

Abstract
Artesunate (AS) is used in combination with amodiaquine (AQ) as first-line treatment for uncomplicated malaria in many countries. We investigated the effect of concomitant AS administration on the pharmacokinetics of AQ and compared concentrations of desethylamodiaquine (DEAQ), the main metabolite of AQ, in plasma between patients with different variants of the cytochrome P4502C8 (CYP2C8) gene. A two-compartment model was fitted to 169 plasma DEAQ concentrations from 103 Ghanaian children aged 1 to 14 years with uncomplicated malaria treated either with AQ alone (n = 15) or with AS plus AQ (n = 88). The population clearance of DEAQ appeared to increase nonlinearly with body weight, and the central volume of distribution of DEAQ was higher (P < 0.001) in the AS-plus-AQ group than in the AQ-only group. The maximum plasma DEAQ concentration was higher (P < 0.001), and the population distribution half-life was shorter (P < 0.01), in the AQ-only group than in the AS-plus-AQ group. The total areas under the plasma DEAQ concentration-time curves (P = 0.68) and elimination half-lives (P = 0.39) were similar for the two groups. There was a high frequency (0.179) of the non-wild-type allele of CYP2C8, but no differences between CYP2C8 genotypes with regard to AQ efficacy or safety were evident. The sample size, however, was limited, so monitoring of AQ toxicity in the study area is still indicated. The nonlinear clearance of DEAQ and the wide variability in kinetic parameters have safety implications for weight-based dosing of higher-body-weight children with AQ. The pharmacokinetics of artemisinin combination therapies should be studied in malaria patients, because the rapid parasite clearance caused by the artemisinin may affect the kinetics of the partner drug and the combination.
Prompt treatment with safe and efficacious antimalarial drugs is an important malaria control strategy. This strategy is being challenged by the escalating resistance of Plasmodium falciparum to the available drugs (33). Artemisinin combination therapy (ACT) is recommended as a rational approach to curb this problem of escalating antimalarial drug resistance (31). Many countries in sub-Saharan Africa have adopted ACT as their first-line malaria treatment policy. The combination of artesunate (AS) with amodiaquine (AQ) is one of the most widely adopted ACT regimens.
AQ is a 4-aminoquinoline that is rapidly absorbed and extensively metabolized upon oral administration to desethylamodiaquine (DEAQ), its main and active metabolite (7, 8). The formation of DEAQ from AQ is catalyzed by cytochrome P4502C8 (CYP2C8), a polymorphic isoform of hepatic cytochrome P450 (18). The gene for CYP2C8 is located in a cluster with the genes for CYP2C9, CYP2C19, and CYP2C18 on chromosome 10q24 (11). CYP2C8*2 and CYP2C8*3 are the two most frequently occurring variant alleles of CYP2C8, and both variants code for enzymes with decreased activity (9, 10, 23), raising the possibility that polymorphisms in the CYP2C8 gene may modulate the metabolism of AQ (5).
AS is a water-soluble hemisuccinate ester derivative of artemisinin, a sesquiterpene lactone peroxide extracted from the leaves of the plant Artemisia annua. After oral administration, AS is rapidly metabolized into dihydroartemisinin, its active metabolite (4). The artemisinin derivatives induce their own elimination, resulting in decreasing concentrations (2), and the hepatic metabolism of artemisinin is mediated in vitro by CYP2B6, as well as by enzymes of the CYP3A subfamily (28). There is evidence for overlapping substrate specificity between CYP2C8 (which is responsible for AQ metabolism) and CYP3A4, an isoform of the CYP3A subfamily (22). Moreover, artemisinin, AQ, and their respective metabolites (DEAQ and dihydroartemisinin) exhibit a range of interactions in vitro (19), and the pharmacokinetics of mefloquine, a quinoline, is altered by concomitant AS administration (16). These considerations raise the possibility of drug-drug interactions between AS and AQ after coadministration. To date, however, there is no report on the pharmacokinetics of AQ after concomitant AS administration in malaria patients, especially in children, the main recipients of antimalarial drugs in areas of endemicity.
The aim of this study was to investigate the effects of concomitant AS administration on the pharmacokinetics of AQ in children with uncomplicated malaria. A secondary objective was to explore and relate polymorphisms in the CYP2C8 gene to DEAQ concentrations in treated subjects. In view of the logistical and medical hazards associated with the repeated blood sampling required for conventional pharmacokinetics studies, as well as the ethical objections to subjecting acutely ill children to such procedures, we used blood samples collected during a clinical trial and employed a population approach (27) to estimate the pharmacokinetic parameters of AQ in the study population.
MATERIALS AND METHODS
Study population.
The data for the study were prospectively collected from children with uncomplicated malaria who participated in a randomized longitudinal trial in Accra, Ghana, in which AQ monotherapy was compared with AS-plus-AQ and artemether-lumefantrine therapies. The full details of the efficacy and safety results of the clinical trial have been reported elsewhere (1). Briefly, children with uncomplicated malaria who met the inclusion and exclusion criteria were recruited after written consent was obtained from the accompanying parent or guardian. Ethical approval for the study was obtained from the Ethical and Protocol Review Committee of the University of Ghana Medical School.
