

Abstract
Bacterial quorum sensing is mediated by low molecular-weight signals and plays a critical role in both the pathogenesis of infectious disease and beneficial symbioses. There is significant interest in the development of synthetic ligands that can intercept bacterial quorum sensing signals and modulate these outcomes. Here, we report the design and comparative analysis of the effects of ~ 90 synthetic N-acylated homoserine lactones (AHLs) on quorum sensing in three Gram negative bacterial species and a critical examination of the structural features of these ligands that dictate agonistic and antagonistic activity, and selectivity for different R protein targets. These studies have revealed the most comprehensive set of structure–activity relationships to date that direct AHL-mediated quorum sensing and a new set of chemical probes with which to study this complex signaling process. Furthermore, this work provides a foundation on which to design next-generation quorum sensing modulators with improved activities and selectivities.
Keywords: combinatorial chemistry, Gram-negative bacteria, homoserine lactones, quorum sensing, structure activity relationships
Introduction
Bacteria use small molecules and peptides to assess their local population densities in a process termed quorum sensing (QS).[1, 2] When they reach a sufficiently high population density (or a “quorum”), bacteria can alter gene expression to behave as a group and initiate processes that play central roles in both pathogenesis and beneficial symbioses.[3, 4] These group behaviors are remarkable in their diversity, ranging from virulence factor and antibiotic production to biofilm formation, root nodulation, and bioluminescence, and have direct and often devastating impacts on the bacterial host. As QS depends on a relatively simple language of low molecular weight compounds, there is intense and growing interest in the design of non-native molecules that can intercept QS signals and modulate these important outcomes.[5, 6] These synthetic ligands would represent valuable molecular probes for studying the fundamental mechanisms of QS and elucidating the roles of this chemical signaling process in host/bacteria interactions. Such studies are essential for the continued evaluation of QS as a new therapeutic target.[3-6]
QS is best characterized in the Gram negative proteobacteria, and thus the majority of research on synthetic modulators of QS has focused on these signaling pathways.[5-8] Proteobacteria use diffusible N-acylated l-homoserine lactones (AHLs) as their primary signaling molecules (Scheme 1); these ligands are produced by AHL synthases (or I proteins) and are sensed by cytoplasmic receptors (or R proteins) that behave as transcription factors. At low cell densities, bacteria constitutively produce the AHL synthase, and thus the AHL ligand, at low levels. As the bacterial colony grows, however, the local concentration of AHL will likewise increase and eventually reach a threshold level at which the AHL will bind to its cognate R protein. Thereafter, the AHL–R protein complex will most often dimerize and bind adjacent to QS promoters to activate the transcription of genes required for bacterial group behaviors. This signaling pathway was first described in the bioluminescent marine symbiont Vibrio fischeri and has been characterized in over 50 different proteobacteria to date.[7, 8] Many of these bacteria are clinically, environmentally, and industrially important, perhaps most notably the opportunistic pathogen Pseudomonas aeruginosa, which uses QS to control virulence factor production and growth into drug impervious biofilms.[3]
Scheme 1.
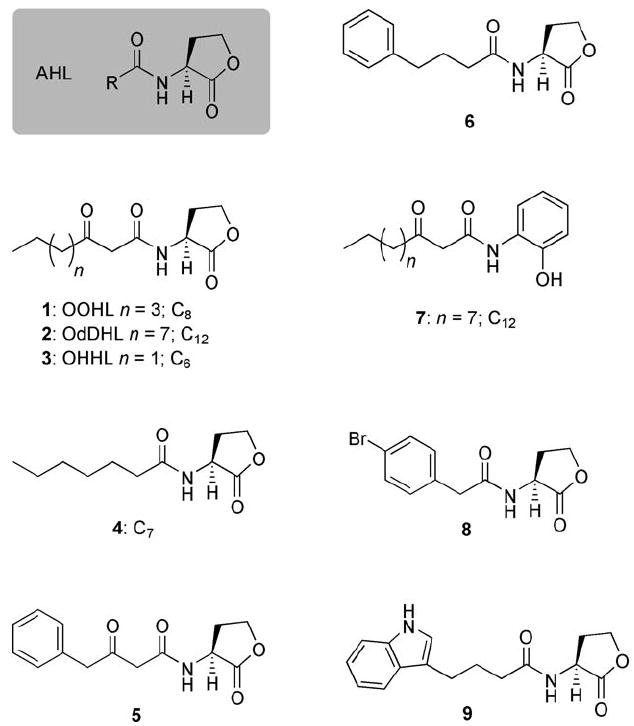
Generic structure of an N-acylated l-homoserine lactone (AHL), and structures of selected native AHL ligands (1–3) and known synthetic antagonists of R protein function (4–9). The number of carbon atoms (C) in selected aliphatic acyl groups is indicated for clarity.
As AHL–R protein binding is an essential event in QS, there has been considerable research on the development of non-native AHLs that can inhibit this ligand–protein interaction. The majority of this work has focused on three of the best characterized AHL–R protein systems (Scheme 1): N-(3-oxo-octanoyl)-l-homoserine lactone (OOHL, 1) and TraR in the plant pathogen Agrobacterium tumefaciens, N-(3-oxo-dodecanoyl)-l-homoserine lactone (OdDHL, 2) and LasR in the animal and plant pathogen P. aeruginosa, and N-(3-oxo-hexanoyl)-l-homoserine lactone (OHHL, 3) and LuxR in the marine symbiont V. fischeri.
AHLs bearing non-native acyl chains represent the most extensively studied class of synthetic QS modulators in these three species.[9-19] Structural modifications to the lactone ring, including inversion of stereochemistry,[20] and replacement of the lactone with different carbo- or heterocycles[9-11, 17, 21-23] have been examined to a lesser degree. Four of the most effective AHL-derived antagonists of TraR, LasR, or LuxR reported to date are shown in Scheme 1: the C7 AHL 4 active against TraR,[13] the 3-oxo-phenylbutanoyl- and phenylbutanoyl HLs (5 and 6) active against LuxR,[14] and the 2-aminophenol analogue of OdDHL 7 active against LasR.[22]
Despite considerable past efforts, potent non-native AHL antagonists of QS remain scarce.[5, 7] Further, as the majority of these ligands have only been tested against one bacterial species, the selectivities of non-native AHLs for different R proteins are largely unknown. Insufficient structure–activity relationship (SAR) data for non-native AHLs within and between different Gram negative bacteria have impeded the design of new ligands with improved activities against and selectivities for R proteins. Likewise, this dearth of SAR data has also protracted the design of non-native AHL activators of QS.[19, 24, 25] The use of different assay procedures to assess agonistic or antagonistic activities against the same R protein has further complicated comparisons between past studies.
To address these challenges, our laboratory has embarked on the design and synthesis of focused, combinatorial libraries of non-native AHLs to identify structural features that engender both antagonistic and agonistic activities toward a range of different R proteins. Our preliminary comparative studies revealed several potent antagonists of both TraR and LasR, most notably 4-bromo phenylacetanoyl HL (PHL 8) and indole AHL 9 (Scheme 1).[18] Recently, we reported the synthesis of four focused AHL libraries and the systematic evaluation of these ligands to modulate R protein activity in A. tumefaciens, P. aeruginosa, and V. fischeri.[26] These studies uncovered some of the most potent synthetic inhibitors and activators of R protein-mediated QS reported to date and provided broad new insights into their mechanism of action. Here, we report full details of the design of the four libraries and a critical analysis of the primary R protein antagonism and agonism data for these ~90 ligands. These studies have afforded an extensive set of SAR data that dictate antagonistic and agonistic activities, and R protein selectivities for AHL ligands in A. tumefaciens, P. aeruginosa, and V. fischeri. Together, these data provide a valuable new roadmap for the design of next-generation ligands for use as chemical probes to study the mechanisms of QS and its complex roles in host/bacteria interactions.
Results and Discussion
Focused AHL libraries—general considerations and synthesis
We sought to construct focused libraries of AHLs that would allow us to probe key features of AHL structure, including 1) acyl chain length, 2) lactone stereochemistry, and 3) functional group diversity in the acyl chain. We designed four AHL libraries that allowed us to investigate these three structural features individually and in tandem (libraries A–D, shown in Scheme 2).[26] An X-ray crystal structure of the ligand-binding site of TraR was also consulted in silico to guide our initial ligand design;[27] as the ligand-binding sites of TraR, LasR, and LuxR have ~70% sequence homology, we reasoned that such analysis was valuable.[8]
Scheme 2.
