugs tested during this assay (n = 9), no false-positive results were observed, indicating that no cross-contamination between antibody plugs occurred. The previously described hybrid method17 could be used to quantitatively screen for cross-contamination. Here, the flow rates were ∼5 times slower than those used in the experiment described in Figure 1, and the total analysis time was less than 5 min.
To demonstrate the applicability of this technology to resolve common blood subtypes, we performed on-chip subtyping of type A RBCs by using a well-established dilution assay with lectin from D. biflorus (lectin DB).1 Subtype A1 RBCs have been shown to agglutinate in the presence of lectin DB indifferent of lectin DB concentration, while subtype A2 RBCs are known to agglutinate only in the presence of a high concentration of lectin DB.1 This selective agglutination is believed to be due to the location of sugar groups on subtypes A1 and A2 RBCs that bind lectin DB. Some B blood types have been reported to agglutinate in response to lectin DB.28 As a control, we also tested two samples of type B RBCs and found that they did not agglutinate in the presence of lectin DB. However, more extensive control testing would be necessary before application in a clinical setting. In this microfluidic system that enables multiple typing reactions simultaneously, this possibility of mistyping can be reduced by specifically testing for type B RBCs before subtyping (Figure 2).
First, we performed a control assay to demonstrate that subtype A2 RBCs agglutinated in the presence of lectin DB in the expected concentration-dependent manor. Here, we used type A2B+ RBCs, and the lectin DB is sensitive to the A2 antigens. Plugs were formed as illustrated in Figure 3A and monitored at five different time points, t1—t5. The concentration of lectin DB was varied by changing the relative flow rates of buffer and lectin streams. As expected, subtype A2B+ RBCs agglutinated in the presence of a high concentration of lectin DB (0.72 mg/mL) (Figure 3C; Figure 3E, blue hollow circles) but not in the presence of a low concentration of lectin DB (0.09 mg/mL) (Figure 3B, Figure 3E, red solid circles). RBC density in Figure 3B appears higher than in Figure 1B and C, because final on-chip concentration of RBCs was two times greater in this experiment. After ∼7 s, agglutination of subtype A2B+ appears to be complete (Figure 3E, blue hollow circles).
Next, we performed the same agglutination assay described above using type A+ RBCs of an unknown subtype. Agglutination of unknown samples of type A+ RBCs did not display dependence on the concentration of lectin DB (Figure 3D; Figure 3F) when compared to subtype A2B+ RBCs (Figure 3E). Agglutination of type A+ RBCs in the presence of lectin DB at both high and low concentrations (Figure 3D; Figure 3F) indicates that these RBCs are subtype A+1. Independent subtyping by using lectin DB-based tube assays by the American Red Cross (Detroit, MI) and the Blood Bank at the University of Chicago Hospital confirmed that these RBCs were subtype A+1.
In addition, these results indicate that subtypes A1 and A2 can be distinguished in as little as 100 s by using this plug-based microfluidic assay, an improvement over the 45 min required for standard tube assays. The kinetics of agglutination for subtype A+1 RBCs were different between low and high concentrations of lectin DB, exhibiting a slight increase in contrast within each plug throughout the duration of the experiment for study at low concentration (Figure 3F, black solid squares). Quantitative analysis by contrast suggests that subtleties in agglutination can be resolved by using in plug-based subtyping. Therefore, this method may be extended to resolve rare weakly agglutinating subtypes of the A or other blood groups in binding avidity studies.
This plug-based microfluidic platform also provides an effective means of comparing relative binding avidity by exposing RBCs to controlled shear rates, a method that can be utilized to resolve less common, weakly agglutinating subtypes of group A RBCs. Binding avidity is correlated with the agglutination response in RBCs. The lectin DB-based dilution assay can easily distinguish common subtypes A1 and A2 with strong binding avidity. However, subtypes with weak binding avidity, including Ax,Ael, and Am, display weak or no agglutination when exposed to lectin DB and therefore cannot be distinguished with the lectin DB dilution assay.
