

Abstract
Background
Porencephaly (cystic cavities of the brain) is caused by perinatal vascular accidents from various causes. Several familial cases have been described and autosomal dominant inheritance linked to chromosome 13q has been suggested. COL4A1 is an essential component in basal membrane stability. Mouse mutants bearing an in‐frame deletion of exon 40 of Col4a1 either die from haemorrhage in the perinatal period or have porencephaly in survivors. A report of inherited mutations in COL4A1 in two families has shown that familial porencephaly may have the same cause in humans.
Objective
To describe three novel COL4A1 mutations.
Results
The three mutations occurred in three unrelated Dutch families. There were two missense mutations of glycine residues predicted to result in abnormal collagen IV assembly, and one mutation predicted to abolish the traditional COL4A1 start codon. The last mutation was also present in an asymptomatic obligate carrier with white matter abnormalities on brain magnetic resonance imaging.
Conclusions
This observation confirms COL4A1 as a major locus for genetic predisposition to perinatal cerebral haemorrhage and porencephaly and suggests variable expression of COL4A1 mutations.
Keywords: porencephaly, stroke, basement membrane, collagen IV, COL4A1
The term porencephaly is used to indicate a fluid filled cavity in the brain. Congenital porencephaly often causes hemiplegia, but also tetraplegia, epilepsy, mental retardation, and dystonia, and optic and pituitary defects when involving deep midline structures.1,2,3
Encephaloclastic (disruptive) porencephaly has been ascribed to a perinatal parenchymal insult to the brain,4 and a familial predisposition with dominant inheritance has been widely documented.5,6,7,8,9,10,11,12,13 Occurrence of other complaints such as stroke, vascular aneurysm, and migraine in pedigrees with familial porencephaly has suggested a genetic predisposition to “vascular liability.”13 It has been suggested that thrombophilia is associated with familial porencephaly,14 but trauma,15,16 maternal disease, and infections are also considered risk factors for sporadic porencephaly.17,18 There has been discussion as to whether the occurrence of porencephaly only at specific periods of intrauterine life (the end of gestation or in general after the 20th gestational week) might be caused by the mutation of a gene specific for brain development.13 A locus for familial porencephaly has recently been described on chromosome 13qter.19 The description of a mouse model where a Col4a1 mutation leads to perinatal cerebral haemorrhage and to porencephaly has shed light on the pathogenesis of this disorder.16 Collagen IV is important for structural integrity and function of basement membrane.20 Together with COL4A2, COL4A1 is the most abundant component of type IV collagen in basement membrane.21,22 These two proteins assemble to form a heterotrimeric triple helix of the type α1.α1.α2(IV), forming an hexamer with itself or with a triple helix α5.α5.α6(IV).23 Mice bearing a heterozygote Col4a1 mutation leading to in‐frame deletion of exon 40, are prone to brain haemorrhage at birth.16 This mutation leads to the synthesis of an abnormal COL4A1 which cannot be properly secreted outside the cell.16 In two families, mutations in conserved glycines of the Gly‐X‐Y repeats in the triple helix domain of COL4A1 have been associated with porencephaly.16
One of the issues raised in familial dominant porencephaly concerns individuals who appear to be non‐manifesting obligate carriers.12,13 It has been suggested that magnetic resonance imaging (MRI) fails to identify asymptomatic carriers.12 In contrast, other observations suggest that subcortical and periventricular white matter lesions resembling gliosis are present in obligate carriers and might aid carrier detection.8,13 Another issue concerns the occurrence of strokes at older age in congenital porencephaly families, as previously reported.13,16
We report three novel mutations in COL4A1 in three unrelated Dutch families with an autosomal dominant predisposition to porencephaly. Clinical and MRI findings of two of these families have been described in detail previously.13
Methods
Mutation analysis of COL4A1
Written informed consent was obtained from all subjects. Genomic DNA was isolated from peripheral blood using standard protocols.
The primers were designed to amplify the 52 exons including at least 50 bases of flanking genomic sequences based on the reference sequence of COL4A1 as deposited in GeneBank (accession number for the mRNA NM_001845 and for the COL4A1 gene Entrez GeneID 1282).