Drug dosing.
A single daily oral dose of AQ (Camoquine; Pfizer, Dakar, Senegal) at 10 mg/kg of body weight was administered either alone or in combination with a single daily oral dose of AS (Plasmotrim; Mepha, Switzerland) at 4 mg/kg of body weight for 3 days (days 0, 1, and 2). Subjects were followed up on days 1, 2, 3, 7, 14, and 28 and then monthly for 1 year.
Blood sampling.
Venous blood (2 to 5 ml) was collected into heparinized polypropylene tubes before treatment and on days 3 and 7 for routine clinical chemistry analyses. The heparinized sample was centrifuged immediately, and the plasma samples were transferred to polypropylene tubes and stored at −20°C until transport on dry ice to Denmark, where they were stored at −20°C until analysis. The exact time of drug administration was recorded for each subject on days 0, 1, and 2, and the exact time of blood sampling was recorded for each subject on days 0, 3, and 7.
HPLC.
Concentrations of AQ, DEAQ and bis-DEAQ in plasma were measured by means of a reverse-phase high-performance liquid chromatography (HPLC) method. Briefly, samples were separated using liquid-liquid extraction and then analyzed on an HPLC system (Agilent 1100 series; Agilent Technologies, Palo Alto, CA) with UV detection at 237 nm. Data were acquired using HPLC Chemstation 10.01 (Agilent Technologies). The coefficients of variation (CV) for interday variations of the assay were <10%, and the CV for intraday variations were <10% at two levels. The limit of detection and limit of quantification (LOQ) were both 10 ng/ml.
DNA genotyping.
Genomic DNA was extracted from filter paper samples using the Chelex-100 method modified into a 96-well format (24). PCR with restriction fragment length polymorphism was used to analyze single-nucleotide polymorphisms in the CYP2C8 gene (5). Briefly, the primer pair 5′-CTT CCG TGC TAC ATG ATG ACG-3′ (sense)-5′-CTG CTG AGA AAG GCA TGA AG-3′ (antisense) was used for PCR amplification, followed by incubation with the BclI endonuclease. Restriction digestion of the PCR product resulted in (i) two completely digested oligonucleotide fragments with molecular weights of 98 bp and 22 bp (wild type), (ii) an undigested fragment with a molecular weight of 120 bp (mutant), or (iii) three oligonucleotide fragments of 98 bp, 22 bp, and 120 bp (heterozygote; due to partial digestion). The fragments were visualized in ethidium bromide-stained gels.
Pharmacokinetic analysis.
The total AQ dose, number of AQ doses administered, sampling and dosing times, plasma DEAQ concentrations, and demographic information (age, sex, and body weight) were available for all subjects. The sampling time was calculated relative to the time of the first drug administration. Drug concentrations below the LOQ were excluded.
Nonlinear mixed-effect modeling was performed using NONMEM software (version V, level 1.1) with NM-TRAN, PREDPP, and the GNU77 Fortran compiler. The data analyzed contained one or two plasma concentration-time points above the LOQ per individual. The plasma samples were drawn approximately 24 to 168 h after the administration of the last AQ dose. The dose of AQ administered was used as the DEAQ dose input for the models.
Two different basic structural models, a one-compartment model and a two-compartment model with first-order absorption, with or without a lag time, were fitted to the plasma DEAQ concentration-time data. The residual variability was modeled as a proportional-error model. The first-order conditional estimation method was used throughout the model-building procedure. The relationships between the structural-model-based Bayesian estimates of the pharmacokinetic parameters and individual covariates were explored graphically. The initial analysis was conducted without including covariates, and subsequently the effects of covariates on the pharmacokinetic parameters were explored. During comparisons of alternative models, the differences in the value of the objective function (ΔOBJ) were approximately chi-square distributed with “n” degrees of freedom. In order to discriminate between two nested models and select significant covariates, a difference in objective function of >7.9 (1 degree of freedom), which corresponds to a significance level (P) of <0.005, was used.
Covariates.
Age, body weight, and concomitant AS administration were explored as covariates. Covariates with continuous variables were centered on their medians so that the population estimates would represent those of an average patient. The actual concentrations of AS were not measured but modeled as a categorical variable, “present” or “absent” (i.e., with or without AS).
The absorption rate constant (Ka) for AQ was fixed at 0.867 h−1 on the basis of data from a previous study (17). However, due to the lack of data on the absorption phase of AQ and the metabolic formation of DEAQ, this fixed Ka could be viewed as a hybrid parameter that includes the Ka of AQ, the volume of distribution (V1) of AQ, and the formation clearance (CL1/f) of DEAQ, as shown in Fig. 1. Thus, a simpler first-order absorption process with a hybrid Ka that compresses information on the Ka of AQ, V1, and CL/f (Fig. 2) was used. Inclusion of a lag time (Tlag) for DEAQ did not contribute to a significant drop in the objective function (ΔOBJ = −0.262). However, it seemed reasonable to include a lag time, because DEAQ, which is a metabolite, needed to be formed from AQ, as suggested previously (17). Thus, Tlag was fixed at 0.84 h.
