

Abstract
Bombesin receptor subtype-3(BRS-3), a G protein-coupled orphan receptor, shares 51% identity with the mammalian bombesin(Bn) receptor for gastrin-releasing peptide(GRPR). There is increasing interest in BRS-3 because it is important in energy metabolism, glucose control,motility and tumor-growth. BRS-3 has low affinity for all Bn-related peptides, however, recently synthetic high-affinity agonists[D-Tyr6/D-Phe6,βAla11,Phe13,Nle14]Bn-(6–14) were described, but they are nonselective for BRS-3 over other Bn-receptors. Based on these peptides, three BRS-3 selective-ligands were developed: peptide#2,[D-Tyr6(R)-Apa11,Phe13,Nle14]Bn(6–14); peptide#3,[D-Tyr6,(R)-Apa11,4Cl-Phe13,Nle14]Bn(6–14); peptide #4,Ac-Phe-Trp-Ala-His(tBzl)-Nip-Gly-Arg-NH2. Their molecular determinants of selectivity/high affinity for BRS-3 are unknown. To address this we used a chimeric/site-mutagenesis approach. Substitution of extracellular domain2(EC2) of BRS-3 by the comparable GRPR domain decreased 26-,4,0-fold affinity for peptides#4,3,2. Substitution of EC3 decreased affinity 4-,11-,0-fold affinity for peptides#2,3,4. Ten point mutations in the EC2 and adjacent transmembrane regions (TM2) 2 and 3 of BRS-3 were made. His107(EC2-BRS-3) for lysine(H107K)(EC2-GRPR), decreased affinity(25-,0-fold) for peptide#4,1; however it could not be activated by either peptide. Its combination with Val101(TM2),Gly112(EC2),Arg127(TM3) resulted in complete loss-of-affinity of peptide#4. Receptor-modeling showed that each of these residues face inward and are within 4Å of the binding-pocket. These results demonstrate [Val101,His107,Gly112,Arg127] in the EC2/adjacent upper TMs of BRS-3 are critical for the high BRS3-selectivity of peptide#4. His107 in EC2 is essential for BRS-3 activation, suggesting amino-aromatic ligand/receptor interactions with peptide#4 are critical for both binding/ activation. Furthermore, these result demonstrate that even though these three BRS-3 selective agonists were developed from the same template peptide,[D-Phe6,βAla11,Phe13,Nle14]Bn-(6–14), their molecular determinants of selectivity/high affinity varied considerably.
Introduction
BRS-3 is an orphan receptor present in both, the central nervous system and peripheral tissues (Fathi, et al., 1993b;Ohki-Hamazaki, et al., 1997;Porcher, et al., 2005;Sano, et al., 2004) whose role in normal physiology or pathological conditions is largely unknown. Because of its 51% and 47% amino acid homology with the human bombesin (Bn) receptors [gastrin-releasing peptide receptor (GRPR) and neuromedin B receptor (NMBR)], it is classified in this receptor family (Fathi, et al., 1993b;Jensen, et al., 2007). BRS-3 is receiving increased attention because its disruption leads to obesity, diabetes and hypertension (Ohki-Hamazaki, et al., 1997). Furthermore, from knockout and expression studies, it has been reported that the BRS-3-receptor is frequently overexpressed by various tumors (Fathi, et al., 1993b;Schulz, et al., 2006;Jensen and Moody, 2006), its presence plays a role in lung tumor invasiveness (Jensen, et al., 2007), plays a role in lung injury (Jensen, et al., 2007), plays a role in regulation of insulin release (Nakamichi, et al., 2004), and may play an important role in regulating gastrointestinal motility (Porcher, et al., 2005).
At present, little was known about BRS-3’s pharmacology or cell biology and almost nothing is known about the molecular determinants of high affinity interaction with the BRS-3-receptor. This occurred because studies demonstrate BRS-3 has low affinity for all occurring natural Bn-related peptides as well as most synthetic bombesin-family member analogues (Mantey, et al., 1997;Jensen, et al., 2007) with NMB and GRP only interacting with BRS-3 in the micromolar concentrations (Mantey, et al., 1997;Jensen, et al., 2007; Fathi, et al., 1993b). However, it has been possible to perform detailed pharmacological studies subsequently, because recently a synthetic Bn analogue [D-Phe6,βAla11,Phe13,Nle14]Bn(6-14)peptide#5 (Table 1) was discovered to have a high affinity for both human (Mantey, et al., 1997;Jensen, et al., 2007;Pradhan, et al., 1998) and monkey (Sano, et al., 2004) BRS-3-receptors. Subsequent studies demonstrated that this synthetic Bn peptide had a unique pharmacology because it had high affinity not only for BRS-3-receptors, but also for human GRPR and NMBR as well as GRPR and NMBR in most species and the frog BB4 receptor (Fathi, et al., 1993b;Pradhan, et al., 1998;Jensen, et al., 2007). Subsequent studies using this synthetic Bn analogue or its Tyr6-derivative demonstrated hBRS-3 activation, stimulated of phospholipase C, phospholipase D and tyrosine kinase cascades, but did not stimulate adenylate cyclase activation (Jensen, et al., 2007;Ryan, et al., 1998).