To investigate the influence of shear rate on the agglutination of RBCs with strong and weak binding avidity, type A RBCs (Figure 4B) and weakly agglutinating type A RBCs of an unknown subtype (Figure 4C) were exposed to anti-A and various flow rates of carrier fluid in a microfluidic device to expose RBCs to various shear rates. The shear rate was controlled by the flow rate of the carrier fluid and the geometry of the device. Shear rate was estimated by dividing the linear, overall flow rate by the diameter of the channel to give an approximation of the average shear rate in a plug.29,30 Plugs were formed by merging anti-A, buffer, and RBCs in the device illustrated in Figure 4A. Here, a winding, bumpy channel was included in the device design to facilitate better mixing and more uniform shear rate.26 While shear is not uniform inside a moving plug, we believe RBCs in a plug can be assumed to experience average shear when chaotic mixing is sufficiently rapid on the time scale of binding and unbinding to rapidly transport the cells through the regions of higher and lower shear.
At the time of plug formation, the shear rate was 13 s-1. At this shear rate, type A RBCs with strong binding avidity agglutinated within 8.9 s (Figure 4B, t1), while type A RBCs with weak binding avidity did not agglutinate after 130 s (Figure 4C, t5). A lack of agglutination at this shear rate would also likely be observed for type O RBCs. Next, the shear rate was reduced to 0 s-1 by stopping all flow rates, and type A RBCs with strong binding avidity remained agglutinated as expected (Figure 4D), while type A RBCs with weak binding avidity formed agglutinins (Figure 4F). These results indicate that a low shear rate of 13 s-1 hinders the agglutination of RBCs with weak binding avidity. In addition, type A RBCs with weak binding avidity require a considerably longer time, ∼10 min, to agglutinate in the absence of shear than type A RBCs with strong binding avidity.
Then, the shear rate was increased from 0 to 530 s-1, and agglutinins of type A RBCs with weak binding avidity broke up (Figure 4G). However, agglutinins of type A RBCs with strong binding avidity remained agglutinated (Figure 4E). In ∼10% of the plugs of type A RBCs with strong binding avidity (representative data in Figure 4E), small fragments of the agglutinin broke away within 10 s at a shear rate of 530 s-1 (data not shown). Our results are consistent with viscometer reports for reversing agglutination, where a shear rate of 600 s-1 breaks up agglutinins.31 Contrast analysis shows that shear has little influence on agglutination of type A RBCs with strong binding avidity (Figure 4H), while shear inhibits the agglutination of type A RBCs with weak binding avidity, illustrated by spike in contrast within plugs of RBCs with weak binding avidity at a shear rate of 0 s-1 in Figure 4I.
Preliminary results demonstrate that this assay is also capable of distinguishing weakly agglutinating type A RBCs from type O RBCs by using shear. A traditional agglutination assay utilizing anti-A or the lectin DB assay may not be effective in distinguishing weakly agglutinating RBCs from type O RBCs, because both types of RBCs do not show an obvious agglutination response to anti-A. However, the binding avidity of these types of RBCs differs—weakly agglutinating type A RBCs with only a few A antigens will have a stronger binding avidity to anti-A than type O RBCs that lack A group antigens completely. We performed the same experiment described above using type O RBCs instead of weakly agglutinating type A RBCs. Type O RBCs did not agglutinate when exposed to an initial shear rate of 10 s-1. Similar to weakly agglutinating type A RBCs, type O RBCs agglutinated when shear was reduced to 0 s-1 for ∼10 min. However, agglutinins of type O RBCs broke up when flow was restored to the initial flow rate of 10 s-1. In contrast, agglutinins of weakly agglutinating type A RBCs persisted at shear rates exceeding 52 s-1 (data not shown), indicating that weakly agglutinating type A RBCs possess some unknown number of A antigens on their surface.
To illustrate the applicability of this technology beyond blood typing and subtyping, we performed an agglutination assay to detect bacteria. The bacteria used here was S. aureus at a concentration of ∼1 × 108 cfu/mL, and latex beads coated with antibody were used instead of RBCs. Plugs were formed as illustrated in Figure 5A, and agglutination was monitored at four time points. Agglutination in the presence of bacteria was visible 20 s after merging (Figure 5B, t1), and agglutinins were obvious after 150 s (Figure 5B, t2). This rapid increase in contrast in plugs with bacteria over the 150-s observation period (Figure 5D, red squares) can likely be attributed to the kinetics of the antibody—bacteria interaction. In the absence of bacteria, contrast in plugs increases minimally over time (Figure 5C; Figure 5D, blue circles), possibly due to nonspecific aggregation or sedimentation of latex particles.