FIG. 1.

Population pharmacokinetic model for AQ and DEAQ, with a Ka that includes the Ka of AQ, the volume of distribution of AQ, and the formation clearance of DEAQ. Q, intercompartmental clearance.
FIG. 2.

Population pharmacokinetic model of a first-order absorption process with a hybrid Ka, which compresses information on the Ka of AQ, the volume of distribution of AQ, and the clearance of DEAQ. Q, intercompartmental clearance.
Model parameters.
A two-compartment model with a fixed first-order Ka described the data better than a one-compartment model, based on the change in the objective function (ΔOBJ = −19.77) and diagnostic plots. The fundamental parameters used to characterize the two-compartment model were clearance (CL/f), central volume of distribution (V2/f), intercompartmental clearance (Q/f), and peripheral volume of distribution (V3/f). The addition of the interindividual variability term for Q/f or V3/f (ηQ/f or ηV3/f) to the two-compartment model did not result in a statistically significant decline in the objective function (ΔOBJ, <7.9); therefore, only covariate effects on the ηCL/f and ηV2/f were examined for the final model including covariates.
Secondary pharmacokinetic parameters.
Using dense sampling points during post hoc estimations of individual predicted concentrations, the maximum concentration (Cmax) and the time to reach maximum concentration (Tmax) of the full concentration-versus-time profiles were read from the individual curves. Each individual's total area under the plasma DEAQ concentration-time curve (AUC) was estimated using the formula 3 × dose/CL/f. The individual distribution half-life (t1/2α) and terminal-phase half-life (t1/2β) were estimated using the following equations:
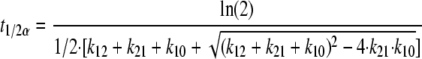 |
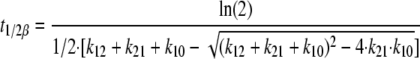 |
where k12 is Q/V2, k21 is Q/V3, and k10 is CL/V2.
RESULTS
Characteristics of subjects.
In total, 103 children (15 receiving AQ alone and 88 receiving AS plus AQ) were included in the analysis. All subjects (except two AQ recipients) responded adequately to treatment. Parasite clearance was more rapid (P < 0.01) in the AS-plus-AQ group than in the AQ-only group. Table 1 shows the baseline characteristics of recruited subjects, including the proportion of subjects who were parasitemic after 48 h, for the two treatment groups. The national malaria treatment policy in Ghana changed from chloroquine to AS-plus-AQ treatment 2 months after the initiation of this trial. This change necessitated discontinuation of the AQ monotherapy arm of the trial for ethical reasons, explaining the low number of subjects in the AQ arm.
TABLE 1.
Selected baseline characteristics of 103 children in the pharmacokinetic study
| Treatment group (no. of patients) | Characteristic
|
||||
|---|---|---|---|---|---|
| Age (yr)a | Sex
|
Body wt (kg)a | Proportion (%)b parasitemic after 48 h | ||
| No. of males | No. of females | ||||
| AQ (15) | 5.3 (3.9-7.6) | 5 | 10 | 18 (12.0-21.8) | 46.6 (7/15) |
| AS + AQ (88) | 6.0 (4-10) | 48 | 40 | 18 (15-26.8) | 10.2 (9/88) |
Expressed as median (interquartile range).
Expressed as percent parasitemic (number parasitemic/total number of patients in the group). P < 0.01.
Plasma DEAQ concentrations and pharmacokinetic parameters.
A total of 169 measurable plasma DEAQ concentration-time points were available for inclusion in the final pharmacokinetic model. There were no detectable concentrations of AQ, DEAQ, or bis-DEAQ in any of the pretreatment (day zero) samples. AQ was not detectable in any of the postdose samples, either, whereas DEAQ was detected in all day 3 and day 7 samples. Measurable bis-DEAQ concentrations were detected in 47 samples on day 3 and in 7 samples on day 7, but these were not included in the pharmacokinetic analysis. The mean (standard deviation [SD]) DEAQ concentration on day 3 was 156.6 (94.7) ng/ml.
Covariates and DEAQ pharmacokinetics.
The population oral clearance of DEAQ correlated nonlinearly with body weight, since incorporation of this covariate significantly reduced the minimum value of the objective function (ΔOBJ = −28.345). The relationship between clearance and body weight, which appeared to increase in a Michaelis-Menten-type kinetics (Fig. 3), could be described by the following equation: population oral clearance = θ1·(BW/median)/[θ2 + (BW/median)], where BW is body weight. This works out to 29.9·(BW/18)/[0.992+ (BW/18)].
FIG. 3.

(A) Relationship between total clearance (CL/f) and the covariate body weight, using the final population model. (B) Goodness of fit evaluated by weighted residuals versus body weight. The lack of a distinct pattern in the plot of weighted residuals versus body weight indicates that the final model appropriately accounts for the influence of body weight on the pharmacokinetic parameter.
