

Abstract
The mitogen-activated protein kinases (MAPKs) are activated by extracellular signals, and translocate to the nucleus where they modulate transcription. Integrin-mediated cell adhesion to extracellular matrix (ECM) proteins is required for efficient transmission of MAPK-based signals initiated by growth factors. However, the modulation of G protein-coupled receptor (GPCR) signaling by adhesion is less well understood. In the present study we assessed the impact of cell adhesion on MAPK activation by muscarinic M3 receptors. The muscarinic agonist carbachol more efficiently promoted stress fiber formation and tyrosine phosphorylation of focal adhesion-associated proteins in M3 receptor-expressing cells adherent to fibronectin or collagen type I, as compared to polylysine. Overall MAPK activation was robust in cells adherent to all three substrata. However, total levels of MAPK and mitogen-activated protein kinase kinase (MEK) in the nucleus were significantly greater in cells adherent to ECM proteins for 2.5 hours, and levels of activated MAPK and MEK in the nuclei of these cells were higher following carbachol stimulation, relative to levels in cells adherent to polylysine. MEK inhibitors did not prevent adhesion-dependent translocation of MAPK and MEK to the nucleus, and increased nuclear phospho-MEK levels in carbachol-stimulated cells. The results suggest that adhesion of cells to ECM triggers the redistribution of MAPK and MEK to the nucleus, possibly as a result of the cytoskeletal rearrangements that accompany cell spreading. This may represent a mechanism for priming the nucleus with MEK and MAPK, leading to more rapid and pronounced increases in intranuclear phospho-MAPK upon GPCR stimulation.
Keywords: integrins, ERK, collagen, fibronectin, focal adhesion
INTRODUCTION
The mitogen-activated protein kinase/ extracellular signal-regulated kinase (MAPK/ERK) signaling module is a key mediator of cellular responses to growth factors, G-protein coupled receptor (GPCR) ligands, and extracellular matrix (ECM) proteins. [Chen et al., 1994; Cobb and Goldsmith, 1995; Widmann et al., 1999]. The classical MAPK cascade triggered by these stimuli involves sequential activation of the small GTPase Ras, the serine/threonine kinase Raf, the dual-specificity MAPK kinases (MEK1/2), and the MAPKs ERK1 and ERK2 [Seger and Krebs, 1995; Widmann et al., 1999; Garrington and Johnson, 1999]. MAPK has multiple substrates in membranes, in the cytoplasm, in the cytoskeletal compartment, and in the nucleus. Many of the nuclear targets of MAPK are transcription factors, and translocation of MAPK to the nucleus is necessary for its effects on transcriptional regulation [Brunet et al., 1999; Kim-Kaneyama et al., 2000]. Most studies indicate that nuclear translocation of MAPK involves active transport of dimerized, phosphorylated MAPK, although passive diffusion of monomers has also been reported [Khokhlatchev et al., 1998; Adachi et al., 1999]. MEK1/2, in contrast, is largely confined to the cytoplasm [Lenormand et al., 1993; Zheng and Guan,1994]. Although MEK1/2 diffuses constitutively into the nucleus, it is rapidly transported out again in a manner dependent on a nuclear export signal located in its N-terminus [Fukuda et al., 1996; Jaaro et al., 1997]. In addition to serving as the upstream activator of MAPK, MEK1/2 may play the additional roles of anchoring inactive MAPK in the cytoplasm, and escorting MAPK out of the nucleus once it is inactivated by intra-nuclear phosphatases [Fukuda et al., 1997; Adachi et al., 2000].
A number of recent studies have demonstrated that growth factor-induced activation of MAPK, and translocation to the nucleus, is dependent on cell adhesion [Miyamoto et al., 1996; Renshaw et al., 1997; Lin et al., 1997; Aplin and Juliano, 1999; Danilkovitch et al., 2000; Aplin and Juliano, 2001; Aplin et al., 2001; Aplin, 2003]. Integrins, the principal mediators of cell adhesion, form clusters upon engagement by ECM proteins, initiating the recruitment of additional structural and signaling proteins. In cultured cells, these attachment sites are particularly large and complex, and are known as focal adhesions. Bundles of actin filaments, or stress fibers, are anchored to these sites via interactions with other components of the complex including talin, α-actinin, tensin, and vinculin [Clark and Brugge, 1995; Burridge and Chrzanowska-Wodnicka, 1996; Burridge et al., 1997]. The mechanism by which cell adhesion regulates the transmission of signals to MAPK from growth factor receptors is still undefined, although cytoskeletal components appear to play a role [Aplin and Juliano, 1999; Juliano, 2002].
Signaling by GPCRs may also be modulated by adhesion to ECM proteins. An earlier report from this laboratory concluded that tyrosine phosphorylation of the focal adhesion-associated proteins paxillin and focal adhesion kinase (FAK) in response to muscarinic M3 receptor stimulation is adhesion dependent, because it was disrupted by soluble peptides containing the amino acid sequence arginine-glycine-aspartic acid-serine (RGDS), which inhibits integrin binding to ECM proteins [Slack, 1998]. Subsequently, it was demonstrated that RGDS-containing peptides block activation of MAPK by lysophosphatidic acid and bradykinin in PC12 cells, and by gonadotropin-releasing hormone in transfected human embryonic kidney (HEK) cells, but not MAPK activation by thrombin and lysophosphatidic acid in rat1 fibroblasts, suggesting that dependence of GPCR signaling on focal adhesion integrity may be cell-type specific [Della Rocca et al., 1999; Davidson et al., 2004]. Coupling of the P2Y class of purinergic receptors to MAPK in endothelial cells was also reported to be adhesion-dependent, and was attenuated in cells maintained in suspension, or replated onto polylysine. Interestingly, upstream elements of this pathway, including phosphatidylinositol turnover and activation of protein kinase C, were adhesion-independent, whereas activation of MEK and MAPK further downstream of the receptor required cell adhesion [Short et al., 2000].
For this study, we examined the effect of cell adhesion on activation of MAPK by muscarinic receptors of the M3 subtype [Peralta et al., 1988]. Adhesion of M3 receptor-expressing HEK cells to the ECM proteins fibronectin or collagen type I, but not to polylysine, resulted in a sustained redistribution of MEK and MAPK to the nucleus that was not affected by MEK inhibitors. Upon stimulation with carbachol, levels of activated MAPK in the nucleus rose to significantly higher levels in cells replated on ECM proteins, and the duration of the increase was noticeably longer in cells replated on collagen type I than in cells adherent to fibronectin. The results suggest that redistribution of MEK and MAPK to the nucleus during cell spreading facilitates the transmission of M3 receptor-initiated signals to the nucleus, and may be an important determinant of the transcriptional effects of GPCR activation.