Amplification reactions (exon 3 to exon 52) were carried out in 21 μl containing 1×polymerase chain reaction (PCR) buffer (Invitrogen, San Diego, California, USA), 1.5 mM MgCl2, 0.01% W‐1, 250 μM of each dNTP, 1 μM forward primer, 1 μM reverse primer, 0.75 units of platinum Taq DNA polymerase (Invitrogen), and 25 ng genomic DNA. Exons 1 and 2 were amplified in 20 μl containing 1×GCII TaKaRa, 400 μM of each dNTP, 1 μM forward primer, 1 μM reverse primer, 1 unit of LA Taq DNA polymerase (TaKaRa), and 25 ng genomic DNA.
Cycle conditions were: seven minutes 30 seconds at 95°C; 10 cycles of 30 seconds denaturation at 94°C, annealing at 68°C minus 1°C per cycle, one minute extension at 72°C, followed by 25 cycles of 30 seconds denaturation at 94°C, annealing at 58°C, one minute extension at 72°C; and final extension of five minutes at 72°C. The designed primer sequences are available upon request (from GB).
Templates for the direct sequencing reactions were cleaned from dNTPs and primers using 2 μl ExoSAP‐IT (US Biochemical, Cleveland, Ohio, USA) during 15 minutes at 37°C, followed by a 15 minutes inactivation step at 80°C. Direct sequencing of both strands was undertaken using Big Dye Terminator chemistry version 3.1 (Applied Biosystems, Foster City, California, USA) as recommended by the manufacturer. Fragments were loaded on an ABI3100 automated sequencer and analysed with DNA Sequencing Analysis (version 3.7) and SeqScape (version 2.1) software (Applied Biosystems).
Templates for the SNaPshot reactions (3389G→A; exon 39, and 4267G→C; exon 48) were cleaned from dNTPs and primers using 2 μl ExoSAP‐IT (US Biochemical) during 15 minutes at 37°C followed by a 15 minute inactivation step at 80°C. About 20 ng of pooled product were used in a primer extension reaction including the primers shown in table 1.
Table 1 Primers used.
| Mutation | Exon | Extension primer 5′→3′ | Strand | Size (bp) |
|---|---|---|---|---|
| 3389G→A | 39 | caaaggcctcccaggattggatggcatccctg | Sense | 32 |
| 4267G→C | 48 | tctcctatcttccaggtccaagaggatttcca | Sense | 32 |
bp, base pair.
Reactions were carried out in 10 μl containing 1 µl SNaPshot multiplex ready reaction mix (Applied Biosystems), 2.5 μM extension primer, and 1 μl ½ term buffer (200 mM Tris HCl; 5 mM MgCl2 pH 9). Additional thermal cycling was carried out (40 cycles of 10 seconds at 95°C; five seconds at 50°C, and 30 seconds at 60°C). Removal of the 5′ phosphoryl groups was achieved using one unit of shrimp alkaline phosphatase (SAP) (Roche Products, Indianapolis, Indiana, USA) for 30 minutes at 37°C. After the addition of 10 μl Hi‐Di formamide (Applied Biosystems) containing GeneScan‐120 LIZ size standard (Applied Biosystems) to 1 μl SnaPshot products, samples were heated for five minutes at 95°C, placed on ice, and loaded on an ABI3100 genetic analyser (Applied Biosystems). Fragments were analysed using GeneMapper V3.0 software (Applied Biosystems).
Digestion of exon 1 PCR product was undertaken by adding 1× NEBuffer 4 (New England Biolabs, Beverly, Massachussets, USA) and 10 units of NcoI, incubating for two hours at 37°C. The 1A→T mutation generates a NcoI restriction size cutting the 539 base pair (bp) sized exon 1 in two fragments of 317 and 222 bases, respectively. Fragments were analysed on a 2% agarose gel.
Case reports
Pedigrees are shown in fig 1. Families A and B have been described previously, under the same title, by Mancini et al.13

Figure 1 Pedigrees of families A, B, and C with respective mutations in COL4A1. Black symbols indicate individuals with neurological impairment and radiological evidence of porencephaly on magnetic resonance imaging (MRI) or computed tomography (CT). Individual A‐II‐2 is indicated by a dot as she is an obligate carrier and has white matter lesions on MRI (see figure 5 in reference 13 for MRI pictures). Similarly, subject B‐II‐1 has pyramidal signs and white matter changes but no porencephaly on MRI. She refused DNA tests. For further explanation see case reports.