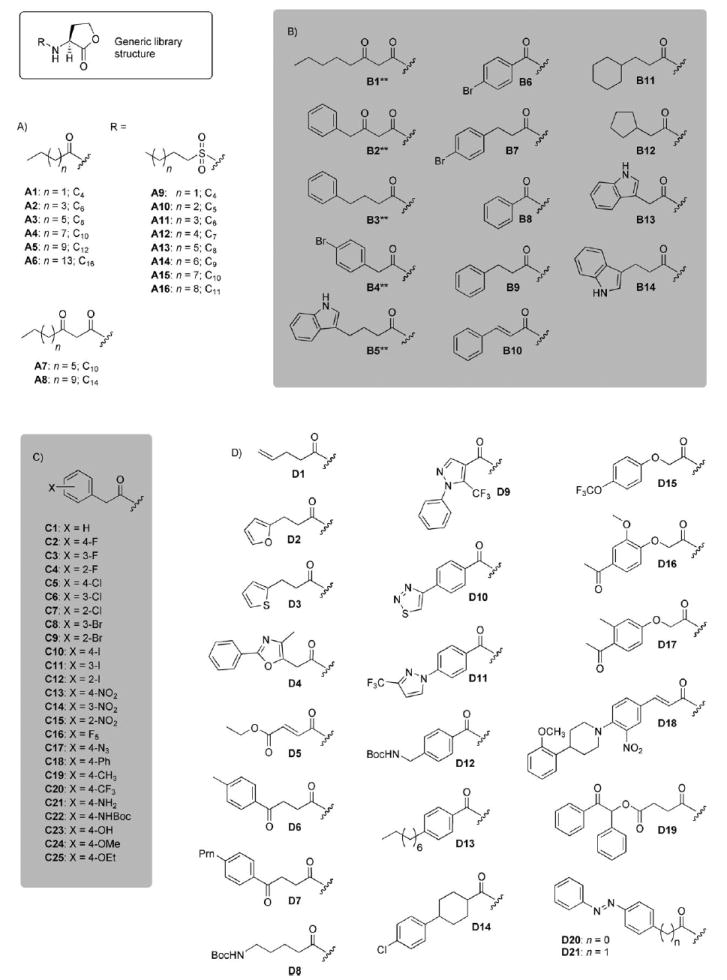
Structures of AHL libraries A–D. The number of carbon atoms (C) in certain aliphatic acyl groups is indicated for clarity. **=Indicates the AHL has d-stereochemistry; all others have l-stereochemistry.
Libraries A–D were synthesized in parallel according to our previously reported solid-phase methods.[18, 19, 26] The ~90 AHLs were isolated in moderate to good yields (55–75%) and with excellent purities (90–99 %).[26] Detailed rationales behind the design of each library are provided below.
Design of AHL library A
Library A was designed to test the effects of different aliphatic acyl, 3-keto acyl, and sulfonyl groups on AHL ligand activity in the three bacterial species. The structures of this 16-member focused library are shown in Scheme 2 A, and represent the most structurally simple AHL derivatives examined in this study. AHLs A1–A8 are naturally occurring AHLs utilized by other Gram negative bacteria for QS,[1, 8] and several have been evaluated in TraR, LasR, and/or LuxR agonism or antagonism assays previously.[9-11, 13] The C4 AHL A1 is also utilized by P. aeruginosa as a secondary signaling molecule for QS (via RhlR).[3] Several of the sulfonyl compounds in library A (A9–A14) were reported by Castang et al. to inhibit LuxR activity at a low to moderate level (in a heterologous E. coli LuxR reporter strain), with activity maximal at a five-carbon (six atom) acyl chain length (i.e., A10).[15] Collectively, however, these ligands have not been examined in the three bacterial strains utilized in this study. Consequently, library A was designed to provide important benchmark data for the comparison of antagonistic and agonistic ligand activities between the strains
Design of AHL library B
The structures of the second AHL library in this study, library B, are shown in Scheme 2B. This library was designed to investigate the roles of the following AHL structural features on R protein antagonism and agonism: 1) lactone stereochemistry, 2) acyl group aromaticity, and 3) alkyl “spacer” length between aromatic groups and the HL ring. We examined these three features by perturbing the structures of five known active compounds: the native agonist OOHL (1), the two phenylbutanoyl HL control antagonists (5 and 6),[14] and our two previously reported antagonists, 4-bromo PHL (8) and indole AHL (9, Scheme 1).[18] The effect of lactone stereochemistry on R protein activation had only been examined for a limited set of native AHLs,[20] and to our knowledge, had yet to be examined in synthetic AHL antagonists.[17] (We note, however, that many synthetic AHLs have been tested in racemic form, or their reported stereochemistry was not explicit, which adds additional complexity to this analysis.[9-11, 13-16]) Lastly, the roles of acyl group aromaticity and spacer length on ligand activity, specifically in our antagonists 8 and 9, were unknown.
Design of AHL library C
The structures of library C are shown in Scheme 2C; each of the 25 library members was designed to systemically test the effects of different functional groups and their positions on the PHL phenyl ring. These functional groups differ significantly in terms of electronics and steric size, and range from halogens to aromatic groups. Library C was inspired in part by the high antagonistic activity of control PHL 8 toward TraR and LasR reported previously by our laboratory.[18] In addition, we recently examined a subset of the PHLs in library C in LuxR antagonism and agonism assays, and identified several potent modulators of LuxR in V. fischeri.[19] These preliminary studies underscore the value of the PHL scaffold for the design of new R protein modulators, and provide a foundation for the systematic examination of PHLs C1–C25 across the three strains in the current work.
Design of AHL library D
Library D contained the most structurally diverse set of synthetic AHLs reported to date (shown in Scheme 2 D), and was designed to examine the effects of a range of different acyl groups on AHL-mediated R protein antagonism and agonism. These acyl groups differ extensively in terms of overall size and the type and placement of functional groups. However, as several active non-native AHLs contain aromatic groups (Scheme 1), we deliberately installed an aromatic functionality (or at least one π-system) in all but one of the acyl groups of library D. For ease of synthesis, we selected acyl groups that could be installed with commercially available carboxylic acids. Despite the higher hydrophobic character of many of the ligands in library D relative to the other ligands in this study (e.g., AHLs D18–D21), we did not encounter problems with compound insolubility in any of the biological assays reported herein (see below).
Bacterial reporter gene assays
R protein inhibition and activation by non-native AHLs is most frequently assessed with bacterial reporter strains.[6, 7] These strains lack their AHL synthase (I) genes, but retain their native R genes. In the presence of exogenously added AHL ligand, the AHL–R protein complex will bind adjacent to a promoter that controls reporter gene expression and activate transcription. Therefore, R protein activity, and consequently ligand activity, can be measured with standard reporter gene readouts. Competitive antagonism assays are performed with synthetic ligand in the presence of native AHL ligand, while agonism assays are performed with synthetic ligand alone.
We utilized three bacterial reporter strains for the TraR, LasR, and LuxR agonism and antagonism assays in this study (see the Experimental Section). The A. tumefaciens strain produces β-galactosidase upon TraR activation and ligand activity can be measured in standard Miller absorbance assays.[28] The E. coli strain harbors LasR from P. aeruginosa and also reports LasR activity by β-galactosidase production. We found that this heterologous E. coli strain provided more reproducible data than related P. aeruginosa reporters, although the differences between R protein antagonists and agonists were somewhat muted relative to the other two strains in this study.[26] Lastly, the V. fischeri strain retains its native lux operon (yet lacks a functional luxI), which allows LuxR activation to be measured as luminescence.[19]
Libraries A–D were systematically screened in R protein antagonism and agonism assays in the three bacterial reporter strains. The native ligands OOHL (1), OdDHL (2), and OHHL (3) and the known R protein antagonists 4–9 (Scheme 1) served as critical controls for these assays (synthesized according to reported procedures).[18, 19, 26] Over 40% of the ligands were potent inhibitors of TraR, LasR, and/or LuxR, with activities either comparable or surpassing that of the controls. In turn, 18% of the ligands were identified as either LasR or LuxR agonists (~25% activation or higher). These ligands represent some of the most potent modulators of R proteins reported to date; a detailed examination of this ligand subset has been reported elsewhere.[26] Here, we provide a critical analysis of all of the primary antagonism and agonism assay data for libraries A–D and delineate broad SAR trends revealed by these data for each library.