Incorporation of AS treatment resulted in further improvement of the objective function (ΔOBJ = −12.572). Furthermore, concomitant AS administration increased (422 liters) the central volume of distribution over that with administration of AQ alone (85.4 liters) (P < 0.001). Incorporation of age as a covariate appeared initially to have an effect on CL/f and V2/f. However, combining this model with the nonlinear variation of body weight with CL/f failed to produce any additional effect. Figure 4 shows the observed versus predicted plasma concentrations and goodness-of-fit plots. Table 2 shows final pharmacokinetic parameter estimates with 95% confidence intervals obtained by 1,000 bootstrap runs.
FIG. 4.

Quality of fit of the pharmacokinetic data. (A) Observed versus predicted typical (population) concentrations, based on the population parameters and covariate information. (B) Observed versus predicted individual Bayesian concentrations based on the values of the parameters for the specific individuals. (C and D) Goodness of fit evaluated by weighted residuals versus the predicted population (C) and individual (D) concentrations, respectively.
TABLE 2.
Parameter estimates of the final population pharmacokinetic model and results using the bootstrap validation procedure
| Parameter | Estimate | RSEa (%) | Median (2.5-97.5% range) for 1,000 bootstrap replicates |
|---|---|---|---|
| Ka (h−1) | 0.867 (fixed) | ||
| Tlag (h) | 0.84 (fixed) | ||
| CL/f (liters/h)b (population mean) | 15.5 | 32.0 | |
| V2/f (liters)b (population mean) | 368 | 34.0 | |
| Q/f (liters/h) | 16.0 | 11.9 | 15.8 (11.8-19.3) |
| V3/f (liters) | 1,060 | 14 | 1,120 (854-2,433) |
| θ1c | 29.9 | 24.7 | 32.5 (20.1-147) |
| θ2c | 0.992 | 45.5 | 1.23 (0.44-10.4) |
| θ3d | 85.4 | 37.2 | 84 (44.1-204) |
| θ4d | 422 | 33.2 | 447 (236-884) |
| Interindividual variability (ω) | |||
| CVCL/f (%) | 31.9 | 41.6 | 31.6 (14.5-46.2) |
| CVV2/f (%) | 40.9 | 64.7 | 44.3 (14.8-82.3) |
| Residual variability (σ) (proportional error [%]) | 31.9 | 18.3 | 30.8 (23.1-37.0) |
| t½α (h)b (population mean) | 7.38 | 35.3 | |
| t½β (h)b (population mean) | 110 | 26.8 |
RSE, relative standard error.
Means and SDs were calculated based on post hoc individual predicted parameters.
Population oral clearance = θ1·(BW/median)/[θ2 + (BW/median)], where BW is body weight. This works out to 29.9·(BW/18)/[0.992 + (BW/18)].
θ3 and θ4 represent the V2/f of the typical individual in the population treated with AQ plus AS and with AQ alone, respectively. The population V2/f is 85.4 liters without AS and 422 liters with AS.
Secondary pharmacokinetic parameters.
The derived secondary pharmacokinetic parameters based on the final model are shown in Table 3. Figure 5 shows the observed plasma DEAQ concentration-versus-time profiles derived from the model for subjects treated with AQ alone or AS plus AQ. The t1/2α and t1/2β derived from the model were 7.38 h (range, 1.6 to 10.9) and 110 h (range, 70.2 to 285 h), respectively. There was a reduction in Cmax (P < 0.001), and an approximately fourfold increase in t1/2α (P < 0.001), in the AS-plus-AQ group compared to the AQ group. The trough concentrations of DEAQ were lower in the AQ group than in the AS-plus-AQ group. The total areas under the plasma DEAQ concentration-time curves (P = 0.68) and elimination half-lives (P = 0.39) (Table 3) were similar for the two groups.
TABLE 3.
Secondary pharmacokinetic parameters based on the final model
| Treatment group (no. of patients) | Mean (SD) value for the following parameter:
|
||||
|---|---|---|---|---|---|
| Cmax (ng/ml) | Tmax (h) | t½α (h) | t½β (h) | AUC (h·ng/ml) | |
| AQ (15) | 1,185 (432) | 47.9 (1.3) | 2.0 (0.3) | 104 (18.2) | 38,516 (14,138) |
| AS + AQ (88) | 537 (244) | 46.9 (7.8) | 8.3 (1.4) | 111 (31.0) | 40,339 (16,021) |
FIG. 5.

Plasma concentration-versus-time profiles of DEAQ based on the final population model. The observed concentrations of DEAQ following administration of AQ only (filled triangles) or AS plus AQ (open squares) are shown. The lines represent the simulated post hoc population estimates for a typical 6-year-old patient weighing 18 kg who was administered AQ only (solid line) or AS plus AQ (red dotted line).
CYP2C8 polymorphisms and plasma DEAQ concentrations.