Family A
Patient A‐IV‐1 showed a left sided hemiparesis and an occipito‐frontal head circumference –1 SD from the normal mean at the age of 1.5 years,. Brain MRI at that age showed cystic dilatation of the right lateral ventricle in the frontal and parietal area, reaching the cortex and surrounded by a thin layer of white matter. The whole periventricular white matter (down to the occipital area) showed bilateral patches of high signal intensity on T2 weighted images, low signal intensity on T1 weighted images, and hyperintensity on proton density imaging, suggesting gliosis (see figure 3 in reference 13). The right internal capsule was thinned and difficult to identify. There was a clear asymmetry of the cerebral peduncles, with signs of wallerian degeneration of the right peduncle. The left ventricle was not enlarged. The corpus callosum was thin. Coagulation status was normal.

Figure 3 Sequence electropherograms of mutations found in COL4A1. Unaffected and affected DNA and amino acid sequences are shown. From top to bottom: Family A, heterozygous A to T transversion (W) at position 1 (1 A→T) resulting in no protein or to a translation initiation site moving upstream or downstream; the absence of translation into an amino acid is indicated by an asterisk; Family B, heterozygous G to A base change (R) at position 3389 (3389 G→A) resulting in a substitution of a glycine by aspartic acid arginine at codon 1130 (G1130D); Family C, heterozygous G to C base change (S) leading to a substitution of a glycine by arginine at codon 1423 (G1423R).
In patient A‐III‐1, neglect for the right arm was noted at the age of five months. At the age of 15 months a right sided hemiplegia was diagnosed. She attended regular high school and has remained healthy except for attacks of common migraine. Brain MRI at the age of 27 years showed irregularity of the edge of the middle part of the left lateral ventricle and a porencephalic cystic dilatation and bilateral disseminated white matter lesions, with a secondary wallerian degeneration of the left cerebral peduncle, which was thinned (see figure 4 in reference 13).

Figure 4 Sequence alignment of COL4A1 orthologues in seven different species, showing conservation of the Gly residues in position 1130 of exon 39 (mutated in family B to an aspartate residue) and in position 1423 of exon 42 (mutated into an arginine residue in family C).
In patient A‐IV‐2, brain MRI was undertaken at the age of four years because of recurrent attacks of vertigo. It was normal.
Patient A‐II‐2 has migraine and presents with a small right infratentorial meningioma. Angiography of the cerebral arteries also revealed an aneurysm on the top of the right carotid artery. Brain MRI studies showed bilateral scattered areas of high signal intensity on T2 weighted images in the periventricular white matter but no porencephaly (see figure 5 in reference 13).
Patients A‐II‐3 and A‐II‐4 are both known to have congenital hemiplegia. Patient A‐II‐3 had a stroke at the age of 49 years. From the medical records, cystic dilatation of the ventricles was present on CT (scan not reviewed by us). Patient A‐II‐4 suffers from migraine attacks. Brain CT of patient A‐II‐4 at the age of 40 years showed diffuse dilatation of the right lateral ventricle, most abnormal in the parietal and occipital areas (see figure 6 in reference 13).
Family B
Patient B‐III‐1 was examined at the age of six years because of unexplained mild left sided hemiparesis and moderate psychomotor retardation. The pregnancy and birth had been uneventful. Brain MRI at the age of eight years showed a thin corpus callosum, smooth dilatation of part of the frontal horn of the right lateral ventricle, surrounded by a thin wall, and asymmetry of the gyral pattern in the cortex covering the cyst, without overt signs of cortical dysplasia or abnormal signal intensity (see figure 7 in reference 13).
Patient B‐III‐2 was examined at the age of two years because of left sided hemiplegia.
At the age of four years she developed focal epilepsy. Brain CT at that age showed cystic dilatation of the frontal horn and the medial part of the right lateral ventricle (data not shown). She also has mild mental handicap.
Neurological examination of patient B‐II‐4 at the age of 38 years showed a mild pyramidal syndrome and positive Babinski sign in the right lower limb. Brain MRI at the same age revealed a cystic cavity adjacent to the frontal horn of the left lateral ventricle, communicating with the ventricle, and contralateral white matter hyperintensity in frontal areas, apparently localised to the frontal watershed area (see figure 8 in reference 13). His father and his half sister (patient B‐II‐1) were known to limp.