CONCLUSIONS
In this paper, we presented a plug-based microfluidic system as proof of concept to perform ABO and D blood typing and subtyping on-chip without cross-contamination. Though these experiments were completed on several different microfluidic devices, these techniques could, in principle, be combined into a single microfluidic device. This method resolves common A1 and A2 subtypes with on-chip dilution and can use shear to distinguish rare, weakly agglutinating A subtypes. Concentration-dependent agglutination of subtypes A1 and A2 is believed to be the result of a difference in the number of antigens on the RBCs of each subtype. While subtype A1 RBCs are believed to have antigens on both terminal and subterminal complex sugars, subtype A2 RBCs are believed to have antigens on only subterminal sugars. We found that rare, weakly agglutinating subtypes of type A RBCs can be distinguished from type O RBCs by using shear rate. The weakly agglutinating type A RBCs are likely one of four less common type A subtypes, such as A3,,Ax,,Am,and Ael, which cannot be distinguished from type O blood by using the lectin DB dilution assay. We believe these results indicate that this technology may be useful in reducing mistyping of these subtypes in the clinical setting. By providing controlled shear, this system also enables acquisition of kinetic data on agglutination to provide a quantitative description of the process. In our studies, shear rate was varied by flow rates, but others have reported that shear rate could be tailored by creating devices with more sophisticated geometries and applying them to studies of binding avidity of RBCs.32
Assuming that the techniques presented here can be successfully combined in a single device, this plug-based method could be used to type blood for all 20 factors known to be present on RBCs by including these factors in preformed antibody arrays. We routinely aspirate arrays of 48 different reagents separated by blank spacer plugs (buffer or gas) to screen many reaction or protein crystallization conditions in a single experiment.14-16 Conversely, many blood samples could be analyzed in the same experiment by aspirating all samples into a single array. This array could be split into many identical arrays of smaller plugs, and each daughter array could be combined with a different antibody.18 Also, by using the hybrid method17 with automation and preformed antibody arrays, multiple antibodies at multiple concentrations could be used to measure the binding avidity between RBCs and agglutinins all on a similar style device. The hybrid approach may also be used to test the compatibility of the donor RBCs with RBCs of perspective recipients with sickle cell anemia, where alloimmunization leads to delayed transfusion reactions.33
Subtyping could be valuable in early detection of hemolytic disease of the newborn (HDN), where the transfer of maternal IgG antibodies to the fetus through the placenta causes hemolysis of fetal RBCs, currently performed with molecular-based applications.34 In HDN detection, minimizing the necessary sample volume is important, and microfluidics provides reduced sample consumption in comparison to standard test tube subtyping methods. Reduced sample consumption is also an important advance for point of care applications when volume is a limiting factor. Assuming this system design works in a clinical setting, we believe point of care applications could also benefit from the adaptation of this system to utilize Teflon tubing instead of PDMS to enable bedside use.
In addition, automation of this system could enable rapid, complete blood typing and subtyping in a single experiment using a single blood sample. While blood banks routinely automate blood typing analysis of single samples, this platform could be extended to analyze multiple samples in a serial fashion with no risk of cross-contamination (prevented by encapsulation of all solutions within plugs), decreasing assay time. Incorporation of the hybrid method with preformed cartridges of plugs would maintain automation while enabling concentration-dependent antibody screening or cross-matching of blood samples.
This plug-based microfluidic agglutination assay is also capable of detecting bacteria. Bacterial contamination of blood used in transfusions is a serious threat to patients with weakened immune systems, and a combined assay that uses a cartridge of plugs containing antibodies to determine the type of blood and also test for the presence of bacteria is an attractive long-term opportunity provided by this microfluidic system. A dramatic improvement in sensitivity would be required to detect 1–10 cfu/mL of bacteria by this method, and it remains to be seen whether this or other microfluidic methods are most suitable for such detection. Plug-based assays could also be extended to immunoassays for detection of viruses. Immunoassays are known to be compatible with rapid-prototyping microfluidics.35 Similar to distinguishing binding avidity of RBCs by using shear, plug-based microfluidics may be useful for detecting changes in antibody avidity after virus infection as a function of shear.36-39 It remains to be seen whether this technique can be extended beyond immunoassays to measure aggregation of red blood cells in occlusive vascular diseases.40 We believe this plug-based platform could become useful for a wide range of agglutination assays with minimal consumption of sample, a high degree of multiplexing, and a high degree of control of interparticle interactions using shear.