MATERIALS AND METHODS
Materials
Antibodies were obtained from the following sources: anti-paxillin, anti-phosphotyrosine (clone PY20), and goat peroxidase-linked anti-mouse IgG from BD Biosciences (San Diego, CA); anti-MEK1/2, anti-phospho-MEK1/2 (Ser217/221), anti-MAPK and anti-phospho-MAPK (Thr202/Tyr204) from Cell Signaling Technology (Beverly, MA); polyclonal anti-phosphotyrosine from Upstate (Charlottesville, VA); anti-histone H1 and peroxidase-linked anti-phosphotyrosine (clone PY99) from Santa Cruz (Santa Cruz, CA), goat peroxidase-linked anti-rabbit IgG from Bio-Rad Laboratories (Hercules, CA). Protein G agarose was purchased from Oncogene Research Products (Cambridge MA) and protein A Sepharose from Amersham Biosciences (Piscataway, NJ). PD98059 was purchased from Calbiochem (San Diego, CA) and U0126 from Cell Signaling Technology. Alexa Fluor 488 phalloidin and Prolong Antifade reagent were obtained from Molecular Probes (Eugene, OR). Culture dishes and cover-slips pre-coated with fibronectin, and acid-solubilized rat-tail collagen type I, were obtained from BD Biosciences. Reagents, equipment and mini-gels for electrophoresis were supplied by Bio-Rad Laboratories (Hercules, CA). Other reagents and supplies were obtained from Sigma Chemical Company (St. Louis MO) or Fisher Scientific (Pittsburgh, PA).
Cell Culture
HEK cells stably transfected with muscarinic M3 receptors (HEK-M3 cells) were maintained in Dulbecco's modified Eagle medium (DMEM)/F12 supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA). Cells were seeded onto 100 mm tissue culture dishes 3 to 4 days prior to an experiment. Cultures were incubated overnight in serum-free DMEM prior to an experiment. All subsequent pharmacological treatments were carried out in serum-free DMEM. For adhesion experiments, cells were grown on tissue culture dishes, incubated overnight in serum-free DMEM, and detached by incubating for 5 minutes at 37 °C in non-enzymatic cell dissociation solution (Sigma).
The cell suspensions were triturated, gently pelleted in a clinical centrifuge, and resuspended in DMEM. When desired, MEK inhibitors or vehicle (DMSO) were added to the cell suspension, and the cells were then re-plated onto culture dishes coated with fibronectin, collagen type I or poly-d-lysine, and allowed to adhere for 2.5 hours. Culture dishes were coated with collagen type I or poly-d-lysine (5 µg/cm2) for 2 hours at room temperature, then rinsed with sterile water and allowed to dry. The dishes were refrigerated until use.
Cell Lysis and Immunoblot Analysis
Following treatment, the medium was removed and cells were collected in 500 µL of lysis buffer A containing 1% Nonidet P-40, 0.05% SDS, 0.5% deoxycholate, 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM sodium orthovanadate, 25 mM NaF, 2 mM 4-(2-aminoethyl)benzenesulfonylfluoride (AEBSF), 1 µg/ml leupeptin and 2 µg/ml aprotinin. Lysates were centrifuged, normalized for protein content, and incubated overnight with immunoprecipitating antibodies, usually at concentrations of 2 to 5 µg/500 µg protein, and protein A or G Agarose (3 mg per sample), centrifuged and washed three times (in a washing buffer containing 0.1 % Triton X-100, 25 mM Tris pH 7.5, 250 mM NaCl, and 1 mM sodium orthovanadate). To make nuclear and cytoplasmic fractions, cells were collected in phosphate-buffered saline (PBS), pH 7.4, supplemented with a protease inhibitor cocktail (Sigma) and fractionated using NE-PER cell fractionation reagents (Pierce Biotechnology Inc., Rockford, IL) according to the manufacturers instructions, except that the CER I and NER reagents were supplemented with 1 mM sodium orthovanadate and protease inhibitor cocktail (Sigma). In some experiments, nuclear and cytosolic fractions were prepared using an alternate method [Bijur and Jope, 2001] with slight modifications. Cells were collected in a lysis buffer (10 mM Tris, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.05% Nonidet P-40, 1 mM EGTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 1 µg/ml okadaic acid, 100 µM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 5 µg/ml pepstatin A) and centrifuged at 2,700 × g for 10 min at 4° The supernatant (cytosolic fraction) was collected and cleared by centrifugation at 20,000 × g for 20 min at 4°C. The nuclei in the pellet were washed twice by resusupending them in 200 µl of a washing buffer (10 mM PIPES, pH 6.8, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 25 mM NaCl, 1 mM sodium orthovanadate, 50 mM sodium fluoride, with protease inhibitors) and centrifuging at 2,700 × g for 5 min. The nuclei were resuspended a third time in 100 µl washing buffer, layered on top of 1 ml of sucrose buffer (1 M sucrose, 1 mM sodium orthovanadate, 50 mM sodium fluoride with protease inhibitors), and centrifuged at 2,700 × g for 10 min at 4 °C. The pellet was washed once with lysis buffer, centrifuged again, resuspended in 100 µl of extraction buffer (20 mM HEPES, pH 7.9, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1 mM β-glycerophosphate, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 1 µg/ml okadaic acid, with protease inhibitors) by mixing vigorously for 15 s on a vortex mixer, then incubated on ice for 30 min. The extracts were cleared by centrifugation at 20,000 × g for 20 min at 4 °C. The protein content of cell lysates and subcellular fractions was determined using the bicinchoninic acid method. Aliquots of immunoprecipitated proteins, cell lysates, and nuclear and cytoplasmic extracts normalized for protein content were size-fractionated by SDS-PAGE Proteins were transferred to polyvinylidene difluoride membranes, and the membranes blocked with 3% gelatin or 5% milk in Tris-buffered saline containing 0.15% Tween-20 (TBST). Membranes were probed with primary antibodies, washed 3 times in TBST, and incubated in peroxidase-linked secondary antibodies. After washing 3 times in TBST, immunoreactive bands were detected using enhanced chemiluminescence reagents (Perkin-Elmer Life-Sciences, Inc., Boston, MA, or Pierce Biotechnology Inc.). Bands were visualized on a Kodak ImageStation 440CF and quantitated using Kodak 1D software.
Immunofluorescence Microscopy
Serum-deprived cells were placed in suspension using non-enzymatic cell dissociation medium, resuspended in serum-free DMEM, and replated onto glass cover-slips coated with fibronectin, poly-d-lysine, or rat tail collagen type I. The cells were allowed to adhere for 2.5 hours, then treated with control or carbachol-containing medium for 20 minutes. The cells were fixed in 3.0% paraformaldehyde in PBS for 5 minutes, permeabilized in 0.1% Triton X-100 in PBS for 5 minutes, and blocked for 60 minutes in 1% BSA in PBS. Cells were incubated for 30 minutes with Alexa Fluor 488 phalloidin conjugate at a final concentration of 5 U/ml, and the cover-slips were mounted on glass slides using Prolong Antifade reagent. The preparations were examined by fluorescence microscopy, photographed with a Spot RT KE camera, and processed using SPOT software (Diagnostic Instruments, Sterling Heights, MI).