Neurological examination of patient B‐II‐1 at the age of 50 years revealed left sided pyramidal signs, while brain MRI showed bilateral patchy areas of abnormal signal intensity of the cerebral white matter on T2 weighted images, but no porencephalic cyst (see figure 9 in reference 13). Patient B‐II‐2 has severe mental retardation, epilepsy, and a normal neurological examination and MRI. Patient B‐II‐3 experienced a stroke at the age of 55 years but refused access to medical records. Individuals B‐II‐1, B‐II‐2, and B‐II‐3 refused DNA tests.
Family C
Family C consists of a two year old boy (C‐III‐1) and his mother, both with congenital right hemiplegia and normal cognition. The pregnancy was uneventful but delivery was characterised by prolonged labour, though the Apgar scores were normal. His mother (C‐II‐2) also had right sided hemiplegia and right hand dystonia with atrophy of the right leg from infancy. The maternal grandmother is known to have a “thin leg” but has not been investigated. Brain MRI of the boy showed dilatation of the left ventricle in the frontoparietal area without cortical or basal ganglia abnormality. MRI of his mother showed left ventricular dilatation and atrophy of the left thalamus (fig 2). Coagulation tests and DNA analysis of factor II and factor V Leiden are normal in both mother and son.
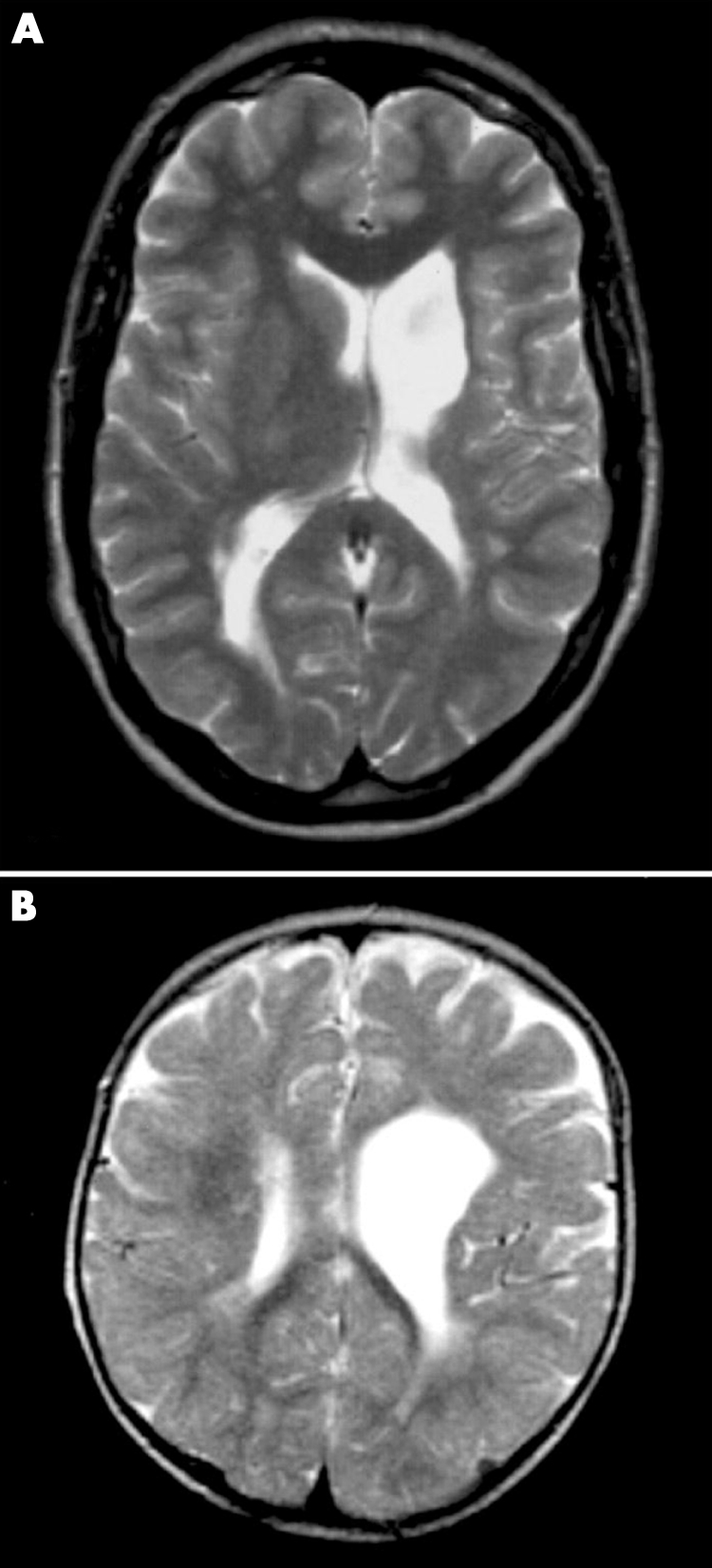
Figure 2 Brain magnetic resonance imaging of patients C‐II‐2 and C‐III‐1. (A) T2 weighted TSE axial image of patient C‐II‐2 at the age of 25 years, showing a porencephalic enlargement of the left ventricle, ipsilateral atrophy of the caudate nucleus and thalamus, and subtle bilateral occipital and right subcortical frontal white matter hyperintensity. (B) T2 weighted TSE axial image of patient C‐III‐1 at the age of one year, showing porencephalic enlargement of the left ventricle in the frontal area. Bilateral occipital white matter signal intensity is interpreted as the normal “end zone” of myelination, but patches of white matter hyperintensity are also visible around the right frontal horn.