Primary assay data for control compounds
The primary assay data for the control compounds largely corroborated those from previously reported experiments (Table 1). Each of the R proteins was inhibited to some degree (15–89%) by control native ligands (1–3) that were close in carbon-chain length to their native AHL (i.e., two or four carbon atoms difference).[9-11, 13] In addition, all of the control antagonists (4–9) showed inhibitory activities in the three strains, albeit at varied levels (18–93 %), with the exception of 2-aminophenol 7, which was surprisingly inactive. Intriguingly, control 7 weakly agonized LasR instead (18 %). This latter result contrasted with previous reports that 7 is a strong inhibitor of LasR activity in similar assays; however, these studies involved a different LasR reporter strain.[22] The C7 AHL 4, phenylbutanoyl HL (6), and 4-bromo PHL (8) were the most active control antagonists across all three strains (~90% in TraR, ~25% in LasR, and ~76% in LuxR). The C7 AHL 4 was also a weak LuxR agonist under the primary agonism assay conditions (23 %; see below).
Table 1.
Antagonism and agonism assay data for library A and controls 1–9 in three bacterial reporter strains.[a]
| Compound | A. tumefaciens–TraR[b] | E. coli–LasR[e] | V. fischeri–LuxR[h] | |||
|---|---|---|---|---|---|---|
| Inhibition [%][c] | Activation [%][d] | Inhibition [%][f] | Activation [%][g] | Inhibition [%][i] | Activation [%][j] | |
| 1: OOHL | – | 100 | 50 | 19 | 63 | 24 |
| 2: OdDHL | 28 | 12 | – | 100 | 86 | 2 |
| 3: OHHL | 89 | 1 | 15 | 1 | – | 100 |
| 4 | 93 | 4 | 28 | 0 | 78 | 23 |
| 5 | 85 | 8 | 18 | 5 | 45 | 2 |
| 6 | 93 | 2 | 20 | 3 | 70 | 3 |
| 7 | 9 | 0 | 4 | 18 | 3 | 1 |
| 8 | 88 | 1 | 28 | 0 | 79 | 3 |
| 9 | 35 | 0 | 36 | 3 | 72 | 3 |
| A1 | 0 | 0 | 0 | 0 | 43 | 2 |
| A2 | 48 | 1 | 11 | 2 | 69 | 25 |
| A3 | 83 | 7 | 43 | 1 | 86 | 12 |
| A4 | 92 | 2 | 34 | 44 | 96 | 1 |
| A5 | 22 | 1 | −10 | 85 | 73 | 2 |
| A6 | 0 | 0 | 5 | 11 | 27 | 2 |
| A7 | 14 | 1 | 8 | 1 | 47 | 2 |
| A8 | 10 | 11 | −18 | 87 | 77 | 2 |
| A9 | 86 | 7 | 16 | 1 | 5 | 4 |
| A10 | 59 | 0 | 12 | 0 | 30 | 2 |
| A11 | 93 | 1 | 10 | 0 | 32 | 2 |
| A12 | 88 | 1 | 12 | 0 | 60 | 2 |
| A13 | 44 | 0 | 10 | 0 | 77 | 3 |
| A14 | 37 | 3 | 15 | 0 | 81 | 2 |
| A15 | 34 | 0 | 17 | 0 | 80 | 2 |
| A16 | 29 | 0 | 21 | 0 | 74 | 1 |
All assays performed in triplicate; error did not exceed ± 10%. Data of significance are highlighted in bold. Negative controls containing no compound were subtracted from each sample to account for background. Negative inhibition values indicate that the compound activates at the tested concentration. See Figures S1–S15 in the Supporting Information for primary assay data in bar graph format.
Strain: A. tumefaciens WCF47 (pCF372). Assay data were normalized with respect to OOHL (1).
Screen was performed by using 10 μm synthetic ligand against 100 nm OOHL (1).
Screen was performed by using 10 μm ligand.
Strain: E. coli DH5α (pJN105 L pSC11). Assay data were normalized with respect to OdDHL (2).
Screen was performed by using 5 μm synthetic ligand against 7.5 nm OdDHL (2).
Screen was performed by using 5 μm ligand.
Strain: V. fischeri ES114 (ΔluxI). Assay data were normalized with respect to OHHL (3).
Screen was performed by using 5 μm synthetic ligand against 5 μm OHHL (3).
Screen was performed by using 200 μm ligand.
Primary assay data and SAR for library A
The simple aliphatic AHLs (A1–A6) in library A displayed inhibitory activity trends against the three R proteins that correlated with increasing carbon number; inhibition was maximal at C8 (A3) for LasR, and C10 (A4) for TraR and LuxR and then decreased thereafter (Table 1). The long chain, 3-oxo AHLs (C10 A7 and C14 A8) exhibited minimal inhibitory activities against TraR and LasR, yet were moderate (47%) to good (77%) inhibitors of LuxR, respectively. Of the three R proteins, LuxR appeared to be the most sensitive to inhibition by 3-oxo AHLs (i.e., by OOHL (1), OdDHL (2), control 6, A7, and A8). Interestingly, the 3-oxo C14 AHL A8 displayed agonistic, as opposed to antagonistic, activity against LasR in this assay (see below).
Antagonism by sulfonyl HLs (A9–A16) against the three R proteins also correlated with carbon number, and the most striking trends in inhibitory activity were observed against TraR and LuxR. Inhibition was maximal at C6 (A11) in TraR, with activity largely increasing up until this carbon length and then decreasing thereafter. Notably, the sulfonyl HL A11, with a seven-atom acyl tail (including the sulfur), displayed analogous inhibitory activity as control C7 AHL 4 (93%); this suggests that seven atoms in AHL acyl tails enhances antagonistic activity in TraR. In LuxR, inhibitory activity for the sulfonyl HLs increased gradually from C4 to C9 and decreased only minimally at the longer acyl chain lengths tested (i.e., in A15 and A16), with C9 (A14) exhibiting the highest inhibitory activity (81 %). These results directly contrasted with those of Castang et al. for sulfonyl HLs in LuxR[15] (see above) and highlight the differences in ligand activity often observable when using different reporter strains. Again, the sulfonyl HL with 10 atoms in its acyl tail (A14) and the C10 AHL (A4) were the most active LuxR inhibitors of their structure classes; this indicates that acyl-chain atom number also plays a role in AHL antagonistic activity against LuxR.
Far fewer synthetic agonists were identified in library A relative to antagonists (Table 1). None of these ligands activated TraR to an appreciable level. This result corroborates screening data reported by Zhu et al. for several related AHL derivatives, from which no TraR agonists were identified.[13] Similarly, only a few ligands activated LuxR, with the C6 AHL A2, the C7 control AHL 4, and OOHL (1) displaying ~25% activation. Thus, within library A, only compounds with structures very closely related to the native LuxR ligand (OHHL, 3) were LuxR agonists. The results from the LasR agonism screen of library A were more striking. Here, we identified two ligands that substantially activated LasR (~85%): C12 AHL A5 and 3-oxo C14 AHL A8 (Table 1). Moreover, these two ligands selectively activated LasR relative to TraR and LuxR. The C10 AHL A4 and OOHL (1) also displayed agonistic activities, albeit reduced (≤44%), indicating that in analogy to LuxR, AHLs in library A with structures most similar to the native LasR ligand (OdDHL, 2) were effective LasR agonists. These data trends correlated with those reported by Passador et al. for the same compounds (yet in an alternate E. coli LasR reporter strain).[11] However, these researchers also reported that 3-oxo C10 AHL (A7) exhibited analogous agonistic activity as 3-oxo C14 A8; the former ligand failed to activate LasR in our assays. This result was unexpected, in view of the structural similarity of this ligand to the other moderate to strong LasR activators that we identified in this study (i.e., A4, A5, and A8), and again exemplifies the disparities that can arise when different reporter strains are utilized for screening QS modulators.