Statistical Analysis
Comparisons based on immunoblotting data were derived from samples processed on the same blot. The statistical significance of differences was estimated by analysis of variance followed by Fisher’s Least-Significant-Difference test. Differences were taken to be significant at p < 0.05.
RESULTS
Fibronectin and Collagen Type I Potentiate M3 Receptor-Evoked Tyrosine Phosphorylation of Focal Adhesion-Associated Proteins
Earlier work from this laboratory demonstrated that stimulation of muscarinic M3 receptors by the acetylcholine analog carbachol evokes tyrosine phosphorylation of the focal adhesion-associated proteins paxillin and FAK, and formation of stress fibers and focal adhesions, in cells adherent to fibronectin. These responses were attenuated in cells attached to polylysine-coated surfaces [Slack, 1998]. Fibronectin is a ligand for the integrin α5β1, which is expressed by HEK cells [Bodary and McLean, 1990]. These cells also express the collagen-binding integrins α1β1 and α2β1 [Bodary and McLean, 1990]. To determine if the ECM protein collagen type I was capable of supporting M3 receptor signaling, HEK-M3 cells were replated on polylysine, fibronectin (used as a positive control), or collagen type I, allowed to adhere for 2.5 hours, and stimulated with carbachol. Tyrosine phosphorylation was then assessed by immunoblot analysis.
Adhesion to fibronectin [Slack, 1998], or to collagen type I (not shown) induced tyrosine phosphorylation of paxillin and FAK. This response peaked within 30 minutes, and declined to near basal levels within 2.5 hours. Subsequent stimulation with carbachol induced robust tyrosine phosphorylation of these proteins in cells replated on fibronectin, whereas phosphorylation was significantly weaker in cells replated on polylysine (Fig. 1A, left panel). Overall, tyrosine phosphorylation of paxillin evoked by carbachol exhibited greater dependence on the presence of ECM proteins than FAK phosphorylation. Like fibronectin, collagen type I supported robust tyrosine phosphorylation of paxillin in response to carbachol, whereas the response was much weaker in cells replated on polylysine (Fig. 1A, right panel). In contrast, activation of MAPK by carbachol, assessed by measuring levels of activated, tyrosine/threonine phosphorylated MAPK (phospho-MAPK) in cell lysates, was of similar magnitude in cells replated on either polylysine, fibronectin, or collagen type I (Fig. 1B). These experiments were repeated at least 5 times with similar results.
Fig. 1.

Effect of ECM proteins on M3 receptor-evoked tyrosine phosphorylation and MAPK activation. Serum-deprived HEK-M3 cells were placed in suspension, and replated onto culture dishes coated with polylysine (pl), fibronectin (fn), or collagen type I (cI). The cells were allowed to adhere for 2.5 hours, then stimulated with control medium (con), or medium containing 100 µM carbachol (carb), for 20 minutes. (A) Tyrosine phosphorylated proteins were immunoprecipitated (IP) with anti-phosphotyrosine antibodies (clone PY20), resolved by SDS-PAGE, and immunoblotted with peroxidase-linked anti-phosphotyrosine antibodies (clone PY99). Arrows indicated paxillin (pax) and focal adhesion kinase (FAK). Note that FAK appears as a doublet in this cell line [Slack, 1998]. (B) Total MAPK and phospho-MAPK were detected by immunoblot analysis of cell lysates.
M3 Receptor Activation Promotes Stress Fiber Formation in Cells Adherent to Fibronectin or Collagen Type I
The ability of fibronectin and collagen type I to promote cell spreading and stress fiber formation was compared in the next series of experiments. Both fibronectin and collagen type I promoted HEK-M3 cell spreading, which was pronounced within 1 hour after replating. At this time, cells adherent to polylysine were still rounded in appearance (Fig. 2A). Activation of M3 receptors was previously reported to stimulate stress fiber formation and focal adhesion formation in cells plated on fibronectin [Slack, 1998]. To compare this response with that observed in cells adherent to collagen type I, HEK-M3 cells plated onto coverslips coated with polylysine or the desired ECM protein were treated with control or carbachol-containing DMEM for 20 min, then fixed and stained with fluorescence-conjugated phalloidin to visualize actin filaments. Serum-deprived cells adhering to polylysine-coated coverslips showed F-actin staining at the cell periphery, but did not form stress fibers in response to carbachol (Fig. 2B). In contrast, carbachol treatment induced stress fiber formation in cells plated on fibronectin or collagen type I (Fig. 2B).
Fig. 2.

Cell spreading and M3 receptor-evoked stress fiber formation is promoted by fibronectin and collagen type I. HEK-M3 cells were serum-deprived overnight, placed in suspension and replated onto dishes coated with polylysine, fibronectin, or collagen type I. (A) Cells were allowed to adhere for 1 hour, and photographed under phase-contrast using a 10X objective. (B) Cells replated on coverslips coated with polylysine, fibronectin, or collagen type I were allowed to adhere for 2.5 hours, then treated with control medium (con), or medium containing 100 µM carbachol (carb), for 20 minutes. Cells were fixed and permeabilized, and immunofluorescence labeling was carried out with Alexa Fluor 488-conjugated phalloidin to detect F-actin. Bar, 10 µM.
Nuclear Localization and Activation of MAPK in Response to Carbachol is Differentially Modulated by ECM Proteins
It has been demonstrated that MAPK translocates to the nucleus upon activation, and that this response, when elicited by growth factors, is adhesion dependent [Aplin et al., 2001; Aplin et al., 2002; Aplin, 2003]. In order to determine if ECM proteins modulate nuclear translocation of phospho-MAPK in response to M3 receptor stimulation, HEK-M3 cells adherent to different substrates were challenged with carbachol for varying periods of time, lysed, and separated into nuclear and cytosolic fractions using the NE-PER method (Pierce). Immunoblot analysis demonstrated that the nuclear fractions were enriched in histone H1, a nuclear protein, whereas cytoplasmic fractions were enriched in paxillin, which is principally located in cytosolic and cytoskeletal compartments (Fig. 3A). Levels of nuclear phospho-MAPK were significantly increased within two minutes following the addition of carbachol to cells adherent to either polylysine or fibronectin, and remained elevated for at least 10 minutes. At two minutes, the response was significantly greater in cells adherent to fibronectin, as compared to polylysine, but it declined rapidly such that by 5 minutes, levels of nuclear phospho-MAPK were similar in both groups (Fig. 3B). Levels of total MAPK in nuclear fractions were significantly elevated at the zero time-point (prior to the addition of carbachol) in cells adherent to fibronectin, relative to levels in cells adherent to polylysine (Fig. 3C), but declined to control levels within five minutes after the addition of carbachol. In cells adherent to polylysine, in contrast, total nuclear MAPK levels were relatively constant over the entire course of carbachol treatment (Fig. 3C).