Results
Clinical and molecular data are summarised in table 2.
Table 2 Clinical and neuroimaging findings in the porencephaly families (modified from reference 13).
| Patients | Laterality of cyst | Localisation | White matter T2 hyperintensity | Signs and symptoms | Vascular area | COL4A1 mutation |
|---|---|---|---|---|---|---|
| A‐IV‐1 | Unilateral | Right parieto‐occipital | Bilateral diffuse gliosis | Left hemiparesis | Vena terminalis | + |
| A‐III‐2 | Unilateral | Left frontotemporal | Bilateral diffuse | Right hemiparesis | + | |
| A‐II‐4 | Unilateral | Right frontoparietal and occipital | Not visible on CT | Left hemiparesis | Vena terminalis | Test refused |
| A‐II‐2 | No cyst | – | Bilateral diffuse, frontal to occipital | None (migraine) | ||
| B‐III‐1 | Unilateral | Right frontotemporal | Left frontotemporal | Left hemiparesis; mental retardation | Vena terminalis | + |
| B‐II‐4 | Unilateral | Left frontal | Right frontal, centrum semiovale (multiple foci) | Right sided pyramidal signs | Right frontal watershed area | + |
| B‐III‐2 | Unilateral | Right frontotemporal | Not visible on CT | Left hemiparesis; focal epilepsy | + | |
| B‐II‐1 | No cyst | – | Bilateral diffuse | Limp; left hyperreflexia | Test refused | |
| C‐III‐1 | Unilateral | Left frontal | Right frontal | Right hemiparesis | Vena terminalis | + |
| C‐II‐2 | Unilateral | Left frontal | Bilateral occipital, right frontal subcortical | Right hemiplegia and hand dystonia | Vena terminalis and branches of the vena cerebri interna | + |
CT, computed tomography.
Genomic sequencing of COL4A1 revealed the base changes detailed in table 3. In family A, an heterozygote mutation in the start codon of the gene1A→T in exon 1 was found cosegregating with the disease in the index patients A‐IV‐1, A‐III‐2, and A‐II‐4 (fig 1A). Subject A‐II‐3 agreed to share his medical records but refused a DNA test. Subject A‐II‐2, considered an obligate carrier, also has the same mutation. The c.1A→T change was not found in more than 350 ethnically matched control chromosomes tested and is predicted to eliminate the ATG start codon of COL4A1 (fig 3A), resulting in no protein or in a translation initiation site moving upstream or downstream. The next in‐frame ATG can be found 193 nucleotides downstream (base 277 in ref seq NM_01845) and, if used, it may lead to the synthesis of a protein lacking 64 N‐terminal amino acids. Although we cannot confirm the synthesis of an abnormal protein in affected family members, disease cosegregation and absence of the mutation in controls suggest pathogenicity.
Table 3 COL4A1 mutations found in this study.
| Family | Exon | DNA | Codon effect | Protein effect |
|---|---|---|---|---|
| A | 1 | 1A→T | ATG‐→TTG | Unknown |
| B | 39 | 3389G→A | GGT→GAT | G1130D |
| C | 48 | 4267G→C | GGT→CGT | G1423R |
Sequencing in family B showed a missense c.3389G→A change in exon 39 (fig 3B), leading to a p.G1130D, which co‐segregates with the disease. This mutation is in one of the Gly‐X‐Y repeats of COL4A1 and was not present in more than 300 control chromosomes and is conserved in seven different species from primates through nematodes (fig 4A). Compared with glycine, the replacing aspartate is a larger amino acid introducing a negatively charged side chain which probably interferes with triple helix assembly.