Primary assay data and SAR for library B
Examination of library B in the reporter gene assays revealed several additional SARs that dictated AHL ligand activity against R proteins (Table 2). First, the d-enantiomer of OOHL (B1) displayed no antagonistic activity in any of the three strains. Likewise, inversion of stereochemistry in control antagonists 5 and 6 (to give d-AHLs B2 and B3) reduced their inhibitory activity by ~40–60% in TraR. A similar ~40% reduction in inhibitory activity was also observed for B3 in LuxR; however, B2 exhibited analogous inhibitory activity as its l-stereoisomer 5 (~45%). The activity trends for d-AHLs B2 and B3 were yet more complex in LasR; here, B2 displayed strong agonistic as opposed to antagonistic activity (see below), while B3 inhibited LasR at a comparable level to its l-stereoisomer 6 (~20%). In contrast to B2 and B3, the d stereoisomers of our control 4-bromo PHL and indole AHL antagonists, B4 and B5, showed uniformly reduced inhibitory activity across all three strains, ranging from ~90% reduction for B4 in TraR to at least 50% for both B4 and B5 in LasR and LuxR. These results suggest that AHL stereochemistry, in concert with acyl chain structure, plays a multifaceted role in AHL-mediated R protein inhibition and activation. One effect is clear, however; inversion of lactone stereochemistry does not completely abolish antagonistic activity for the ligands examined in this study.
Table 2.
Antagonism and agonism assay data for library B and selected controls in three bacterial reporter strains.[a]
| Compound | A. tumefaciens–TraR | E. coli–LasR | V. fischeri–LuxR | |||
|---|---|---|---|---|---|---|
| Inhibition [%] | Activation [%] | Inhibition [%] | Activation [%] | Inhibition [%] | Activation [%] | |
| 1: OOHL | – | 100 | 50 | 19 | 63 | 24 |
| 2: OdDHL | 28 | 12 | – | 100 | 86 | 2 |
| 3: OHHL | 89 | 1 | 15 | 1 | – | 100 |
| 5 | 85 | 8 | 18 | 5 | 45 | 2 |
| 6 | 93 | 2 | 20 | 3 | 70 | 3 |
| 8 | 88 | 1 | 28 | 0 | 79 | 3 |
| 9 | 35 | 0 | 36 | 3 | 72 | 3 |
| B1 | 6 | 4 | 16 | 0 | 7 | 2 |
| B2 | 50 | 2 | −7 | 84 | 46 | 3 |
| B3 | 31 | 0 | 16 | 0 | 41 | 3 |
| B4 | 9 | 0 | 13 | 1 | 34 | 2 |
| B5 | 9 | 0 | 13 | 0 | 29 | 2 |
| B6 | 9 | 0 | 13 | 0 | 40 | 2 |
| B7 | 93 | 2 | 52 | 11 | 80 | 2 |
| B8 | 8 | 0 | 14 | 1 | 34 | 2 |
| B9 | 16 | 0 | 21 | 0 | 44 | 3 |
| B10 | 7 | 3 | 22 | 2 | 48 | 3 |
| B11 | 25 | 0 | 36 | 0 | 82 | 12 |
| B12 | 11 | 0 | 19 | 0 | 57 | 3 |
| B13 | 20 | 0 | 21 | 0 | 42 | 3 |
| B14 | 21 | 0 | 48 | 1 | 47 | 2 |
See footnotes for Table 1.
The remaining members of library B were designed to probe the role of acyl chain structure on antagonistic activity for control antagonists 8 and 9. Shortening the alkyl spacer in 4-bromo PHL 8 by one carbon (to give benzoyl AHL B6) dramatically reduced its inhibitory activity in all three R proteins; the reduction ranged from 90% in TraR to ~50% in LasR and LuxR (Table 2). However, lengthening the alkyl spacer by one carbon produced a ligand (B7) with equivalent inhibitory activity to control 4-bromo PHL 8 in TraR and LuxR, and twofold higher inhibitory activity in LasR. Notably, B7 was also almost twofold as active as the potent, control antagonist 9 (52 and 36%, respectively), and amongst the most potent inhibitors of LasR identified in these primary assays.
Removing the 4-bromide substituent from benzoyl AHL B6 (to give B8) had little effect on an already low antagonistic activity, while removing the 4-bromide from the potent antagonist B7 (to give B9) had a more significant impact and reduced inhibition by at least 50% across all three strains. In turn, the cyclohexyl analogue of B9, AHL B11, displayed slightly enhanced antagonistic activity in TraR and LasR relative to B9, and activity against LuxR, analogous to the most potent non-native AHL inhibitor in library B, B7 (~80%). Finally, shortening the alkyl spacer of control indole AHL 9 by one or two carbons (B14 and B13, respectively) had only a minor effect on inhibitory activity in TraR, while these shorter indole analogues were ~40% less active than control 9 in LuxR. In contrast, the one-carbon shorter indole analogue B14 exhibited heightened activity in LasR relative to control 9 (48 and 36%, respectively), and was one of the most potent LasR inhibitors identified in this study.
These results for library B reveal several trends in antagonistic activity for synthetic AHLs: 1) a flexible carbon spacer of at least one carbon and a 4-bromo substituent are necessary for appreciable activity in ligands structurally related to 4-bromo PHL 8, with AHL B7 being the most active inhibitor across the three R proteins, 2) an aromatic functionality is not essential for LuxR inhibition in ligands related to control PHL 8 (e.g., AHL B11), and 3) a three-carbon spacer is optimal for TraR and LuxR inhibition in ligands structurally related to control indole AHL 9, while a two-carbon spacer is optimal for inhibition of LasR (i.e., AHL B14).
In analogy to library A, very few agonists were identified in library B (Table 2). Indeed, only one ligand with considerable agonistic activity against one R protein, LasR, was identified: the d-enantiomer of control antagonist 5, d-AHL B2. This ligand was capable of activating LasR at 84% relative to the native ligand OdDHL (2) at equal concentrations. AHL B2 is unique, as this d-AHL displays strong agonistic activity and its l-stereoisomer, control AHL 5, is virtually inactive in LasR (but is a moderate to strong antagonist in LuxR and TraR, respectively). This trend is opposite to what has been observed for native AHL ligands, for which the l-stereoisomer is an active agonist and the d-stereoisomer is almost inactive;[17, 20] we observed this latter trend in the current study for OOHL (1). The reasons behind this trend reversal for B2 remain unclear, and in view of the complex antagonistic activity trends displayed by the limited set of d enantiomers in library B, suggest that lactone stereochemistry will be an important feature to probe in the future design of AHL-derived QS modulators.
Primary assay data and SAR for library C
The antagonism and agonism primary screening data for library C are listed in Table 3 and reveal the largest percentage of potent antagonists and agonists in this study (37% of the library have activities of ≥50% in at least one strain). This result serves to validate the PHL structure as a scaffold for the design of potent modulators of R protein function.
Table 3.
Antagonism and agonism assay data for library C and selected controls in three bacterial reporter strains.[a]
| Compound | A. tumefaciens–TraR | E. coli–LasR | V. fischeri–LuxR | |||
|---|---|---|---|---|---|---|
| Inhibition [%] | Activation [%] | Inhibition [%] | Activation [%] | Inhibition [%] | Activation [%] | |
| 1: OOHL | – | 100 | 50 | 19 | 63 | 24 |
| 2: OdDHL | 28 | 12 | – | 100 | 86 | 2 |
| 3: OHHL | 89 | 1 | 15 | 1 | – | 100 |
| 8 | 88 | 1 | 28 | 0 | 79 | 3 |
| C1 | 4 | 0 | 15 | 0 | 14 | 2 |
| C2 | 25 | 0 | 26 | 1 | 59 | 2 |
| C3 | 11 | 0 | 27 | 0 | 63 | 8 |
| C4 | 4 | 0 | 19 | 0 | 49 | 2 |
| C5 | 74 | 0 | 29 | 0 | 75 | 3 |
| C6 | 50 | 0 | 41 | 0 | 65 | 61 |
| C7 | 6 | 0 | 10 | 0 | 53 | 6 |
| C8 | 56 | 0 | 45 | 3 | 58 | 70 |
| C9 | 12 | 0 | 12 | 0 | 38 | 12 |
| C10 | 93 | 1 | 36 | 0 | 85 | 4 |
| C11 | 70 | 0 | 57 | 3 | 78 | 28 |
| C12 | 5 | 0 | 17 | 0 | 63 | 7 |
| C13 | 88 | 1 | 27 | 0 | 47 | 24 |
| C14 | 46 | 0 | 54 | 11 | −15 | 129 |
| C15 | 1 | 0 | 17 | 0 | 23 | 4 |
| C16 | 2 | 0 | 19 | 2 | 40 | 2 |
| C17 | 70 | 0 | 19 | 0 | 66 | 5 |
| C18 | 67 | 0 | 20 | 0 | 79 | 7 |
| C19 | 21 | 3 | 11 | 0 | 58 | 1 |
| C20 | 92 | 2 | 26 | 0 | 78 | 3 |
| C21 | 5 | 0 | 8 | 0 | 12 | 3 |
| C22 | 11 | 0 | 10 | 27 | 33 | 2 |
| C23 | 3 | 0 | 7 | 0 | 12 | 2 |
| C24 | 26 | 0 | 16 | 0 | 57 | 3 |
| C25 | 35 | 0 | 15 | 0 | 55 | 2 |
See footnotes for Table 1.