Table 1.
Peptide number and structure of peptides studied.
| Bn | pGlu-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 |
|---|---|
| 1 | [D-Tyr6, βAla11,Phe13,Nle14]Bn(6–14) |
| 2 | [D-Tyr6,(R)-Apa11,Phe13,Nle14] Bn(6–14) [compound 14 in(Mantey, et al., 2001)] |
| 3 | [D-Tyr6,(R)-Apa11,4Cl-Phe13, Nle14]Bn(6–14) [compound 7 in (Mantey, et al., 2004)] |
| 4 | Ac-Phe-Trp-Ala-His(tBzl)-Nip-Gly-Arg-NH2 [compound 34 in (Boyle, et al., 2005)] |
| 5 | [D-Phe6, βAla11,Phe13,Nle14]Bn(6–14) |
Abbreviations: Bn, bombesin, Ac, acetyl; βAla, β-alanine; Nle, norleucine; Bzl, Benzyl; Nip, piperidine-3 carboxylic acid; Apa, 3-amino-propionic acid.
Because of its potential importance in a number of physiological and pathological processes, recently a number of studies have attended to develop BRS-3-receptor selective ligands, using either [D-Phe6, βAla11,Phe13,Nle14]Bn(6–14) or [D-Tyr6, βAla11,Phe13,Nle14]Bn(6–14)as a starting point. Detailed structure-function studies identified three related peptides, which have selectivity for BRS-3 over the GRPR or the NMB receptor (Mantey, et al., 2001;Mantey, et al., 2004;Boyle, et al., 2005;Mantey, et al., 2006;Jensen, et al., 2007). However, nothing is known of the molecular basis of binding, affinity or selectivity of these peptide agonists for BRS-3 or the molecular determinants of high affinity interaction of any peptides with this unique orphan receptor.
To gain insight into the molecular basis of high affinity for the BRS-3 and determinants of receptor selectivity, in the present study we have investigated the ability of various nonselective and BRS-3-receptor selective agonists to interact with this receptor using both a chimeric receptor approach and site-directed mutagenesis of involved receptor regions.
Materials and methods
Materials
The following cells and materials were obtained from the sources indicated: Balb 3T3 (mouse fibroblast) cells from the American Type Culture Collection, (Rockville, MD); CHOP cells (Polyoma large T antigen-expressing Chinese hamster ovary cells) were gift from James W. Dennis (Samuel Lunenfeld Research Institute, Toronto, Canada); the mammalian expression vectors, pcDNA3 and pcDNA3.1, custom primers, restriction endonucleases (SmaI, and EcoRI), penicillin-streptomycin, Lipofectamine PLUS Reagent, OPTI-MEM I Reduced-Serum Medium and GENETICIN selective antibiotic (G418 Sulfate) from Invitrogen (Carlsbad, CA); QuikChange Site-Directed Mutagenesis Kit from Stratagene (La Jolla, CA); Dulbecco’s minimum essential medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum (FBS) and trypsin/versene solution from Biosource International (Camarillo, CA); Na125I (2,200 Ci/mmol) from Amersham Biosciences (Piscataway, NJ); 1,3,4,6-tetrachloro-3α, 6α-diphenylglycouril (IODO-GEN), dithiotreitol (DTT), and Bond-Breaker TCEP solution from Pierce Biotechnology Inc (Rockford, IL); myo-[2-3H]Inositol (20 Ci/mmol) from Amersham Pharmacia Biotech (Piscataway, NJ); formic acid, ammonium formate, disodium tetraborate, soybean trypsin inhibitor, bacitracin and AG 1-X8 resin from Bio-Rad, (Richmond, CA); bovine serum albumin fraction V (BSA) and N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) from ICN Pharmaceutical, Inc. (Aurora, OH). All other chemicals were of the highest purity commercially available.