In family C we observed a missense c.4267G→C in exon 48 leading to a p.G1423R substitution in patients C‐III‐1 and C‐II‐2. This mutation was not present in the unaffected father (C‐II‐1) or in 370 ethnically matched control chromosomes. The mutation also disrupts a highly conserved glycine in a Gly‐X‐Y repeat (fig 4B), introducing a large positively charged amino acid. Gly→Ser and Gly→Arg changes in Gly‐X‐Y repeats have previously been associated with familial porencephaly.16 We therefore conclude that these mutations at Gly‐X‐Y sites are likely to be pathogenic.
Discussion
We report three new mutations in COL4A1 in three unrelated Dutch families with dominant porencephaly. Two of the mutations affect highly conserved glycines of Gly‐X‐Y repeats within the collagenous domain of the protein which is known to interact with COL4A2 to form a collagen IV triple helix.
A mouse model for perinatal cerebral haemorrhage and porencephaly shows an in‐frame deletion of exon 40 of Col4a1. Mutations in conserved Gly‐X‐Y repeats of COL4A1 were subsequently found in two other families with autosomal dominant porencephaly.16 Immunohistochemistry and electronmicroscopy of Col4a1 mutants indicate impaired secretion of both COL4A1 and COL4A2. Similarly, a mutation of the C elegans orthologue of Col4a1 (let‐2) results in impaired secretion and intracellular accumulation of both let‐2 and the Col4a2 orthologue emb‐9.24 Mice homozygous for Col4a1Δex4016 or a null allele20 are not viable, and heterozygous null mice have no apparent phenotype.20 However, 50% of heterozygous Col4a1+/Δex40 mice die following parturition and 18% of the survivors show obvious porencephalic lesions. This suggests that synthesis of a mutant protein in Col4a1+/Δex40 mice has a negative effect on survival. Experimentally, a negative effect of Col4a1+/Δex40 mutation was demonstrated on collagen IV triple helix assembly and secretion.16 Mutations in highly conserved Gly‐X‐Y domains have been shown in several collagen proteins, leading to a dominant negative effect.25,26 Based on these observations, synthesis of an abnormal protein can be predicted in our families B and C.
The consequence of the mutation in family A is more difficult to predict. One possibility is that this is an effective null allele and a transcript is only produced from the normal allele. However, evidence from model organisms shows no obvious phenotype in heterozygotes for null alleles and suggests that dominant interfering proteins are necessary for pathogenesis. Thus a null mutation is not expected to be pathogenic and it is unlikely that the mutation in family A is a null allele. Following the conserved initiation codon, the next in‐frame start codon contains a pyrimidine at the ‐3 position, which is highly conserved with respect to translation initiation sites and suggests that this start site might be used.27 Initiation of translation at this second site would result in synthesis of a protein with a 64 amino acid N‐terminal truncation but with an intact NC1 domain and collagenous domain. We predict that the NC1 domain of the mutant trimer is able to initiate assembly of heterotrimers but that the heterotrimers would be structurally or functionally abnormal because of the N‐terminal truncation. Further insights into the effect of this mutation will result from the introduction of this start codon mutation in mice, or functional studies on the COL4A1/2 protein in patients from family A. Future in vitro expression studies of the mutant protein by our group are also aimed at confirming this assumption.
The gliotic lesions in our families indicate that the time of onset of porencephaly is related to hypoxic‐ischaemic events occurring in a late stage of pregnancy (after the 20th week).13 This is compatible with the specific function of COL4A1 in the formation of α1.α1.α2.(IV) protomer, which is expressed in early embryonic development (from the 32 to 64 cell stage of the mouse embryo) but is not essential for basement membrane deposition. Instead, its essential function involves the maintenance of the structural integrity of the basement membranes at later stages—that is, later fetal life.20 Its total ablation leads to embryonic lethality only at E10.5–11.5 because of impaired basement membrane stability.20 In this respect, localisation of the white matter lesions in areas draining from the vena terminalis in our families A and C and in the watershed area between the anterior and medial cerebral arteries in patient B‐II‐413 (table 2) are compatible with abnormalities of the vascular basement membrane as a result of COL4A1 mutations.