Fig. 3.
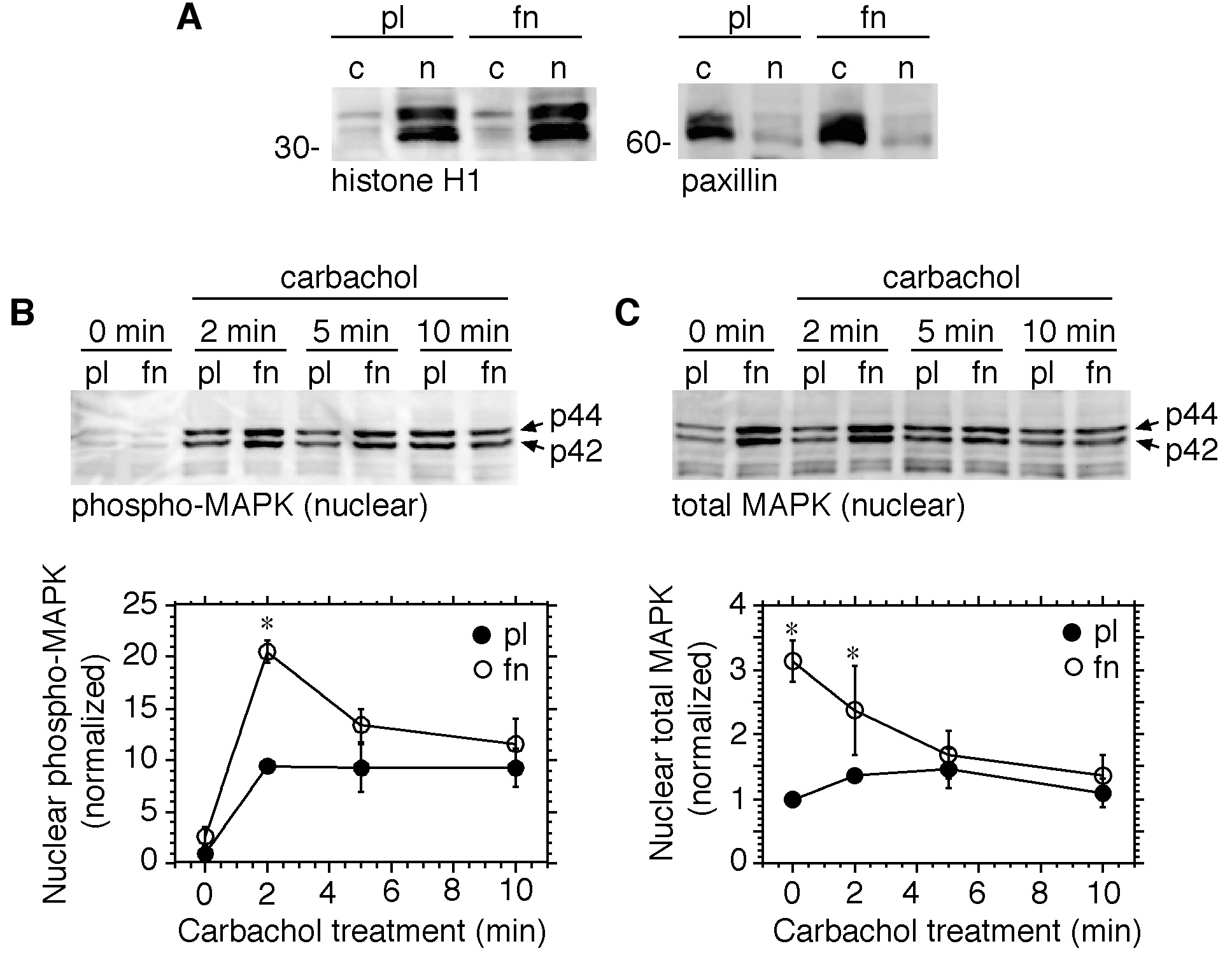
M3 receptor-mediated activation of nuclear MAPK is increased in cells adherent to fibronectin. Serum-deprived HEK-M3 cells were placed in suspension, and replated onto dishes coated with polylysine (pl) or fibronectin (fn). After 2.5 hours, the cells were stimulated with carbachol (100 µM) for 0 to 10 minutes. Nuclear and cytosolic fractions were prepared and analyzed by immunoblotting. (A) Cytosolic (c) and nuclear (n) fractions were immunoblotted with antibodies to histone H1 (left panel) and paxillin (right panel) to assess the purity of the fractions. Nuclear fractions were assayed for phospho-MAPK (B) or total MAPK (C). Bands were quantitated by densitometry and values were normalized to those derived from cells adherent to polylysine at 0 minutes (carbachol treatment). Results are expressed as means ± s.e.m. from 3–4 experiments. *, Significantly different from corresponding time-matched control group.
When cells adherent to collagen type I were compared to cells replated on polylysine, a somewhat different pattern emerged. Again, nuclear levels of phospho-MAPK were significantly increased by carbachol within 2 minutes in both groups. However, in cells plated on collagen type I, nuclear phospho-MAPK levels remained significantly elevated above levels in the polylysine controls for at least 10 minutes following carbachol treatment (Fig. 4A). Levels of total MAPK in the nucleus were elevated in collagen type I-adherent cells prior to the addition of carbachol, and remained significantly higher than levels observed in polylysine controls for 5 minutes after the start of stimulation, before declining toward control levels at 10 minutes (Fig. 4B). When levels of nuclear phospho-MAPK were normalized to total nuclear MAPK, the increases induced by carbachol were of similar magnitude whether cells were adherent to polylysine or collagen type I (Fig. 4C). Thus, although total nuclear MAPK was increased by adhesion to collagen, the proportion of the nuclear MAPK pool that became phosphorylated in response to carbachol remained constant. Similar results were obtained when comparing cells adherent to polylysine and fibronectin (not shown). Because carbachol-evoked increases in nuclear phospho-MAPK were more persistent in cells adherent to collagen type I, relative to fibronectin, subsequent experiments were focussed on the modulation by collagen type I of M3 receptor signaling, using polylysine as a control substrate.
Fig. 4.