As observed in libraries A and B, the majority of the active ligands in library C were antagonists. Replacement of the 4-bromide of control PHL 8 with a hydrogen atom in C1 largely abolished inhibitory activity across the three strains (Table 3), in analogy to what was observed for the one-carbon-longer analogues B7 and B9 in library B (see above). The monohalogen (C2–C12) and nitro series (C13–C15) exhibited remarkable trends in inhibitory activity against all three R proteins. These trends were most pronounced in TraR. Namely, inhibition dramatically increased (from ~1 to 90%) as the halogen or nitro substituents were moved from the 2- to the 3- to the 4-position on the PHL phenyl ring. Inhibition also increased with substituent size, with 4-iodo PHL (C10) and 4-nitro PHL (C13) inhibiting at the highest level in this series (~90%). The monohalogenated PHLs displayed the same trends in antagonistic activity in LuxR, albeit slightly muted within each series. However, the nitro series (C13–C15) displayed a more complicated activity pattern, with 4-nitro PHL (C13) only moderately inhibiting LuxR (47%) and, more notably, 3-nitro PHL (C14) dramatically activating LuxR (see below). These assay data indicate that both antagonistic and agonistic activities are exquisitely affected by the nature and position of the substituents on the PHL phenyl ring.
Uniform antagonistic activity trends were also observed for the monohalogen and nitro PHL series in LasR (Table 3). Here, in contrast to TraR and LuxR, the 3-substituted PHLs displayed the highest inhibitory activities, followed by the 4- and 2-substituted derivatives. Antagonism still increased with increasing substituent size, in analogy to TraR, with the 3-iodo (C11) and 3-nitro (C14) PHLs exhibiting the highest antagonistic activities in LasR for the series (~55%). Moreover, these two ligands were the most potent LasR inhibitors identified in these primary assays overall. A final halogenated PHL, pentafluoroaromatic PHL (C16), was designed to examine whether its reversed aromatic quadrupole could enhance PHL-mediated R protein modulation (potentially through favorable π-stacking interactions).[29] Analogous to its nonfluorinated analogue C1, this ligand displayed minimal inhibitory activities in TraR and LasR, and only low inhibitory activity (40%) against LuxR.
The remaining PHLs in library C were designed to probe the effects of different substituents in the 4-position of the phenyl ring on R protein modulation. Both the 4-azido PHL (C17) and 4-phenyl PHL (C18) were moderate to strong inhibitors of TraR and LuxR (~70%; Table 3). The activity of 4-azido PHL (C17) is particularly notable as the azido moiety renders this inhibitor photoactive, and thus C17 could have value as a potential photoaffinity labeling tool for R proteins and provide insights into the ligand-binding site for PHLs.[30] Likewise, the activity of 4-phenyl PHL (C18) was significant, as it instructed us that sterically demanding groups could be tolerated on the phenyl ring of PHL-derived R protein antagonists.
The 4-methyl and 4-trifluoromethyl PHLs (C19 and C20) exhibited markedly different activities in the antagonism assays. The 4-methyl PHL C19 was only a weak to moderate inhibitor of all three R proteins (Table 3), and inhibited at a two- to fourfold lower level relative to the 4-bromo PHL control (8). As a methyl group is roughly equivalent in steric size to a bromide, this activity trend indicated that substituent size alone does not dictate inhibitory activity for 4-substituted PHLs. In contrast, the 4-trifluoromethyl PHL C20 displayed equivalent antagonistic activity as control 4-bromo PHL 8 in all three strains. This result suggests, along with the other antagonism data outlined above for library C, that electron-withdrawing and lipophilic groups in the 4-position enhance PHL inhibitory activity against R proteins. This hypothesis is further corroborated by the low to moderate antagonistic activities displayed by PHLs C21–C25, all of which contain electron-donating groups in the 4-position of the phenyl ring. In addition, the two PHLs in this set with hydrogen bond donors in the 4-position (i.e., 4-amino (C21) and 4-hydroxy (C23) PHLs) were amongst the weakest inhibitors in library C (~7%), with activities comparable to C1.
Turning next to agonism assays, six PHLs were identified in library C that were capable of activating R proteins by ≥25% (Table 3). The most potent agonists were highly selective for LuxR, and we focus on these compounds here. Again, we observed striking trends in the activities for PHLs with halogen and nitro groups. In contrast to the antagonism data for these PHLs in LuxR, the 3-substituted compound in each series showed the strongest activity relative to the 2- and 4-substituted derivatives, with the 3-chloro (C6), 3-bromo (C8), and 3-nitro (C14) PHLs exhibiting at least 60% luminescence induction relative to the native ligand OHHL (3) at equal concentrations. When substituents were moved on the PHL phenyl ring by a single carbon (from the 4- to the 3-position), the ligands were converted from LuxR antagonists to LuxR agonists. Moreover, 3-nitro PHL (C14) was able to induce 29% higher luminescence than OHHL (3) in this primary assay. This result was remarkable, and explained the unusual inhibition trends for the nitro PHL series in LuxR (C13–C15; see above). Few superactivators of R proteins have been reported;[24, 25] therefore, our discovery of 3-nitro PHL C14 as a superactivator of LuxR is significant. Additional studies in our laboratory have shown that PHL C14 can also superactivate LuxR in wildtype V. fischeri and is tolerated in invertebrate model systems; this suggests that this compound could have considerable value as a probe to study V. fischeri–host symbioses.[19]
Overall, the screening data for library C indicate that the PHL structure is a highly versatile scaffold for the design of both R protein antagonists and agonists, and that seemingly simple structural modifications to the PHL phenyl ring can have major effects on ligand activity. Most notably, these structural modifications can convert potent antagonists into agonists (over the concentration ranges tested). These primary assays revealed some of the most potent and selective R protein modulators in this study, including 4-iodo PHL (C10) that inhibits all three R proteins, 3-nitro PHL (C14) that strongly inhibits LasR but also superactivates LuxR, and 4-phenyl PHL (C18) and 4-trifluoromethyl PHL (C20) that strongly inhibit TraR and LuxR but are considerably less active against LasR.
Primary assay data and SAR for library D
Library D also contained several new and potent synthetic modulators of TraR, LasR, and LuxR (Table 4). The most active compounds or those displaying interesting SAR trends are described here. AHLs D1–D5 displayed negligible inhibitory activity against TraR, and only low to modest inhibitory activity against LasR and LuxR; this suggests that their compact, unsaturated, and/or heterocyclic acyl groups significantly reduced activity against these three R proteins. AHL D6, in contrast, was a strong inhibitor of TraR (90 %), a moderate inhibitor of LuxR (68 %), and a relatively weak inhibitor of LasR (28 %). A clear rationale for the heightened antagonistic activity of D6 relative to D1–D5 was not obvious, except potentially its higher structural similarity to the potent control antagonists 5, 6, and 8. Interestingly, enlarging the substituent in the 4-position of the aromatic ring from a methyl group in D6 to an n-propyl group in D7 halved the inhibitory activity in TraR and LasR, yet had no effect against LuxR.
Table 4.