ACKNOWLEDGMENT
This work was supported in part by the NIH and the NIBIB (Grant R01 EB001903) the Camille Dreyfus Teacher-Scholar Award. M.K.R. was supported in part by Burroughs Wellcome Fund Interfaces I.D. 1001447. R.F.I. is a Cottrell Scholar of the Research Corporation and an A. P. Sloan Research Fellow. Some of this work was performed at the MRSEC microfluidics facility (funded by the NSF). We thank Rebecca R. Pompano for helpful discussions and Jessica M. Price for assistance in writing and editing the manuscript.
References
- (1).Klein HG, Anstee DJ, editors. Mollison’s Blood Transfusions in Clinical Medicine. 11th Blackwell Publishing Ltd; Malden, MA: 2005. [Google Scholar]
- (2).Lapierre Y, Rigal D, Adam J, Josef D, Meyer F, Greber S, Drot C. Transfusion. 1990;30:109–113. doi: 10.1046/j.1537-2995.1990.30290162894.x. [DOI] [PubMed] [Google Scholar]
- (3).Defigueiredo M, Lima M, Morais S, Porto G, Justica B. Transfus. Med. 1992;2:115–118. doi: 10.1111/j.1365-3148.1992.tb00144.x. [DOI] [PubMed] [Google Scholar]
- (4).Kim DS, Lee SH, Ahn CH, Lee JY, Kwon TH. Lab Chip. 2006;6:794–802. doi: 10.1039/b516495h. [DOI] [PubMed] [Google Scholar]
- (5).Ahn CH, Choi JW, Beaucage G, Nevin JH, Lee JB, Puntambekar A, Lee JY. Proc. IEEE. 2004;92:154–173. [Google Scholar]
- (6).Avent ND. Transfus. Clin. Biol. 2007;14:10–15. doi: 10.1016/j.tracli.2007.04.011. [DOI] [PubMed] [Google Scholar]
- (7).Dada A, Beck D, Schmitz G. Transfus. Med. Hemother. 2007;34:341–346. [Google Scholar]
- (8).Titlestad K, Georgsen J, Andersen H, Kristensen T. Vox Sang. 1997;73:246–251. doi: 10.1046/j.1423-0410.1997.7340246.x. [DOI] [PubMed] [Google Scholar]
- (9).Butch SH, Judd WJ, Steiner EA, Stoe M, Oberman HA. Transfusion. 1994;34:105–109. doi: 10.1046/j.1537-2995.1994.34294143935.x. [DOI] [PubMed] [Google Scholar]
- (10).Linden JV, Paul B, Dressler KP. Transfusion. 1992;32:601–606. doi: 10.1046/j.1537-2995.1992.32792391030.x. [DOI] [PubMed] [Google Scholar]
- (11).Boldt J, Walz G, Triem J, Suttner S, Kumle B. Intensive Care Med. 1998;24:1187–1193. doi: 10.1007/s001340050743. [DOI] [PubMed] [Google Scholar]
- (12).Brito VO, Raimundo IM. Anal. Chim. Acta. 1998;371:317–324. [Google Scholar]
- (13).Song H, Li HW, Munson MS, Van Ha TG, Ismagilov RF. Anal. Chem. 2006;78:4839–4849. doi: 10.1021/ac0601718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zheng B, Roach LS, Ismagilov RF. J. Am. Chem. Soc. 2003;125:11170–11171. doi: 10.1021/ja037166v. [DOI] [PubMed] [Google Scholar]
- (15).Zheng B, Tice JD, Roach LS, Ismagilov RF. Angew. Chem., Int. Ed. 2004;43:2508–2511. doi: 10.1002/anie.200453974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zheng B, Ismagilov RF. Angew. Chem., Int. Ed. 2005;44:2520–2523. doi: 10.1002/anie.200462857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li L, Mustafi D, Fu Q, Tereshko V, Chen DLL, Tice JD, Ismagilov RF. Proc. Natl. Acad. Sci. U. S. A. 2006;103:19243–19248. doi: 10.1073/pnas.0607502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Adamson DN, Mustafi D, Zhang JXJ, Zheng B, Ismagilov RF. Lab Chip. 2006;6:1178–1186. doi: 10.1039/b604993a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Song H, Ismagilov RF. J. Am. Chem. Soc. 2003;125:14613–14619. doi: 10.1021/ja0354566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Song H, Tice JD, Ismagilov RF. Angew. Chem., Int. Ed. 2003;42:768–772. doi: 10.1002/anie.200390203. [DOI] [PubMed] [Google Scholar]