The allele frequencies of the main CYP2C8 genotypes were in Hardy-Weinberg equilibrium (Table 4), and the frequency of the non-wild-type allele was 0.179. Plasma DEAQ concentrations were slightly but insignificantly lower (Fig. 6) in subjects with mutant CYP2C8 genotypes than in those with wild-type alleles or heterozygotes (P = 0.58). The CYP2C8 genotype did not contribute to the differences in DEAQ concentrations between subjects who received AQ alone and subjects who received AS plus AQ. Furthermore, a plot of parameter estimates (V2 and CL) against the various CYP2C8 genotypes failed to show any significant trends in DEAQ concentrations (data not shown).
TABLE 4.
Distribution of CYP2C8 genotypes in 92 children from southern Ghana
| CYP2C8 genotypea | No. of patients receiving:
|
Prevalence of genotype (%) | No. of patients withb:
|
|||||
|---|---|---|---|---|---|---|---|---|
| AS + AQ | AQ only | ANC of <1,000 | Elevated LFT result | Pruritus | HR of <60 | Excessive fatigue | ||
| *1/*1 | 53 | 11 | 69.6 | 11 | 3 | 3 | 2 | 3 |
| *1/*2 | 23 | 0 | 25 | 1 | 0 | 0 | 0 | 0 |
| *2*/2 | 5 | 0 | 5.4 | 0 | 0 | 0 | 0 | 0 |
*1/*1, wild type (homozygote); *1/*2, heterozygote; *2*/2, mutant (homozygote).
ANC, absolute neutrophil count; LFT, liver function test; HR, heart rate.
FIG. 6.

Box plot showing the distribution of plasma DEAQ concentrations in subjects with different CYP2C8 genotypes. The horizontal line within each box is the median concentration. The lower and higher horizontal lines are the 25th and 75th quartile values of the distribution, respectively. The ends of the vertical lines extending from each box represent the lower and upper limit of the distribution. Concentrations outside the lower and upper limits, indicated by dots, are outliers.
CYP2C8 polymorphisms, efficacy, and adverse effects.
Two subjects treated with AQ monotherapy failed to respond adequately to treatment. The plasma DEAQ concentrations in these two subjects on day 3 were 18 ng/ml and 52 ng/ml, respectively. Laboratory-detected or self-reported adverse events considered possibly or probably drug related were neutropenia, elevated serum aminotransferase levels, bradycardia, pruritus, and excessive sleepiness (Table 4).
DISCUSSION
In this study, we have used a population approach to estimate the pharmacokinetics of AQ administered alone or combined with AS, using data from one or two blood samples collected from children with uncomplicated malaria participating in a clinical trial. Although a recent report of the pharmacokinetics of AQ in children with uncomplicated malaria included a subset cotreated with AS (14), data on the effect of concomitant AS administration on the pharmacokinetics of AQ were not available. This is one of the few studies, therefore, to report on the multiple-dose kinetics of AQ and, to our knowledge, the only available report on the pharmacokinetics of the AS-AQ combination in pediatric malaria patients.
The data showed a trend toward a change in the central volume of distribution of DEAQ after concomitant AS administration, findings similar to those from a previous study that showed an alteration in the pharmacokinetics of mefloquine after concomitant AS administration (16). This effect of AS on AQ is likely to be an indirect effect of AS on disease, possibly through accelerated parasite clearance and rapid clinical recovery, with an indirect effect on the volume of distribution, or on clearance. This effect of AS on AQ could also result from drug-drug interaction, possibly through competition for protein binding sites between AQ and AS, since AQ and AS are both highly protein bound (25). The possibility of drug-drug interaction between AQ and AS, or between the metabolites of these two antimalarials, may have implications for the safety of the combination, especially in the context of coadministration of this regimen with other drugs, such as an antiretroviral drug, as has recently been reported (12).
The data showed wide interindividual variation in the pharmacokinetic parameters of AQ, which is consistent with previous reports on AQ metabolism (25, 30). In this study, however, the wide interindividual variation in AQ pharmacokinetics could reflect the maturational and physiological heterogeneity of this childhood population, which spanned most of the childhood age groups, each of which may be associated with distinct developmental and possibly distinct drug-metabolizing characteristics.
The data showed a nonlinear relation between the elimination of DEAQ and body weight, with body weights above 29.9 kg tending to approach a constant clearance. This suggests a high total exposure to DEAQ for subjects with low body weight; exposure tends to decline toward a constant level above a body weight of 29.9 kg. While this finding lends some support to the current recommendation of basing AQ dosing on weight in children, it raises questions about possible drug accumulation in older children, the subgroup that lacks representative data on weight-based AQ dosing (29).
The mean plasma DEAQ concentration on day 3 in this study was comparable to those reported for African children (3) or adults (32) but different from day 3 DEAQ concentrations in whole (capillary) blood obtained from Papua-New Guinean children with malaria (15). The discrepancy between our findings and those of the latter study is likely to be due to the blood subtype analyzed, since it is suggested that effective AQ concentrations should be analyzed in plasma rather than in whole blood (17). The plasma DEAQ concentrations of two subjects who did not respond to treatment were below the predicted in vitro DEAQ MIC (6) and the in vivo DEAQ concentration of 135 ng/ml that has been suggested to be the minimum concentration required for adequate treatment efficacy (3). The clinical responses of these two subjects therefore likely resulted from inadequate drug levels rather than from parasite resistance.