Our findings also confirm that in asymptomatic carriers white matter lesions on MRI can be considered an expression of (and perhaps a risk factor for) COL4A1 mutation.
A history of strokes in middle age in patients A‐II‐3 and B‐II‐2 could not be correlated with COL4A1 abnormalities as these patients refused DNA tests. However, recurrent strokes in COL4A1 related porencephaly have also been observed by Gould et al,16 and additional patients need to be tested to prove this relation. Further evidence is needed to determine whether other neurological complaints in our families, such as the carotid artery aneurysm in patient A‐II‐2,13 recurrent migraine in family A, and mental retardation in family B, are also related to COL4A1 mutations.
Porencephaly in our families occurs in areas where traumatic perinatal arterial bleeding (watershed areas)14,15 or venous thrombotic events also occur,13 and white matter lesions are seen in asymptomatic carriers of COL4A1 mutations. This suggests that trauma28,29 and thrombophilia14,30 could be factors influencing the occurrence of cerebral bleeding in COL4A1 mutants. In this case genetic testing for COL4A1 mutations in families at risk could aid counselling or suggest the need for additional perinatal care to avoid a traumatic delivery.
Acknowledgements
We thank the families participating in this study and referring physicians Dr P W J van Mossevelde and Dr N J Langendoen. We also thank Dr P Govaert for useful discussions.
Footnotes
Conflicts of interest: none declared
References
- 1.Zeki S M, Hollman A S, Dutton G N. Neuroradiological features of patients with optic nerve hypoplasia. J Pediatr Ophthalmol Strabismus 199229107–112. [DOI] [PubMed] [Google Scholar]
- 2.Barkovich A J, Kjos B O. Schizencephaly: correlation of clinical findings with MRI characteristics. Am J Neuroradiol 19921385–94. [PMC free article] [PubMed] [Google Scholar]
- 3.Lubinsky M S. Hypothesis: septo‐optic dysplasia is a vascular disruption sequence. Am J Med Genet 199769235–236. [PubMed] [Google Scholar]
- 4.Cross J H, Harrison C J, Preston P R, Rushton D I, Newell S J, Morgan M E, Durbin G M. Postnatal encephaloclastic porencephaly – a new lesion? Arch Dis Child 199267307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haar L, Dyken P. Hereditary nonprogressive athetotic hemiplegia: a new syndrome. Neurology 197727849–854. [DOI] [PubMed] [Google Scholar]
- 6.Berg R A, Aleck K A, Kaplan A M. Familial porencephaly. Arch Neurol 198340567–569. [DOI] [PubMed] [Google Scholar]
- 7.Airaksinen E. Familial porencephaly. Clin Genet 198426236–238. [Google Scholar]
- 8.Smit L M, Barth P G, Valk J, Njiokiktjien C. Familial porencephalic white matter disease in two generations. Brain Dev 1984654–58. [DOI] [PubMed] [Google Scholar]
- 9.Zonana J, Adornato B T, Glass S T, Webb M J. Familial porencephaly and congenital hemiplegia. J Pediatr 1986109671–674. [DOI] [PubMed] [Google Scholar]
- 10.Sensi A, Cerruti S, Calzolari E, Vesce F. Familial porencephaly. Clin Genet 199038396–400. [PubMed] [Google Scholar]
- 11.Herranz Fernandez J L, Moreno Belzue C, Arce Garcia J L, Arteaga Manjon‐Cabeza R. [Congenital familial hemiparesis and familial porencephaly]. An Esp Pediatr 199237431–433. [PubMed] [Google Scholar]
- 12.Vilain C, Van Regemorter N, Verloes A, David P, Van Bogaert P. Neuroimaging fails to identify asymptomatic carriers of familial porencephaly. Am J Med Genet 2002112198–202. [DOI] [PubMed] [Google Scholar]
- 13.Mancini G M, de Coo I F, Lequin M H, Arts W F. Hereditary porencephaly: clinical and MRI findings in two Dutch families. Eur J Paediatr Neurol 2004845–54. [DOI] [PubMed] [Google Scholar]