- (21).Chen DLL, Li L, Reyes S, Adamson DN, Ismagilov RF. Langmuir. 2007;23:2255–2260. doi: 10.1021/la062152z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Anal. Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- (23).McDonald JC, Duffy DC, Anderson JR, Chiu DT, Wu HK, Schueller OJA, Whitesides GM. Electrophoresis. 2000;21:27–40. doi: 10.1002/(SICI)1522-2683(20000101)21:1<27::AID-ELPS27>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- (24).Xia YN, Whitesides GM. Angew. Chem., Int. Ed. Engl. 1998;37:551–575. doi: 10.1002/(SICI)1521-3773(19980316)37:5<550::AID-ANIE550>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- (25).Roach LS, Song H, Ismagilov RF. Anal. Chem. 2005;77:785–796. doi: 10.1021/ac049061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liau A, Karnik R, Majumdar A, Cate JHD. Anal. Chem. 2005;77:7618–7625. doi: 10.1021/ac050827h. [DOI] [PubMed] [Google Scholar]
- (27).Song H, Bringer MR, Tice JD, Gerdts CJ, Ismagilov RF. Appl. Phys. Lett. 2003;83:4664–4666. doi: 10.1063/1.1630378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yamaguch H, Okubo Y, Ogawa Y, Tanaka M. Vox Sang. 1973;25:361–369. doi: 10.1111/j.1423-0410.1973.tb04384.x. [DOI] [PubMed] [Google Scholar]
- (29).Handique K, Burns MA. J. Micromech. Microeng. 2001;11:548–554. [Google Scholar]
- (30).King C, Walsh E, Grimes R. Microfluidics Nanofluidics. 2007;3:463–472. [Google Scholar]
- (31).de Isla N, Rasia RJ, Valverde JR, Stoltz JF. Transfus. Med. Hemother. 2004;31:41–48. [Google Scholar]
- (32).Abkarian M, Faivre M, Stone HA. Proc. Natl. Acad. Sci. U. S. A. 2006;103:538–542. doi: 10.1073/pnas.0507171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Davies SC, RobertsHarewood M. Blood Rev. 1997;11:57–71. doi: 10.1016/s0268-960x(97)90012-6. [DOI] [PubMed] [Google Scholar]
- (34).Reid ME. Transfusion. 2003;43:1748–1757. doi: 10.1111/j.0041-1132.2003.00597.x. [DOI] [PubMed] [Google Scholar]
- (35).Linder V, Sia SK, Whitesides GM. Anal. Chem. 2005;77:64–71. doi: 10.1021/ac049071x. [DOI] [PubMed] [Google Scholar]
- (36).Inouye S, Hasegawa A, Matsuno S, Katow S. J. Clin. Microbiol. 1984;20:525–529. doi: 10.1128/jcm.20.3.525-529.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Deory F, Casas I, Domingo CJ, Echevarria JM. Clin. Diagn. Virol. 1995;3:323–332. doi: 10.1016/0928-0197(94)00045-v. [DOI] [PubMed] [Google Scholar]
- (38).Whitby D, Howard MR, Tenantflowers M, Brink NS, Copas A, Boshoff C, Hatzioannou T, Suggett FEA, Aldam DM, Denton AS, Miller RF, Weller IVD, Weiss RA, Tedder RS, Schulz TF. Lancet. 1995;346:799–802. doi: 10.1016/s0140-6736(95)91619-9. [DOI] [PubMed] [Google Scholar]
- (39).Gassmann C, Bauer G. J. Med. Virol. 1997;51:242–251. [PubMed] [Google Scholar]
- (40).Vaya A, Martinez M, Carmena R, Aznar J. Thromb. Res. 1993;72:119–126. doi: 10.1016/0049-3848(93)90213-8. [DOI] [PubMed] [Google Scholar]