The 17.9% frequency of the non-wild-type CYP2C8 genotype in this southern Ghanaian population was similar to that found in a previous report (16.8%) from northern Ghana (26) and comparable to those in reports from other areas in sub-Saharan Africa, such as Zanzibar (13.9%) (5) and Burkina Faso (11.5%) (23), or in reports on African-Americans (18.3%) (9). We observed no differences in efficacy or side effect occurrence between subjects with different CYP2C8 genotypes, but this was likely due to the limited number of subjects. However, the potential impact of CYP2C8 polymorphisms on AQ efficacy may be influenced by other factors, such as linkage disequilibrium with other alleles, such as CYP2C9 (34), the possibility of extrahepatic metabolism of AQ to an as yet uncharacterized metabolite (18), and the fact that AQ and DEAQ are both pharmacologically active. On the other hand, the potential impact of the prevalence of the CYP2C8 allele on the safety of AQ could be significant. This is because the reactive quinone imine metabolite of AQ produced as a result of bioactivation is more toxic than the respective DEAQ quinone imine (13, 20), while DEAQ quinone imine is less likely to be formed than AQ quinone imine. This implies that a process that may slow the conversion of AQ to DEAQ may increase the likelihood of AQ quinone imine formation and, consequently, the potential for increased toxicity. We were unable to make associations between side effect occurrence and CYP2C8*2 prevalence in this study, partly because of the limited sample size. It is possible that the lower—albeit insignificant—DEAQ concentrations observed in subjects with CYP2C8 mutant alleles in this study could reflect a slow metabolism phenotype, but this hypothesis requires a larger sample for confirmation.
Recently, a rare CYP2C8 variant allele (CYP2C8*3), which occurs at 15 to 20% prevalence in Caucasian populations but is almost nonexistent in non-Caucasian populations, has been associated with defective metabolism of AQ to an extent higher than that for CYP2C8*2 (23). The CYP2C8*3 variant allele was not found in a previous study in northern Ghana (26). It is possible that this variant allele could have been responsible for cases of AQ-associated agranulocytosis and hepatitis, effects reported almost exclusively for nonimmune, non-African subjects (13, 21).
The main conclusions of this study are that the pharmacokinetics of antimalarials used in combination with artemisinin derivatives should not be extrapolated from the pharmacokinetics of the monotherapy, since the rapid parasite clearance by the artemisinin derivative may influence the pharmacokinetics of the partner drug and the combination. Furthermore, the dosing of AQ for children with uncomplicated malaria should be based on body weight up to 30 kg, and then AQ should be administered as a fixed dose to children of higher body weight. However, our findings were based on one to two data points per patient and thus, while providing potentially important therapeutic information, may have limited accuracy. Further studies based on intense sampling, especially within the distribution phase of AQ, as well as pharmacokinetic studies using measured concentrations of AS, would be an added advantage. Population-based studies to characterize CYP2C8 alleles would be beneficial for safety monitoring of AQ-based antimalarial combinations in areas of endemicity.
Acknowledgments
This study received support from the Danish Council for Development Research (grant 91199) and from the Global Fund for AIDS, TB, and Malaria through the National Malaria Control Programme, Ghana (GHN-202-M03-M-00).
We acknowledge the assistance of the clinical personnel at the malaria research laboratory at the Department of Child Health, Korle Bu Teaching Hospital, and the Korle Bu Polyclinic. We are grateful to Ulla Abildrup and W. J. Asaku for the laboratory analyses. We thank George Amofa, Director of Public Health, Ghana Health Service, and Constance Bart-Plange, Manager, National Malaria Control Programme, Ghana, for support.
Footnotes
Published ahead of print on 8 September 2008.