Antagonism and agonism assay data for library D and selected controls in three bacterial reporter strains.[a]
| Compound | A. tumefaciens–TraR | E. coli–LasR | V. fischeri–LuxR | |||
|---|---|---|---|---|---|---|
| Inhibition [%] | Activation [%] | Inhibition [%] | Activation [%] | Inhibition [%] | Activation [%] | |
| 1: OOHL | – | 100 | 50 | 19 | 63 | 24 |
| 2: OdDHL | 28 | 12 | – | 100 | 86 | 2 |
| 3: OHHL | 89 | 1 | 15 | 1 | – | 100 |
| 4 | 93 | 4 | 28 | 0 | 78 | 23 |
| 5 | 85 | 8 | 18 | 5 | 45 | 2 |
| 6 | 93 | 2 | 20 | 3 | 70 | 3 |
| 8 | 88 | 1 | 28 | 0 | 79 | 3 |
| 9 | 35 | 0 | 36 | 3 | 72 | 3 |
| D1 | 10 | 0 | 15 | 3 | 47 | 2 |
| D2 | 9 | 0 | 18 | 0 | 15 | 3 |
| D3 | 8 | 0 | 22 | 0 | 38 | 2 |
| D4 | 12 | 2 | 18 | 0 | 7 | 2 |
| D5 | 3 | 0 | 13 | 0 | 37 | 1 |
| D6 | 90 | 3 | 28 | 0 | 68 | 2 |
| D7 | 59 | 1 | 13 | 0 | 69 | 2 |
| D8 | 5 | 0 | 12 | 0 | 18 | 2 |
| D9 | 8 | 0 | 16 | 4 | 7 | 2 |
| D10 | 8 | 0 | 9 | 0 | 35 | 2 |
| D11 | 9 | 0 | 10 | 0 | 56 | 2 |
| D12 | 13 | 0 | 12 | 0 | 35 | 1 |
| D13 | 8 | 0 | 27 | 3 | 59 | 1 |
| D14 | 11 | 0 | 36 | 36 | 45 | 1 |
| D15 | 90 | 4 | 49 | 30 | 39 | 2 |
| D16 | 11 | 0 | 18 | 0 | 40 | 1 |
| D17 | 92 | 1 | 26 | 0 | 50 | 2 |
| D18 | 16 | 0 | 34 | 32 | 19 | 2 |
| D19 | 11 | 0 | 17 | 7 | 30 | 1 |
| D20 | 4 | 0 | 12 | 0 | 46 | 2 |
| D21 | 9 | 0 | 13 | 1 | 63 | 3 |
See footnotes for Table 1.
The AHLs in library D with aromatic (D9–D13) or carbocyclic functionality (D14) directly adjacent to the carbonyl in the acyl group exhibited minimal inhibitory activity against TraR (Table 4). Only two AHLs in this group (D11 and D13) were reasonably strong inhibitors of LuxR (~60%); notably, these two AHLs both contained benzoyl functionalities and had the most extended acyl chains of this ligand set. AHL D13 was also a modest inhibitor of LasR (27 %), while D11 was weakly active. The cyclohexyl AHL derivative D14, however, was a relatively strong inhibitor of LasR, with activity analogous to that of the indole AHL control 9 (36 %).
The three AHLs in library D with phenyl ether functionality in their acyl chains (D15–D17) displayed clear inhibition trends across the three strains (Table 4). Notably, these three compounds had two-atom spacers between the carbonyl groups and the aromatic rings in their acyl chains, analogous to the potent inhibitors B7 and B14 identified in library B (see above). All three of these phenyl ether AHLs were only modest inhibitors of LuxR (~45%). However, 4-trifluoromethyl phenyl ether AHL D15 was a potent inhibitor of TraR and the strongest inhibitor of LasR identified in library D (90 and 49% inhibition, respectively). The two structurally similar 4-keto phenyl ether AHLs (D16 and D17) exhibited contrasting activities in both TraR and LasR: D16 was virtually inactive against TraR, while D17 was similar in activity to D15 and one of the most potent inhibitors of TraR (92%) uncovered in these assays overall. Likewise, D17 was 50% more active against LasR relative to D16. Interestingly, compounds D16 and D17 only differ in the placement of a substituent on the aromatic ring of the acyl group (2-methoxy and 3-methyl, respectively; Scheme 2 D). This result suggests that, similar to the PHL series in library C, inhibitory activity can increase in this phenyl ether AHL series when substituents on the aromatic ring are placed closer to the 4-position.
The remaining four AHLs in library D (D18–D21) contained the most sterically bulky acyl chains examined to date. These four AHLs exhibited minimal inhibitory activity against TraR; this is analogous to the low inhibitory activity observed for the relatively bulky AHLs D9–D14 (Table 4). In contrast, the most sterically bulky of this ligand set (D18) was a relatively strong inhibitor of LasR and the most active of the four. Finally, the azobenzene AHL derivatives D20 and D21 displayed medium to moderately strong inhibitory activity against LuxR (46 and 63%, respectively). These compounds are of interest because of the photoisomerization ability of the azobenzene moiety.[30] For example, their inhibitory activity could be altered upon cis/trans isomerization, as this conformational shift may cause the ligand to dislodge from (or bind differently in) the ligand-binding site. Therefore, these azobenzene AHL ligands (D20 and D21), along with the 4-azido PHL antagonist (C17) identified in library C, could represent novel photoactive tools for the study of R protein function.
Similar to libraries A–C, the agonism screen of library D revealed few synthetic agonists (Table 4). Indeed, no library members were agonists of TraR and LuxR. Three ligands (D14, D15, and D18), however, were weak activators of LasR (~33 %). The structures of these AHLs were not highly similar, but each had a relatively bulky acyl chain containing aromatic functionality, most notably D18. Intriguingly, these three ligands were also the most potent antagonists of LasR identified in library D (see above). Moreover, their percent antagonistic activities were approximately equivalent to their percent agonistic activities. Additional studies of AHLs D14, D15, and D18 suggest that these ligands are not antagonists of LasR, but rather can behave as partial agonists (see below);[26] such a mechanism of action would explain these conflicting primary assay data. Ongoing work in our laboratory is directed at fully understanding the mechanism of R protein modulation by these and related AHL ligands.
Summary of SAR trends for libraries A–D
Overall, we found that subtle changes to the AHL acyl group, some as simple as the addition or removal of one carbon or halogen atom, had dramatic effects on ligand activity in each of the three bacterial strains in this study. In general, AHLs with acyl groups of moderate size (up to eight atoms long) and containing either aromatic functionality with electron-withdrawing groups or straight-chain aliphatic functionality can antagonize TraR, LasR, and LuxR over the concentrations tested in this study. AHL B7 epitomizes such a broad-spectrum antagonist, and was one of the most active antagonists identified. Within this class, sulfonyl groups can replace carbonyl groups on aliphatic AHL TraR and LuxR antagonists without significant loss in activity. Likewise, AHLs bearing sterically bulky, aromatic acyl groups can selectively inhibit and activate LasR (e.g., D18). Of the AHLs analyzed herein, the PHL appears to be the most unique scaffold for R protein modulation, as members of this structure class display a wide range of antagonistic and agonistic activities across all three R proteins in this study. The 4- and 3-substituted PHLs display the most remarkable trends in activity, ranging from a potent antagonist of all three R proteins (C10) to a superactivator of only LuxR (C14). Finally, inversion of lactone stereochemistry (from l to d) was not found to fully abolish activity for the AHLs examined herein; indeed, one d-AHL (B2) was shown to strongly activate LasR.
Pharmacophore modeling
To obtain a better understanding of how different structural features of AHLs engender antagonistic and agonistic activities, we generated computational pharmacophore models for the AHL modulators of each of the three R proteins. Preliminary studies from our laboratory suggest that many of the most potent “antagonists” identified in libraries A–D may elicit their activities via a partial agonism mechanism (e.g., PHLs C20 in TraR, C14 in LasR, and C13 in LuxR, and bulky AHLs D14, D15, and D18 in LasR).[26] Therefore, these ligands do not appear to inhibit R protein activity; rather, they simply are unable to activate the R protein to the same level as the native ligand. In view of these new mechanistic data, all of the primary antagonism and agonism assay data in this study were utilized to calculate AHL pharmacophore models for TraR, LasR, and LuxR (see the Experimental Section).