M3 receptor-mediated activation of nuclear MAPK is increased in cells adherent to collagen type I. Serum-deprived cells were placed in suspension, and replated onto dishes coated with polylysine (pl) or collagen type I (cI). After 2.5 hours, the cells were stimulated with carbachol (100 µM) for 0 to 10 minutes. Nuclear fractions were prepared and analyzed by immunoblotting for phospho-MAPK (A) or total MAPK (B). Bands were quantitated by densitometry and values were normalized to those derived from cells adherent to polylysine at 0 minutes (carbachol treatment). Results are expressed as means ± s.e.m. from 3–4 experiments. *, Significantly different from corresponding time-matched control group. (C) The ratio of phospho-MAPK to total MAPK at each time point was calculated. The resulting values are expressed as means ± s.e.m. from 3 experiments.
Cell Adhesion to Collagen Type I Causes Nuclear Translocation of MEK1/2
It has been reported that MEK, as well as MAPK, is transported to the nucleus upon mitogenic stimulation [Jaaro et al., 1997; Yao et al., 2001], and interactions between MEK and MAPK may regulate the subcellular localization of MAPK [Fukuda et al., 1997; Adachi et al., 2000; Pouysségur et al., 2002]. Levels of MEK and phospho-MEK were measured in nuclear extracts from cells allowed to adhere to collagen type I or polylysine, and then stimulated with carbachol for 5 min. Nuclear levels of total MEK1/2 were significantly higher in cells adherent to collagen type I relative to those replated on polylysine, at 2.5 hours after re-plating (Fig. 5A, control), whereas phospho-MEK1/2 was barely detectable in cells on either substrate (Fig. 5B, control). Carbachol treatment for 5 minutes rapidly increased levels of phospho-MEK1/2 in the nucleus in cells adherent to either substrate, but the maximum response was significantly greater in cells adherent to collagen type I (Fig. 5B, carbachol).
Fig. 5.

Activation of MEK1/2 by M3 receptor stimulation is increased in cells adherent to collagen type I. Serum-deprived cells were placed in suspension, and re-plated onto dishes coated with polylysine (pl) or collagen type I (cI). After 2.5 hours, the cells were stimulated with carbachol (100 µM) for 5 minutes or left untreated (control). Nuclear fractions were analyzed by immunoblotting for total MEK1/2 (A) or phospho-MEK1/2 (B). Bands were quantitated by densitometry. Values were normalized to the control/polylysine treatment group (A) or the carbachol/polylysine group (B) and expressed as means ± s.e.m. from 6–9 experiments. *, Significantly different from the corresponding treatment group on polylysine; **, significantly different from the control group on the same substrate.
Adhesion-Dependent Nuclear Translocation of MAPK and MEK1/2 is Not Prevented by MEK Inhibitors
The results show that several hours after cells are replated onto fibronectin or collagen type I, increased levels of non-phosphorylated MAPK and MEK1/2 are observed in the nucleus. In order to determine if activation of MAPK or MEK1/2 is necessary for nuclear translocation to occur, cells were placed in suspension, then replated in the presence of the MEK inhibitor U0126 (10 µM), or the vehicle DMSO. After 2.5 hours, cells were treated for 5 minutes with carbachol-containing or control DMEM. As before, carbachol increased nuclear phospho-MAPK levels in cells plated on both substrates, and the inhibitor significantly reduced this response (Fig. 6A, left panel). However, the MEK inhibitor did not prevent the increase in total nuclear MAPK observed in cells replated onto collagen type I (Fig. 6A, right panel). Levels of total MEK1/2 in the nucleus were also increased in cells plated on collagen type I, and were unaffected by the presence of U0126 or by subsequent treatment with carbachol alone, although in the combined presence of carbachol and U0126, MEK1/2 levels in the nucleus were significantly lower (Fig. 6B, right panel). As noted earlier, carbachol more than doubled levels of phospho-MEK1/2 in cells replated on either substrate (Fig. 6B, left panel). However, the effect of carbachol was strikingly potentiated in the presence of U1026 in cells plated on polylysine. Moreover, the inhibitor alone caused a comparable potentiation of MEK1/2 phosphorylation in cells adherent to collagen type I, which was increased still further by the addition of carbachol. (Fig. 6B, left panel). Qualitatively similar results were obtained with the MEK inhibitor PD98059, although the observed increases in phospho-MEK were of lesser magnitude than those that occurred in cells treated with U0126. This difference probably reflects the ability of U0126 to inhibit both MEK1 and MEK2, whereas PD98059 preferentially inhibits MEK1 [Alessi et al., 1995; Favata et al., 1998].
Fig. 6.

A MEK inhibitor enhances accumulation of phospho-MEK1/2 in the nucleus. Serum-deprived cells were placed in suspension, treated with DMSO or U0126 (10 µM), and transferred to dishes coated with polylysine (pl) or collagen type I (cI). After 2.5 hours the cells were stimulated with control medium (con), or medium containing 100 µM carbachol (carb), for 5 minutes. Nuclear fractions were prepared and analyzed by immunoblotting for phospho-MAPK (A, left panel), total MAPK (A, right panel), phospho-MEK1/2 (B, left panel) or total MEK1/2 (B, right panel). Bands were quantitated by densitometry. Values were normalized to the control/polylysine group, and expressed as means ± s.e.m. *, Significantly different from the corresponding treatment group on polylysine; **, significantly different from the control group on the same substrate; ***, significantly different from the carbachol-treated group on the same substrate.
Activation of nuclear but not cytosolic MAPK by carbachol is enhanced in cells adherent to collagen type I
In order to verify the results described in the preceding section, an alternate method [Bijur and Jope, 2001] was used to prepare nuclear and cytosolic fractions (see Materials and Methods). Cells were allowed to adhere to polylysine or collagen type I in the presence of DMSO or U0126, then stimulated with carbachol-containing or control medium for 5 min. Immunoblot analysis of nuclear and cytosolic fractions was carried out to show the relative purity of the preparations (Fig. 7A). As before, unphosphorylated MAPK accumulated in the nuclear fraction of cells adherent to collagen type I, and levels of nuclear phospho-MAPK induced by carbachol were greater in cells adherent to collagen than in cells replated on polylysine (Fig. 7B). In contrast, levels of total and phospho-MAPK in the cytosol were not influenced by adhesion of cells to ECM (Fig. 7C).
Fig. 7.

Activation of nuclear but not cytosolic MAPK by carbachol is enhanced in cells adherent to collagen type I. Serum-deprived cells were placed in suspension, treated with DMSO or U0126 (10 µM), and transferred to dishes coated with polylysine (pl) or collagen type I (cI). After 2.5 hours the cells were stimulated with control medium (con), or medium containing 100 µM carbachol (carb), for 5 minutes. Nuclear and cytosolic fractions were prepared according to the method of Bijur and Jope (2001). (A) Cytosolic (c) and nuclear (n) fractions were immunoblotted with antibodies to histone H1 (upper panel) and paxillin (lower panel) to assess the purity of the fractions. Nuclear (B) and cytosolic (C) fractions were analyzed by immunoblotting for phospho-MAPK and total MAPK. This experiment was repeated 4 times with similar results.