REFERENCES
- 1.Adjei, G. O., J. A. Kurtzhals, O. P. Rodrigues, M. Alifrangis, L. C. Hoegberg, E. D. Kitcher, E. V. Badoe, R. Lamptey, and B. Q. Goka. 2008. Amodiaquine-artesunate vs artemether-lumefantrine for uncomplicated malaria in Ghanaian children: a randomized efficacy and safety trial with one year follow-up. Malar. J. 7:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashton, M., T. N. Hai, N. D. Sy, D. X. Huong, H. N. Van, N. T. Nieu, and L. D. Cong. 1998. Artemisinin pharmacokinetics is time-dependent during repeated oral administration in healthy male adults. Drug Metab. Dispos. 26:25-27. [PubMed] [Google Scholar]
- 3.Aubouy, A., M. Bakary, A. Keundjian, B. Mbomat, J. R. Makita, F. Migot-Nabias, M. Cot, B. J. Le, and P. Deloron. 2003. Combination of drug level measurement and parasite genotyping data for improved assessment of amodiaquine and sulfadoxine-pyrimethamine efficacies in treating Plasmodium falciparum malaria in Gabonese children. Antimicrob. Agents Chemother. 47:231-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batty, K. T., L. T. Thu, T. M. Davis, K. F. Ilett, T. X. Mai, N. C. Hung, N. P. Tien, S. M. Powell, H. V. Thien, T. Q. Binh, and N. V. Kim. 1998. A pharmacokinetic and pharmacodynamic study of intravenous vs oral artesunate in uncomplicated falciparum malaria. Br. J. Clin. Pharmacol. 45:123-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavaco, I., J. Stromberg-Norklit, A. Kaneko, M. I. Msellem, M. Dahoma, V. L. Ribeiro, A. Bjorkman, and J. P. Gil. 2005. CYP2C8 polymorphism frequencies among malaria patients in Zanzibar. Eur. J. Clin. Pharmacol. 61:15-18. [DOI] [PubMed] [Google Scholar]
- 6.Childs, G. E., E. F. Boudreau, W. K. Milhous, T. Wimonwattratee, N. Pooyindee, L. Pang, and D. E. Davidson, Jr. 1989. A comparison of the in vitro activities of amodiaquine and desethylamodiaquine against isolates of Plasmodium falciparum. Am. J. Trop. Med. Hyg. 40:7-11. [DOI] [PubMed] [Google Scholar]
- 7.Churchill, F. C., D. L. Mount, L. C. Patchen, and A. Bjorkman. 1986. Isolation, characterization and standardization of a major metabolite of amodiaquine by chromatographic and spectroscopic methods. J. Chromatogr. 377:307-318. [DOI] [PubMed] [Google Scholar]
- 8.Churchill, F. C., L. C. Patchen, C. C. Campbell, I. K. Schwartz, P. Nguyen-Dinh, and C. M. Dickinson. 1985. Amodiaquine as a prodrug: importance of metabolite(s) in the antimalarial effect of amodiaquine in humans. Life Sci. 36:53-62. [DOI] [PubMed] [Google Scholar]
- 9.Dai, D. 2001. Allelic frequencies of human CYP2C8 and genetic linkage among different ethnic populations. FASEB J. 15:A575. [Google Scholar]
- 10.Dai, D., D. C. Zeldin, J. A. Blaisdell, B. Chanas, S. J. Coulter, B. I. Ghanayem, and J. A. Goldstein. 2001. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 11:597-607. [DOI] [PubMed] [Google Scholar]
- 11.Finta, C., and P. G. Zaphiropoulos. 2000. The human CYP2C locus: a prototype for intergenic and exon repetition splicing events. Genomics 63:433-438. [DOI] [PubMed] [Google Scholar]
- 12.German, P., B. Greenhouse, C. Coates, G. Dorsey, P. J. Rosenthal, E. Charlebois, N. Lindegardh, D. Havlir, and F. T. Aweeka. 2007. Hepatotoxicity due to a drug interaction between amodiaquine plus artesunate and efavirenz. Clin. Infect. Dis. 44:889-891. [DOI] [PubMed] [Google Scholar]
- 13.Hatton, C. S., T. E. Peto, C. Bunch, G. Pasvol, S. J. Russell, C. R. Singer, G. Edwards, and P. Winstanley. 1986. Frequency of severe neutropenia associated with amodiaquine prophylaxis against malaria. Lancet i:411-414. [DOI] [PubMed] [Google Scholar]
- 14.Hietala, S. F., A. Bhattarai, M. Msellem, D. Roshammar, A. S. Ali, J. Stromberg, F. W. Hombhanje, A. Kaneko, A. Bjorkman, and M. Ashton. 2007. Population pharmacokinetics of amodiaquine and desethylamodiaquine in pediatric patients with uncomplicated falciparum malaria. J. Pharmacokinet. Pharmacodyn. 34:669-686. [DOI] [PubMed] [Google Scholar]
- 15.Hombhanje, F. W., I. Hwaihwanje, T. Tsukahara, J. Saruwatari, M. Nakagawa, H. Osawa, M. M. Paniu, N. Takahashi, J. K. Lum, B. Aumora, A. Masta, M. Sapuri, T. Kobayakawa, A. Kaneko, and T. Ishizaki. 2005. The disposition of oral amodiaquine in Papua New Guinean children with falciparum malaria. Br. J. Clin. Pharmacol. 59:298-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karbwang, J., B. K. Na, A. Thanavibul, D. J. Back, D. Bunnag, and T. Harinasuta. 1994. Pharmacokinetics of mefloquine alone or in combination with artesunate. Bull. W. H. O. 72:83-87. [PMC free article] [PubMed] [Google Scholar]