Methods
Preparation of peptides
The peptides were synthesized using standard solid-phase methods as described previously (Mantey, et al., 2006). In brief, solid-phase syntheses of peptide amides were carried out using Boc chemistry on methylbenzhydrylamine resin (Advanced ChemTech, Louisville, KY) followed by hydrogen fluoride-cleavage of free peptide amides. The crude peptides were purified by preparative high-performance liquid chromatography on columns (2.5 x 50 cm) of Vydac C18 silica (10 pm), which was eluted, with linear gradients of acetonitrile in 0.1% (v/v) trifluoroacetic acid. Homogeneity of the peptides was assessed by analytical reverse-phase high-performance liquid chromatography, and the purity was usually 97% or higher. Amino acid analysis (only amino acids with primary amino acid groups were quantitated) gave the expected amino acid ratios. Peptide molecular masses were obtained by matrix-assisted laser desorption mass spectrometry spectrometry using a Voyager-DE Pro machine (Applied Biosystems, Foster City, CA) and all corresponded well with calculated values.
Strategy for Construction of mutant BRS-3-receptors and GRPR receptors
Previous studies demonstrate the nonselective agonists, [D-Phe6,βAla11,Phe13,Nle14]Bn-(6-14)peptide#5 or [D-Tyr6,βAla11,Phe13,Nle14]Bn-(6–14)(peptide#1) were equipotent, each had high affinity for BRS-3, GRPR and NMBR, and the selective BRS-3 ligands (peptide#2, #3, #4, Table 1) had the greatest difficulty in distinguishing BRS-3 from GRPR(Mantey, et al., 2001;Mantey, et al., 1997;Mantey, et al., 2006;Mantey, et al., 2004) therefore we examined the molecular basis of affinity of these peptides for BRS-3 and GRPR. Because the molecular basis for the selectivity of various BRS-3 preferring ligands was being investigated, both BRS-3-receptor loss-of-affinity and GRPR gain-of-affinity mutants for selective BRS-3 compounds were made, as well as, various point mutants and combination point mutants in BRS-3. Because the expression level and affinity of BRS-3 for some radiolabeled and unlabeled Bn peptides can be so low that pharmacological studies are limited (Mantey, et al., 1997), an arginine in position 127 was substituted for glutamine (the comparable amino acid from GRPR) in transmembrane region 3 of BRS-3, which has been shown to increase GRP affinity and/or receptor expression allowing radioligand studies to be done (Akeson, et al., 1997;Nakagawa, et al., 2005). This receptor [R127Q]BRS-3 (BRS-3*) was used in all studies with BRS-3.
To construct loss-of-affinity mutants the cDNA of human BRS-3 (BRS-3) in pCD2 was used which was a gift from Eduardo Sainz and James F. Battey (Laboratory of Molecular Biology, NIDCD, National Institutes of Health). The human hBRS-3 cDNA was inserted into pcDNA3.1 (+) at the EcoR I site and BRS-3 mutant receptors were constructed by using the QuikChange Site-Directed Mutagenesis Kit, following the manufacturer’s instructions, with minor modifications. The mouse GRPR (GRPR) used in this study was identical to that described previously (Fathi, et al., 1993a;Akeson, et al., 1997). Nucleotide sequence analysis of the entire coding region was performed. Chimeric receptors were made using human BRS-3 and mouse GRPR, which has been used extensively in several mutagenesis studies and allows comparison with previous results (Akeson, et al., 1997;Nakagawa, et al., 2005; Fathi, et al., 1993a; Tokita, et al., 2001b). The mouse GRP-R has 89% amino acid homology to the human GRPR with most of the differences occurring in the amino terminus of the two receptors, an area which has previously been shown not to be important for high affinity agonist peptide ligand binding (Fathi, et al., 1993a; Tokita, et al., 2001b). In the critical areas of the EC2 and surrounding domains examined in this study in detail, the mouse and human GRPR have 97% homology and the two amino acid differences were conservative changes (i.e., mouse had a V92 instead of I in the human and a K101 instead of R in the comparable position in the human GRPR).
Growth and maintenance of cells
Balb 3T3 cells were grown in DMEM containing 10% (v/v) FBS, 100 units/ml of penicillin and 100 μg/ml of streptomycin. CHOP cells were grown in DMEM containing 10% (v/v) FBS, 100 units/ml of penicillin, 100 μg/ml of streptomycin and 200 μg/ml of G418. All cells were maintained at 37°C in a 5% CO2 atmosphere. Cells were split every 3–4 days at confluence after detaching the cells with Trypsin/Versene Solution.