DISCUSSION
Cell stimulation by growth factors or GPCR ligands, or upon binding of integrins to ECM proteins, activates MAPK via sequential activation of Ras, Raf, and MEK. Although growth factors activate Ras even in suspended cells, transmission of the signal to the more distal components of the cascade, e.g. MEK, or, in some cases, Raf, requires integrin-dependent cell adhesion [Miyamoto et al., 1996; Renshaw et al., 1997; Lin et al., 1997]. Similarly, activation of MEK and MAPK by G protein-coupled P2Y receptors is integrin-dependent, whereas upstream events, including production of inositol phosphates, and release of calcium from internal stores, are not [Short et al., 2000]. In growth factor-stimulated fibroblasts, activated MAPK must be translocated to the nucleus in order to promote cell division [Brunet et al., 1999], and this process has also been shown to require cell adhesion [Danilkovitch et al., 2000; Aplin et al., 2001]. Once activated, phosphorylated MAPK forms dimers, which promotes its translocation into the nucleus via an active transport mechanism [Khokhlatchev et al., 1998; Adachi et al., 1999; Cobb and Goldsmith, 2000]. However, activated monomeric MAPK can also enter the nucleus via passive diffusion, albeit less rapidly [Adachi et al., 1999; Volmat et al., 2001]. MEK, the upstream activator of MAPK, is primarily localized in the cytoplasm, where it interacts with MAPK, and, in resting cells, serves to anchor it in the cytoplasm [Fukuda et al., 1997]. In stimulated cells, MAPK dissociates from MEK in a tyrosine phosphorylation-dependent fashion [Adachi et al., 1999] and translocates to the nucleus. It is subsequently dephosphorylated and then transported from the nucleus in a complex with MEK [Adachi et al., 2000].
Our own results provide evidence for an additional mechanism by which cell adhesion may modulate signal-dependent MAPK activation. Although focal adhesion formation in response to carbachol, assessed by tyrosine phosphorylation of paxillin and FAK, and formation of stress fibers, was clearly attenuated in cells replated on polylysine (Fig. 1 and Fig. 2), we found that overall activation of MAPK by carbachol, as measured in cell lysates, was comparable in cells adherent to polylysine, fibronectin, and collagen type I. It was only upon examining cellular subfractions did it become clear that activation of nuclear, but not cytosolic, MAPK by carbachol was reduced in cells replated on polylysine. Earlier work from other laboratories showed that activation of MAPK by growth factors, as measured in whole cell lysates, was significantly reduced in cells maintained in suspension, or replated on polylysine [Renshaw et al., 1997; Lin et al., 1997], indicating that transmission of the signal along the MAPK pathway was interrupted in the absence of integrin-mediated adhesion. Our results, in contrast, suggest that cell adhesion to ECM proteins served principally to modulate the subcellular distribution of MAPK prior to M3 receptor activation, and was not required for receptor-evoked activation of the MAPK cascade per se. This may reflect the fact that MAPK stimulation by carbachol in HEK-M3 cells exhibits a substantial PKC-dependent component [Slack, 2000]. Both phosphoinositide hydrolysis, which generates the PKC activator diacylglycerol, and release of intracellular calcium, which is required for activation of conventional PKCs, are triggered in response to GPCR stimulation, and are reported to be independent of adhesion [Short et al., 2000]. PKC may in turn directly activate Ras or Raf, or even MEK [Hawes et al., 1995; Ueda et al., 1996; Schöwasser et al., 1998; Marais et al., 1998] in an adhesion–independent manner.
Although many studies suggest that activation of MAPK must precede translocation to the nucleus [Cobb and Goldsmith, 2000], the nuclear export inhibitor leptomycin B caused nuclear MAPK levels to increase even in quiescent cells that were treated with U0126 to remove residual MEK activity [Volmat et al., 2001]. Overexpression of MAPK also results in gradual accumulation in the nucleus in the absence of an activating stimulus [Lenormand et al., 1993; Wolf et al., 2001]. The latter occurrence was attributed to saturation of a limited number of cytoplasmic anchoring sites, and, in fact, concomitant overexpression of MEK causes retention of exogenous MAPK in the cytoplasm [Wolf et al., 2001]. Other cytosolic anchoring sites for MAPK include microtubules, and MAPK phosphatases [Reszka et al., 1995; Fukuda et al., 1997; Brunet et al., 1999]. If a significant proportion of MAPK in HEK cells is anchored to cytoskeletal elements, our results could be explained by positing that the cytoskeletal rearrangements that occur upon integrin activation and cell spreading on ECM proteins are sufficient to displace MAPK from some of its cytosolic anchoring sites, and cause its translocation to the nucleus, even in the presence of a MEK inhibitor [Aplin and Juliano, 2001]. During long-term stimulation with a mitogen, activated, translocated MAPKs become inactive, yet are retained in the nucleus by a mechanism that requires synthesis of nuclear anchoring proteins; candidates for the latter include MAPK phosphatases [Volmat et al., 2001]. Most interesting was the observation that inactive MAPKs retained in the nucleus after prolonged stimulation could be reactivated by treatment with the tyrosine phosphatase inhibitor oxovanadate, but not by selective re-activation of the MAPK pathway [Volmat et al., 2001]. Taken together, the results suggest that MAPK that is sequestered in the nucleus in an inactive form may be rapidly reactivated by certain stimuli. Thus, prolonged exposure to growth factors, or, as the present study suggests, interactions with the ECM that lead to cell spreading, may act as a priming mechanism, loading the nucleus with MAPK that can be rapidly activated by specific stimuli which may differ from those regulating the classical MAPK cascade. Consistent with this hypothesis, the ratio of phospho- to total MAPK in the nucleus of carbachol-stimulated cells was the same whether cells were plated on polylysine or collagen (Fig. 4C); that is, the increase in nuclear phospho-MAPK appeared to be correlated with an elevation in total nuclear MAPK in cells adherent to collagen type I.
Most of the available evidence indicates that MEK is primarily located in the cytoplasm. Although MEK moves from the cytoplasm to the nucleus upon mitogenic stimulation, it is rapidly exported via a nuclear export signal (NES) at its N-terminus [Fukuda et al., 1996; Jaaro et al., 1997; Tolwinski et al., 1999]. Thus, even after stimulation [Lenormand et al., 1993; Pouysségur et al., 2002], or in cells transfected with activated MEK1 [Aplin et al., 2001], significant accumulation of MEK in the nucleus was not observed. Although some studies indicated that nuclear translocation of MEK requires activation [Jaaro et al., 1997; Lenormand et al., 1998; Tolwinski et al., 1999], other work indicates that inactive MEK also diffuses constantly into the nucleus in unstimulated cells, before being rapidly exported via active transport [Adachi et al., 2000; Yao et al., 2001].