Views of the pharmacophore models are shown in Figure 1, and they reveal several different structural features between the R proteins. For example, each model contains regions of hydrophobic/aromatic functionality, H-bond donors, and H-bond acceptors, yet the relative size and positions of these groups on each of the models vary significantly. Notably, these differences could not be fully ascertained through analysis of the primary data presented above; the pharmacophores provide a more global, 3D synopsis of these data. In general, the TraR pharmacophore exhibits an almost equal balance of hydrophobic functionality and H-bond acceptors and is relatively compact. The LuxR pharmacophore is similarly compact, yet has fewer H-bond acceptors relative to TraR. In contrast, the LasR pharmacophore is noticeably larger and exhibits an extensive hydrophobic surface. These differences in size for TraR and LasR are congruent with the compact and expanded AHL-binding sites indicated by the TraR and LasR X-ray structures, respectively, assuming that these non-native AHLs target the same site.[27, 31] While a structure for LuxR is yet to be reported, using this same reasoning, the LuxR pharmacophore reflects a ligand-binding site that is more similar to TraR than LasR. However, it is challenging to fully rationalize the selectivity profiles for the AHLs in this study using these calculated pharmacophores. Structural studies of the R proteins with various ligands (e.g., by X-ray crystallography) will better illuminate the differences in activities, and are ongoing in our laboratory. These pharmacophore models are significant nonetheless as—to our knowledge—they are the first reported for AHL-derived QS modulators. We anticipate that these models, along with the extensive SAR data outlined above, will guide the design of new QS modulators with improved activities and selectivities, and provide new avenues to study the chemistry and biology of bacterial communication.
Figure 1.
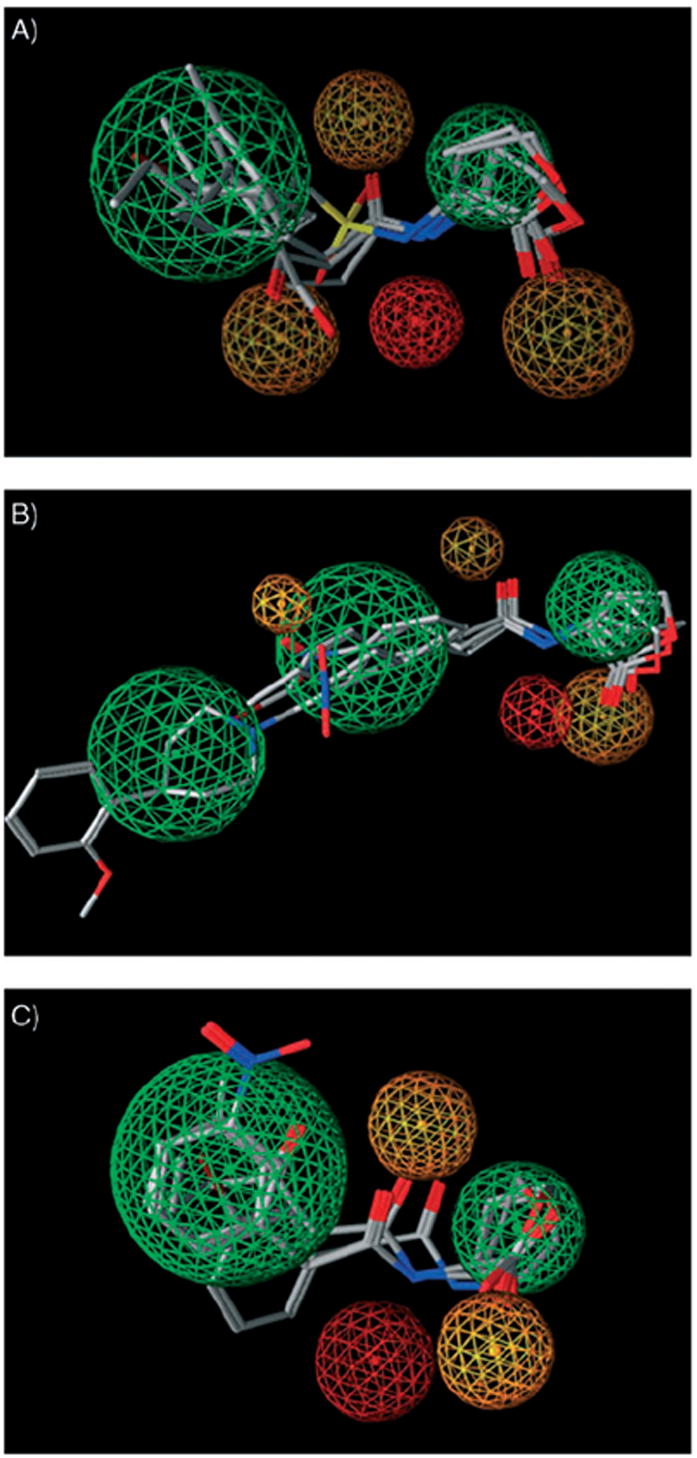
Pharmacophore models for AHL modulators of A) TraR, B) LasR, and C) LuxR calculated from primary screening data for libraries A–D. Green represents hydrophobic/aromatic features, red represents hydrogen-bond donor features, and orange represents hydrogen-bond acceptor features of the pharmacophore. Each pharmacophore is depicted with a selected set of active AHLs to highlight the varying features between the three pharmacophore models: OOHL (1), A11, C10, and D6 for TraR, OdDHL (2), C10, C14, and D18 for LasR, and OHHL (3), C6, C10, and C14 for LuxR.
Conclusions
Synthetic modulators of quorum sensing (QS) represent valuable chemical tools for fundamental studies of bacterial cell–cell signaling and for future biomedical and environmental applications. The majority of the known QS modulators in Gram negative bacteria are N-acylated homoserine lactones (AHLs). In this study, we have delineated key structural features of synthetic AHLs that render these ligands antagonists and agonists of QS in three species: A. tumefaciens, P. aeruginosa, and V. fischeri. These structure–activity relationships (SARs) were determined by the design and synthesis of four focused libraries of AHLs, and the systematic screening of these libraries in bacterial reporter strains. Both species-selective and multispecies modulators of QS were identified.
This work is significant, as it represents the first comparative study of AHL-derived QS modulators across different bacterial species. Moreover, this work provides a foundation on which to design next-generation AHLs, and synthetic ligands in general, with improved activities and selectivities for QS. The pharmacophores reported herein for AHL modulators of TraR, LasR, and LuxR will shape these future design efforts. Lastly, we have discovered several of the most potent synthetic antagonists and agonists of QS known (e.g., A4, B7, C10, and C14), as well as a set of photoactive AHL probes (C17, D20, and D21), which serves to further underscore the utility of focused combinatorial libraries for the identification of QS modulators. Ongoing work is directed at fully elucidating the mechanisms of QS agonism and antagonism by these synthetic AHLs and designing new AHL and non-AHL derived ligands; these studies will be reported in due course.
Experimental Section
Reporter gene assays
The three bacterial reporter strains used in this study were: A. tumefaciens WCF47 (ΔtraI) harboring a plasmid-born traI–lacZ fusion (pCF372),[13] E. coli DH5α harboring the LasR expression vector pJN105L and a plasmid-born lasI–lacZ fusion (pSC11),[32] and V. fischeri ES114 (ΔluxI).[33] The TraR, LasR, and LuxR antagonism and agonism assays were performed as previously reported.[19, 26] Positive controls for antagonism assays (native ligand at its EC50 value) and for agonism assays (native ligand at concentrations that gave maximal activity) were set to 100%. The concentrations of synthetic AHL ligand used in the antagonism and agonism assays, and the relative ratios of synthetic ligand to native ligand (1:1 to ~100:1) in the antagonism assays, were chosen to provide the most obvious differences between inhibitors and activators for each bacterial reporter strain (see Table 1 for details).[26] None of the control compounds (1–9) or library members was observed to be insoluble or affect bacterial growth over the time course of these assays. In addition, no ligand was found to degrade (by lactonolysis or reaction with biological reagents) over the time course of these assays (as determined by LC–MS or GC–MS; data not shown).
Pharmacophore calculations
All computational experiments were performed using the MOE software suite (v. 2006.08; Chemical Computing Group of Canada). Pharmacophores (PH4s) were calculated according to established methods.[34, 35] In brief, a database containing all of the AHL structures in libraries A–D was created by importing ChemDraw (v. 10.0, Std.; CambridgeSoft) .sdf files for each ligand into MOE. These compounds then were minimized using the MMFF force field to an energy gradient of <0.01 to create a 3D structural database. After minimization, a conformational import was performed in MOE to create a second database that retained 100 of the lowest energy conformations for each member of libraries A–D. A field for activity was created in this database, and each ligand was designated as either active (1) or inactive (0). This assignment was based on the primary antagonism and agonism data for the three bacterial strains investigated in this study. All of the ligands that showed either ≥50% inhibition or ≥25% activation of TraR, LasR, or LuxR were designated as active (1); those with lower activities were designated inactive (0). Separate PH4s were created for each R protein, which took into account both active and inactive ligands, by using the pharmacophore elucidator in MOE (see the Supporting Information for parameter details).