- 17.Laurent, F., S. Saivin, P. Chretien, J. F. Magnaval, F. Peyron, A. Sqalli, A. E. Tufenkji, Y. Coulais, H. Baba, G. Campistron, P. Regis, P. Ambrooise-Thomas, A. Bryskier, and G. Houin. 1993. Pharmacokinetic and pharmacodynamic study of amodiaquine and its two metabolites after a single oral dose in human volunteers. Arzneimittelforschung 43:612-616. [PubMed] [Google Scholar]
- 18.Li, X. Q., A. Bjorkman, T. B. Andersson, M. Ridderstrom, and C. M. Masimirembwa. 2002. Amodiaquine clearance and its metabolism to N-desethylamodiaquine is mediated by CYP2C8: a new high affinity and turnover enzyme-specific probe substrate. J. Pharmacol. Exp. Ther. 300:399-407. [DOI] [PubMed] [Google Scholar]
- 19.Mariga, S. T., J. P. Gil, W. H. Wernsdorfer, and A. Bjorkman. 2005. Pharmacodynamic interactions of amodiaquine and its major metabolite desethylamodiaquine with artemisinin, quinine and atovaquone in Plasmodium falciparum in vitro. Acta Trop. 93:221-231. [DOI] [PubMed] [Google Scholar]
- 20.Naisbitt, D. J., J. E. Ruscoe, D. Williams, P. M. O'Neill, M. Pirmohamed, and B. K. Park. 1997. Disposition of amodiaquine and related antimalarial agents in human neutrophils: implications for drug design. J. Pharmacol. Exp. Ther. 280:884-893. [PubMed] [Google Scholar]
- 21.Neftel, K. A., W. Woodtly, M. Schmid, P. G. Frick, and J. Fehr. 1986. Amodiaquine induced agranulocytosis and liver damage. Br. Med. J. (Clin. Res. Ed.) 292:721-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ong, C. E., S. Coulter, D. J. Birkett, C. R. Bhasker, and J. O. Miners. 2000. The xenobiotic inhibitor profile of cytochrome P4502C8. Br. J. Clin. Pharmacol. 50:573-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parikh, S., J. B. Ouedraogo, J. A. Goldstein, P. J. Rosenthal, and D. L. Kroetz. 2007. Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin. Pharmacol. Ther. 82:197-203. [DOI] [PubMed] [Google Scholar]
- 24.Pearce, R. J., C. Drakeley, D. Chandramohan, F. Mosha, and C. Roper. 2003. Molecular determination of point mutation haplotypes in the dihydrofolate reductase and dihydropteroate synthase of Plasmodium falciparum in three districts of northern Tanzania. Antimicrob. Agents Chemother. 47:1347-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pussard, E., and F. Verdier. 1994. Antimalarial 4-aminoquinolines: mode of action and pharmacokinetics. Fundam. Clin. Pharmacol. 8:1-17. [DOI] [PubMed] [Google Scholar]
- 26.Röwer, S., U. Bienzle, A. Weise, U. Lambertz, T. Forst, R. N. Otchwemah, A. Pfutzner, and F. P. Mockenhaupt. 2005. High prevalence of the cytochrome P450 2C8*2 mutation in Northern Ghana. Trop. Med. Int. Health 10:1271-1273. [DOI] [PubMed] [Google Scholar]
- 27.Simpson, J. A., L. Aarons, and N. J. White. 2001. How can we do pharmacokinetic studies in the tropics? Trans. R. Soc. Trop. Med. Hyg. 95:347-351. [DOI] [PubMed] [Google Scholar]
- 28.Svensson, U. S., and M. Ashton. 1999. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br. J. Clin. Pharmacol. 48:528-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor, W. R., D. J. Terlouw, P. L. Olliaro, N. J. White, P. Brasseur, and F. O. ter Kuile. 2006. Use of weight-for-age-data to optimize tablet strength and dosing regimens for a new fixed-dose artesunate-amodiaquine combination for treating falciparum malaria. Bull. W. H. O. 84:956-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White, N. J., S. Looareesuwan, G. Edwards, R. E. Phillips, J. Karbwang, D. D. Nicholl, C. Bunch, and D. A. Warrell. 1987. Pharmacokinetics of intravenous amodiaquine. Br. J. Clin. Pharmacol. 23:127-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White, N. J., F. Nosten, S. Looareesuwan, W. M. Watkins, K. Marsh, R. W. Snow, G. Kokwaro, J. Ouma, T. T. Hien, M. E. Molyneux, T. E. Taylor, C. I. Newbold, T. K. Ruebush, M. Danis, B. M. Greenwood, R. M. Anderson, and P. Olliaro. 1999. Averting a malaria disaster. Lancet 353:1965-1967. [DOI] [PubMed] [Google Scholar]
- 32.Winstanley, P. A., O. Simooya, J. M. Kofi-Ekue, O. Walker, L. A. Salako, G. Edwards, M. L. Orme, and A. M. Breckenridge. 1990. The disposition of amodiaquine in Zambians and Nigerians with malaria. Br. J. Clin. Pharmacol. 29:695-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wongsrichanalai, C., A. L. Pickard, W. H. Wernsdorfer, and S. R. Meshnick. 2002. Epidemiology of drug-resistant malaria. Lancet Infect. Dis. 2:209-218. [DOI] [PubMed] [Google Scholar]
- 34.Yasar, U., S. Lundgren, E. Eliasson, A. Bennet, B. Wiman, U. de Faire, and A. Rane. 2002. Linkage between the CYP2C8 and CYP2C9 genetic polymorphisms. Biochem. Biophys. Res. Commun. 299:25-28. [DOI] [PubMed] [Google Scholar]