Our results show that integrin-mediated cell spreading on collagen type I was associated with the accumulation of unphosphorylated MEK1/2 in the nucleus (Fig. 5). At 2.5 hours after replating on collagen type I, phospho-MEK levels in the nucleus were low, but increased upon carbachol stimulation of cells adherent to collagen I. Both total nuclear MEK levels, and phosphorylation of nuclear MEK by carbachol, were reduced in cells replated on polylysine. Total MEK levels in the nucleus were unaffected by short-term stimulation with carbachol (Fig. 6B, right panel), which is consistent with the possibility that M3 receptor stimulation results in phosphorylation of nuclear MEK, rather than translocation of phospho-MEK to the nucleus. Also interesting was our observation that in the presence of a MEK inhibitor, nuclear accumulation of phospho-MEK increased. This response was magnified in cells replated on collagen type I, and was further increased upon the addition of carbachol (Fig. 6B, left panel). Although the MEK1 inhibitor PD98059 and the MEK1/2 inhibitor U0126 had qualitatively similar effects, the overall increase in phospho-MEK levels seen in the presence of U0126 was significantly greater, suggesting that both MEK1 and MEK2 contributed to the response. The potentiation of MEK phosphorylation by MEK inhibitors might indicate that, in the absence of an inhibitor, activated MEK is dephosphorylated in a MAPK-dependent manner.
One documented circumstance under which wild type MEK is relocalized to the nucleus occurs during early prophase in mitotic cells growing in the presence of 10% fetal bovine serum. The translocation precedes nuclear envelope breakdown, and is reduced in MEK mutants in which the two regulatory serine residues are mutated to alanine [Tolwinski et al., 1999]. The results suggest that, early in mitosis, export of MEK from the nucleus is retarded. Our results suggest that a similar effect could occur during cell spreading, a process that, like mitosis [Cassimeris, 1999], is characterized by extensive rearrangements of cytoskeletal elements [Burridge et al., 1997; Kaverina et al., 1998; Fuchs and Karakesisoglou, 2001], but this remains to be proven.
MAPK has multiple targets located in both cytoplasmic and nuclear compartments of the cell, and mediates a variety of cellular responses, including mitosis and differentiation. Factors that influence the nature of the response to MAPK activation include its subcellular localization [Brunet et al., 1999] and duration of activation [Marshall, 1995]. M3 receptor stimulation caused a transient increase in nuclear phospho-MAPK in cells replated on fibronectin, which peaked at 2 minutes and then rapidly declined. This was accompanied by a decrease in total nuclear MAPK levels (Fig. 3), indicating that MAPK was rapidly exported from the nucleus after stimulation. In contrast, in cells replated onto collagen type I, the carbachol-evoked increase in nuclear phospho-MAPK was sustained for at least 10 minutes (Fig. 4), raising the possibility that activation of nuclear targets, such as transcription factors, might be differentially affected in a manner dependent on ECM composition.
In conclusion, the results demonstrate that ECM composition modulates signaling by M3 muscarinic receptors at the level of focal adhesion formation, and nuclear localization of activated MAPK, and that the time-course of the latter is differentially influenced by specific ECM proteins. ECM-specific modulation of M3 receptor-evoked MAPK activation represents an additional mechanism for fine-tuning cellular responses to neurotransmitter stimulation.
ACKNOWLEDGEMENTS
We thank Dr. Jan K. Blusztajn for critical reading of the manuscript.
Grant support: NIH (to BES); Grant numbers R01 NS30791 and R01 MH59775
Abbreviations used
- ECM
extracellular matrix
- ERK
extracellular signal-regulated kinase
- GPCR
G protein-coupled receptor
- FAK
focal adhesion kinase
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
REFERENCES
- Adachi M, Fukuda M, Nishida E. Two co-existing mechanisms for nuclear import of MAP kinase: passive diffusion of a monomer and active transport of a dimer. EMBO J. 1999;18:5347–5358. doi: 10.1093/emboj/18.19.5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi M, Fukuda M, Nishida E. Nuclear export of MAP kinase (ERK) involves a MAP kinase kinase (MEK)-dependent active transport mechanism. J. Cell Biol. 2000;148:849–856. doi: 10.1083/jcb.148.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Aplin AE. Cell adhesion molecule regulation of nucleocytoplasmic trafficking. FEBS Lett. 2003;534:11–14. doi: 10.1016/s0014-5793(02)03840-1. [DOI] [PubMed] [Google Scholar]
- Aplin AE, Hogan BP, Tomeu J, Juliano RL. Cell adhesion differentially regulates the nucleocytoplasmic distribution of active MAP kinases. J. Cell Sci. 2002;115:2781–2790. doi: 10.1242/jcs.115.13.2781. [DOI] [PubMed] [Google Scholar]
- Aplin AE, Juliano RL. Integrin and cytoskeletal regulation of growth factor signaling to the MAP kinase pathway. J. Cell Sci. 1999;112:695–706. doi: 10.1242/jcs.112.5.695. [DOI] [PubMed] [Google Scholar]
- Aplin AE, Juliano RL. Regulation of nucleocytoplasmic trafficking by cell adhesion receptors and the cytoskeleton. J. Cell Biol. 2001;155:187–191. doi: 10.1083/jcb.200107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aplin AE, Stewart SA, Assoian RK, Juliano RL. Integrin-mediated adhesion regulates ERK nuclear translocation and phosphorylation of Elk-1. J. Cell Biol. 2001;153:273–281. doi: 10.1083/jcb.153.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3β. J. Biol. Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodary SC, McLean JW. The integrin β1 subunit associates with the vitronectin receptor αv subunit to form a novel vitronectin receptor in a human embryonic kidney cell line. J. Biol. Chem. 1990;265:5938–5941. [PubMed] [Google Scholar]
- Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouysségur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M, Zhong C. Focal adhesion assembly. V Trends Cell Biol. 1997;7:342–347. doi: 10.1016/S0962-8924(97)01127-6. [DOI] [PubMed] [Google Scholar]
- Cassimeris L. Accessory protein regulation of microtubule dynamics throughout the cell cycle. Curr. Opin. Cell Biol. 1999;11:134–141. doi: 10.1016/s0955-0674(99)80017-9. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J. Biol. Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: The road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J. Biol. Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. Dimerization in MAP-kinase signaling. Trends Biochem. Sci. 2000;25:7–9. doi: 10.1016/s0968-0004(99)01508-x. [DOI] [PubMed] [Google Scholar]