Each PH4 was examined for best score of accuracy (acc) in MOE, which was designated acc1 for active compounds and acc0 for inactive compounds. The three PH4s reported in this study were selected based on an acc1 value >0.50 (50%) and an acc0 value >0.50 (50 %). This selection was made such that >50% of the active compounds were able to match the PH4, while >50% of the inactive compounds were unable to match the PH4. Based on the overall structural similarity of the compounds in libraries A–D, this designation allowed for the determination of a PH4 that best describes the overall properties of an active AHL modulator for each R protein. The graphical representations of the PH4s shown in Figure 1 were generated in MOE by using the pharmacophore query editor with no modifications to the PH4s performed.
Acknowledgments
Financial support for this work was provided by the NIH (AI063326–01), Greater Milwaukee Foundation Shaw Scientist Program, Burroughs Wellcome Foundation, Camille & Henry Dreyfus Foundation, Research Corporation, Johnson & Johnson, DuPont, and UW–Madison. H.E.B. is an Alfred P. Sloan Foundation Fellow. G.D.G. was supported by an ACS Division of Medicinal Chemistry predoctoral fellowship. J.C.O. was supported by a Novartis Graduate Fellowship in Organic Chemistry. D.M.M. and R.J.W. were supported by traineeships from the UW–Madison NIH Chemistry–Biology Interface and Biotechnology training grants, respectively. We gratefully acknowledge Professors Stephen Winans (Cornell University), Barbara Iglewski (University of Rochester), Peter Greenberg (University of Washington), and Edward Ruby (UW–Madison) for generous donations of bacterial strains and advice on their manipulation, and Dr. Matthew Bowman for his assistance in the synthesis.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Waters CM, Bassler BL. Ann Rev Cell Dev Biol. 2005;21:319. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 2.Bassler BL, Losick R. Cell. 2006;125:237. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 3.de Kievit TR, Iglewski BH. Infect Immun. 2000;68:4839. doi: 10.1128/iai.68.9.4839-4849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg EP. Microbial Ecology and Infectious Disease. In: Rosenberg E, editor. American Society for Microbiology. Washington, D.C.: 1999. pp. 112–122. [Google Scholar]
- 5.Lyon GJ, Muir TW. Chem Biol. 2003;10:1007. doi: 10.1016/j.chembiol.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen TB, Givskov M. Microbiology. 2006;152:895. doi: 10.1099/mic.0.28601-0. [DOI] [PubMed] [Google Scholar]
- 7.Welch M, Mikkelsen H, Swatton JE, Smith D, Thomas GL, Glansdorp FG, Spring DR. Mol Biosyst. 2005;1:196. doi: 10.1039/b505796p. [DOI] [PubMed] [Google Scholar]
- 8.Fuqua C, Greenberg EP. Nat Rev Mol Cell Biol. 2002;3:685. doi: 10.1038/nrm907. [DOI] [PubMed] [Google Scholar]
- 9.Eberhard A, Widrig CA, McBath P, Schineller JB. Arch Microbiol. 1986;146:35. doi: 10.1007/BF00690155. [DOI] [PubMed] [Google Scholar]
- 10.Schaefer AL, Hanzelka BL, Eberhard A, Greenberg EP. J Bacteriol. 1996;178:2897. doi: 10.1128/jb.178.10.2897-2901.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Passador L, Tucker KD, Guertin KR, Journet MP, Kende AS, Iglewski BH. J Bacteriol. 1996;178:5995. doi: 10.1128/jb.178.20.5995-6000.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kline T, Bowman J, Iglewski BH, de Kievit T, Kakai Y, Passador L. Bioorg Med Chem Lett. 1999;9:3447. doi: 10.1016/s0960-894x(99)00626-5. [DOI] [PubMed] [Google Scholar]
- 13.Zhu J, Beaber JW, More MI, Fuqua C, Eberhard A, Winans SC. J Bacteriol. 1998;180:5398. doi: 10.1128/jb.180.20.5398-5405.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reverchon S, Chantegrel B, Deshayes C, Doutheau A, Cotte-Pattat N. Bioorg Med Chem Lett. 2002;12:1153. doi: 10.1016/s0960-894x(02)00124-5. [DOI] [PubMed] [Google Scholar]
- 15.Castang S, Chantegrel B, Deshayes C, Dolmazon R, Gouet P, Haser R, Reverchon S, Nasser W, Hugouvieux-Cotte-Pattat N, Doutheau A. Bioorg Med Chem Lett. 2004;14:5145. doi: 10.1016/j.bmcl.2004.07.088. [DOI] [PubMed] [Google Scholar]
- 16.Frezza M, Castang S, Estephane J, Soulere L, Deshayes C, Chantegrel B, Nasser W, Queneau Y, Reverchon S, Doutheau A. Bioorg Med Chem. 2006;14:4781. doi: 10.1016/j.bmc.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Glansdorp FG, Thomas GL, Lee JJK, Dutton JM, Salmond GPC, Welch M, Spring DR. Org Biomol Chem. 2004;2:3329. doi: 10.1039/B412802H. [DOI] [PubMed] [Google Scholar]
- 18.Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. J Am Chem Soc. 2005;127:12762. doi: 10.1021/ja0530321. [DOI] [PubMed] [Google Scholar]
- 19.Geske GD, O’Neill JC, Blackwell HE. ACS Chem Biol. 2007;2:315. doi: 10.1021/cb700036x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikeda T, Kajiyama K, Kita T, Takiguchi N, Kuroda A, Kato J, Ohtake H. Chem Lett. 2001:314. [Google Scholar]
- 21.Smith KM, Bu YG, Suga H. Chem Biol. 2003;10:81. doi: 10.1016/s1074-5521(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 22.Smith KM, Bu YG, Suga H. Chem Biol. 2003;10:563. doi: 10.1016/s1074-5521(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 23.Jog GJ, Igarashi J, Suga H. Chem Biol. 2006;13:123. doi: 10.1016/j.chembiol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Muh U, Hare BJ, Duerkop BA, Schuster M, Hanzelka BL, Heim R, Olson ER, Greenberg EP. Proc Natl Acad Sci USA. 2006;103:16948. doi: 10.1073/pnas.0608348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janssens JC, Metzger K, Daniels R, Ptacek D, Verhoeven T, Habel LW, Vanderleyden J, De Vos DE, De Keersmaecker SC. Appl Environ Microbiol. 2007;73:535. doi: 10.1128/AEM.01451-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geske GD, O’Neill JC, Miller DM, Mattmann ME, Blackwell HE. J Am Chem Soc. 2007;129:13613. doi: 10.1021/ja074135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang RG, Pappas T, Brace JL, Miller PC, Oulmassov T, Molyneaux JM, Anderson JC, Bashkin JK, Winans SC, Joachimiak A. Nature. 2002;417:971. doi: 10.1038/nature00833. [DOI] [PubMed] [Google Scholar]
- 28.Miller JH. Experiments in Molecular Genetics. 1972 Cold Spring;:352–355. [Google Scholar]
- 29.Meyer EA, Castellano RK, Diederich F. Angew Chem. 2003;115:1244. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:1210. [Google Scholar]
- 30.Fleming SA. Tetrahedron. 1995;51:12479. [Google Scholar]
- 31.Bottomley MJ, Muraglia E, Bazzo R, Carfi A. J Biol Chem. 2007;282:13592. doi: 10.1074/jbc.M700556200. [DOI] [PubMed] [Google Scholar]
- 32.Lee JH, Lequette Y, Greenberg EP. Mol Microbiol. 2006;59:602. doi: 10.1111/j.1365-2958.2005.04960.x. [DOI] [PubMed] [Google Scholar]
- 33.Lupp C, Urbanowski M, Greenberg EP, Ruby EG. Mol Microbiol. 2003;50:319. doi: 10.1046/j.1365-2958.2003.t01-1-03585.x. [DOI] [PubMed] [Google Scholar]
- 34.Meagher KL, Lerner MG, Carlson HA. J Med Chem. 2006;49:3478. doi: 10.1021/jm050755m. [DOI] [PubMed] [Google Scholar]
- 35.Jensen AA, Begum N, Vogensen SB, Knapp KM, Gundertofte K, Dzyuba SV, Ishii H, Nakanishi K, Kristiansen U, Stromgaard K. J Med Chem. 2007;50:1610. doi: 10.1021/jm070003n. [DOI] [PubMed] [Google Scholar]