- 14.Debus O, Koch H G, Kurlemann G, Strater R, Vielhaber H, Weber P, Nowak‐Gottl U. Factor V Leiden and genetic defects of thrombophilia in childhood porencephaly. Arch Dis Child Fetal Neonatal Ed 199878F121–F124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasternak J F, Mantovani J F, Volpe J J. Porencephaly from periventricular intracerebral hemorrhage in a premature infant. Am J Dis Child 1981134673–675. [DOI] [PubMed] [Google Scholar]
- 16.Gould D B, Campbell Phalan F, van der Knaap M S, van Mil S, Schimenti J C, Aguglia U, Breedveld G J, Heutink P, John S W. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 20053081167–1171. [DOI] [PubMed] [Google Scholar]
- 17.Miller S P, Ramaswamy V, Michelson D, Barkovich A J, Holshouser B, Wycliffe N, Glidden D V, Deming D, Partridge J C, Wu Y W, Ashwal S, Ferriero D M. Patterns of brain injury in term neonatal encephalopathy. J Pediatr 2005146453–460. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Croen L A, Backstrand K H, Yoshida C K, Henning L H, Lindan C, Ferriero D M, Fullerton H J, Barkovich A J, Wu Y W. Maternal and infant characteristics associated with perinatal arterial stroke in the infant. JAMA 2005293723–729. [DOI] [PubMed] [Google Scholar]
- 19.Aguglia U, Gambardella A, Breedveld G J, Oliveri R L, Le Piane E, Messina D, Quattrone A, Heutink P. Suggestive evidence for linkage to chromosome 13qter for autosomal dominant type 1 porencephaly. Neurology 2004621613–1615. [DOI] [PubMed] [Google Scholar]
- 20.Poschl E, Schlotzer‐Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 20041311619–1628. [DOI] [PubMed] [Google Scholar]
- 21.Urabe N, Naito I, Saito K, Yonezawa T, Sado Y, Yoshioka H, Kusachi S, Tsuji T, Ohtsuka A, Taguchi T, Murakami T, Ninomiya Y. Basement membrane type IV collagen molecules in the choroid plexus, pia mater and capillaries in the mouse brain. Arch Histol Cytol 200265133–143. [DOI] [PubMed] [Google Scholar]
- 22.Sado Y, Kagawa M, Naito I, Ueki Y, Seki T, Momota R, Oohashi T, Ninomiya Y. Organization and expression of basement membrane collagen IV genes and their roles in human disorders. J Biochem (Tokyo) 1998123767–776. [DOI] [PubMed] [Google Scholar]
- 23.Borza D B, Bondar O, Ninomiya Y, Sado Y, Naito I, Todd P, Hudson B G. The NC1 domain of collagen IV encodes a novel network composed of the alpha 1, alpha 2, alpha 5, and alpha 6 chains in smooth muscle basement membranes. J Biol Chem 200127628532–28540. [DOI] [PubMed] [Google Scholar]
- 24.Gupta M C, Graham P L, Kramer J M. Characterization of alpha1(IV) collagen mutations in Caenorhabditis elegans and the effects of alpha1 and alpha2(IV) mutations on type IV collagen distribution. J Cell Biol 19971371185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brodsky B, Shah N K. Protein motifs. 8. The triple‐helix motif in proteins. FASEB J 199591537–1546. [DOI] [PubMed] [Google Scholar]
- 26.Gaiser K G, Maddox B K, Bann J G, Boswell B A, Keene D R, Garofalo S, Horton W A. Y‐position collagen II mutation disrupts cartilage formation and skeletal development in a transgenic mouse model of spondyloepiphyseal dysplasia. J Bone Miner Res 20021739–47. [DOI] [PubMed] [Google Scholar]
- 27.Kozak M. An analysis of 5′‐noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 1987158125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viljoen D L. Porencephaly and transverse limb defects following severe maternal trauma in early pregnancy. Clin Dysmorphol 1995475–78. [PubMed] [Google Scholar]
- 29.Sherer D M, Anyaegbunam A, Onyeije C. Antepartum fetal intracranial hemorrhage, predisposing factors and prenatal sonography: a review. Am J Perinatol 199815431–441. [DOI] [PubMed] [Google Scholar]
- 30.Debus O M, Kosch A, Strater R, Rossi R, Nowak‐Gottl U. The factor V G1691A mutation is a risk for porencephaly: a case‐control study. Ann Neurol 200456287–290. [DOI] [PubMed] [Google Scholar]