- Danilkovitch A, Donley S, Skeel A, Leonard EJ. Two independent signaling pathways mediate the antiapoptotic action of macrophage-stimulating protein on epithelial cells. Molec. Cell. Biol. 2000;20:2218–2227. doi: 10.1128/mcb.20.6.2218-2227.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, Pawson AJ, Millar RP, Maudsley S. Cytoskeletal reorganization dependence of signaling by the gonadotropin-releasing hormone receptor. J. Biol. Chem. 2004;279:1980–1993. doi: 10.1074/jbc.M309827200. [DOI] [PubMed] [Google Scholar]
- Della Rocca GJ, Maudsley S, Daaka Y, Lefkowitz RJ, Luttrell LM. Pleiotropic coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. Role of focal adhesions and receptor tyrosine kinases. J. Biol. Chem. 1999;274:13978–13984. doi: 10.1074/jbc.274.20.13978. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Karakesisoglou I. Bridging cytoskeletal intersections. Genes Dev. 2001;15:1–14. doi: 10.1101/gad.861501. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Gotoh I, Gotoh Y, Nishida E. Cytoplasmic localization of mitogen-activated protein kinase kinase directed by its NH2-terminal, leucine-rich short amino acid sequence, which acts as a nuclear export signal. J. Biol. Chem. 1996;271:20024–20028. doi: 10.1074/jbc.271.33.20024. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Gotoh Y, Nishida E. Interaction of MAP kinase with MAP kinase kinase: Its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J. 1997;16:1901–1908. doi: 10.1093/emboj/16.8.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- Hawes BE, Van Biesen T, Koch WJ, Luttrell LM, Lefkowitz RJ. Distinct pathways of Gi- and Gq-mediated mitogen-activated protein kinase activation. J. Biol. Chem. 1995;270:17148–17153. doi: 10.1074/jbc.270.29.17148. [DOI] [PubMed] [Google Scholar]
- Jaaro H, Rubinfeld H, Hanoch T, Seger R. Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc. Natl. Acad. Sci. USA. 1997;94:3742–3747. doi: 10.1073/pnas.94.8.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano RL. Signal transduction by cell adhesion receptors and the cytoskeleton: Functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members. Annu. Rev. Pharmacol. Toxicol. 2002;42:283–323. doi: 10.1146/annurev.pharmtox.42.090401.151133. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Rottner K, Small JV. Targeting, capture, and stabilization of microtubules at early focal adhesions. J. Cell Biol. 1998;142:181–190. doi: 10.1083/jcb.142.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev AV, Canagarajah B, Wilsbacher J, Robinson M, Atkinson M, Goldsmith E, Cobb MH. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 1998;93:605–615. doi: 10.1016/s0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- Kim-Kaneyama JR, Nose K, Shibanuma M. Significance of nuclear relocalization of ERK1/2 in reactivation of c-fos transcription and DNA synthesis in senescent fibroblasts. J. Biol. Chem. 2000;275:20685–20692. doi: 10.1074/jbc.M908723199. [DOI] [PubMed] [Google Scholar]
- Lenormand P, Brondello JM, Brunet A, Pouysségur J. Growth factor-induced p42/p44 MAPK nuclear translocation and retention requires both MAPK activation and neosynthesis of nuclear anchoring proteins. J. Cell Biol. 1998;142:625–633. doi: 10.1083/jcb.142.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenormand P, Sardet C, Pages G, L'Allemain G, Brunet A, Pouysségur J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TH, Chen QM, Howe A, Juliano RL. Cell anchorage permits efficient signal transduction between Ras and its downstream kinases. J. Biol. Chem. 1997;272:8849–8852. [PubMed] [Google Scholar]
- Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science. 1998;280:109–112. doi: 10.1126/science.280.5360.109. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Gutkind JS, Yamada KM. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: Roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta EG, Ashkenazi A, Winslow JW, Ramachandran J, Capon DJ. Differential regulation of PI hydrolysis and adenylyl cyclase by muscarinic receptor subtypes. Nature. 1988;334:434–437. doi: 10.1038/334434a0. [DOI] [PubMed] [Google Scholar]
- Pouysségur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem. Pharmacol. 2002;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- Renshaw MW, Ren XD, Schwartz MA. Growth factor activation of MAP kinase requires cell adhesion. EMBO J. 1997;16:5592–5599. doi: 10.1093/emboj/16.18.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reszka AA, Seger R, Diltz CD, Krebs EG, Fischer EH. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc. Natl. Acad. Sci. USA. 1995;92:8881–8885. doi: 10.1073/pnas.92.19.8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol. Cell. Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- Short SM, Boyer JL, Juliano RL. Integrins regulate the linkage between upstream and downstream events in G protein-coupled receptor signaling to mitogen-activated protein kinase. J. Biol. Chem. 2000;275:12970–12977. doi: 10.1074/jbc.275.17.12970. [DOI] [PubMed] [Google Scholar]
- Slack BE. Tyrosine phosphorylation of paxillin and focal adhesion kinase by activation of muscarinic m3 receptors is dependent on integrin engagement by the extracellular matrix. Proc. Natl. Acad. Sci. USA. 1998;95:7281–7286. doi: 10.1073/pnas.95.13.7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack BE. The m3 muscarinic acetylcholine receptor is coupled to mitogen-activated protein kinase via protein kinase C and epidermal growth factor receptor kinase. Biochem. J. 2000;348:381–387. [PMC free article] [PubMed] [Google Scholar]
- Tolwinski NS, Shapiro PS, Goueli S, Ahn NG. Nuclear localization of mitogen-activated protein kinase kinase 1 (MKK1) is promoted by serum stimulation and G2-M progression - Requirement for phosphorylation at the activation lip and signaling downstream of MKK. J. Biol. Chem. 1999;274:6168–6174. doi: 10.1074/jbc.274.10.6168. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S. Protein kinase C δ activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J. Biol. Chem. 1996;271:23512–23519. doi: 10.1074/jbc.271.38.23512. [DOI] [PubMed] [Google Scholar]
- Volmat W, Camps M, Arkinstall S, Pouysségur J, Lenormand P. The nucleus, a site for signal termination by sequestration and inactivation of p42/p44 MAP kinases. J. Cell Sci. 2001;114:3433–3443. doi: 10.1242/jcs.114.19.3433. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Wolf I, Rubinfeld H, Yoon S, Marmor G, Hanoch T, Seger R. Involvement of the activation loop of ERK in the detachment from cytosolic anchoring. J. Biol. Chem. 2001;276:24490–24497. doi: 10.1074/jbc.M103352200. [DOI] [PubMed] [Google Scholar]
- Yao Z, Flash I, Raviv Z, Yung Y, Asscher Y, Pleban S, Seger R. Non-regulated and stimulated mechanisms cooperate in the nuclear accumulation of MEK1. Oncogene. 2001;20:7588–7596. doi: 10.1038/sj.onc.1204963. [DOI] [PubMed] [Google Scholar]
- Zheng C-F, Guan K-L. Cytoplasmic localization of the mitogen-activated protein kinase activator MEK. J. Biol. Chem. 1994;269:19947–19952. [PubMed] [Google Scholar]