Cell transfections and isolation of Stable Cell lines
CHOP cells, which contain no native GRP receptors, were used for transient transfection studies and Balb 3T3 for stable transfection studies, because previous studies demonstrated bombesin receptors have similar pharmacology and cell biology to wild-type receptors when express in these cells (Mantey, et al., 1997). CHOP and Balb 3T3 all were seeded in a 10-cm tissue culture dish at a density of 0.8 and 0.5 × 106 cells/dish, respectively, and grown overnight at 37°C in growth medium. On the following morning, 5 μg of plasmid DNA was transfected to by cationic lipid-mediated method using 30 μl of Lipofectamine reagent and 20 μl of PLUS reagent in OPTI-MEM I Reduced-Serum Medium for 3 h at 37°C. At the end of the incubation period, the medium was replaced with each growth medium. CHOP cells were maintained at 37°C in a 5% CO2 atmosphere and were used 48 h later for binding assays. Transfected Balb 3T3 cells were selected for resistance to aminoglycoside G418 as described previously (Mantey, et al., 1997), and were cultured in DMEM supplemented with 10% fetal bovine serum with 200 μg/ml G418.
Preparation of 125I-[D-Tyr6, βAla11,Phe13,Nle14]Bn-(6–14) [125I-Peptide#1]
125I-[Peptide#1] (Table 1) ) has a Tyr and could be radiolabeled, therefore it was used in all binding studies at a specific activity of 2,200 Ci/mmol and was prepared by a modification of methods described previously (Mantey, et al., 1997). Briefly, 0.8 μg of IODO-GEN (in 0.01 μg/ml chloroform) was transferred to a vial, dried under a stream of nitrogen, and washed with 100 μl of KH2PO4 (pH 7.4). To the reaction vial, 20 μl of 0.5 M KH2PO4 (pH 7.4), 8 μg of peptide in 4 μL of water, and 2 mCi (20 μl) Na 125I were added, mixed gently, and incubated at room temperature for 6 min. The incubation was stopped by the addition of 100 μl of distilled water. Radiolabeled peptide was separated using a Sep-Pak (Waters Associates) and high-performance liquid chromatography as described previously (Tokita, et al., 2001a;Mantey, et al., 1997). Radioligand was stored with 0.5% BSA at −20°C.
Whole cell radioligand binding assays
Binding studies to expressed plasmid DNAs were performed as described previously (Tokita, et al., 2001a; Tokita, et al., 2001b). Briefly, 48 h after transient transfection with Lipofectamine disaggregated transfected cells were incubated for 1 h at 21°C in 250 μl of binding buffer containing 24.5 mM HEPES (pH 7.4), 98 mM NaCl, 6 mM KCl, 2.5 mM KH2PO4, 5 mM sodium pyruvate, 5 mM sodium fumarate, 5 mM sodium glutamate, 2 mM glutamine, 11.5 mM glucose, 0.5 mM CaCl2, 1.0 mM MgCl2, 0.01% (w/v) soybean trypsin inhibitor, 0.2 % (v/v) amino acid mixture, 0.2% (w/v) BSA, and 0.05% (w/v) bacitracin with 50 pM 125I-[Peptide#1] (2,200 Ci/mmol) (Table 1) in the presence of the indicated concentration of unlabeled peptides. Although GRPR expression varying as much as 160-fold (Jensen, et al., 2007) has been shown not to alter receptor affinity or potency under the experimental conditions used in this study, as an added precaution to correct for any differences in ligand bound by different mutant GRPR, binding results with each mutant receptor were compared only to results with wild type GRPR or BRS-3-receptor-containing cells binding similar amounts of ligand. This was accomplished by varying the cell concentration between 0.05 – 4 × 106 cells/ml for each mutant receptor so that < 20% of the total added radioactive ligand was bound during the incubation and the results compared to cells transfected with wild type GRPR or BRS-3-receptor adjusted in concentration to bind a similar amount of ligand. After the incubation, 100 μl aliquot were added to 400 μl microfuge tubes (PGC Scientific, Frederick, MD), which contained 100 μl of binding buffer to determine the total radioactivity. The bound tracer was separated from unbound tracer by pelleting the cells through the binding buffer by centrifugation at 10,000 × g in a Microfuge E (Beckman, Fullerton, CA) for 3 min. The supernatant was aspirated and the pelleted cells were rinsed twice with a washing buffer which contained 1% (w/v) BSA in PBS. The amount of radioactivity bound to the cells was measured in a Cobra II Gamma counter (Packard Instruments, Meriden, CT). Binding was expressed as the percentage of total radioactivity that was associated with the cell pellet. All binding values represented saturable binding (i.e., total binding minus nonsaturable binding). Nonsaturable binding was defined as the amount of binding that occurred with 1 μM Bn or 1 μM Peptide#1 (Table 1) in the incubation solution. Nonsaturable binding was <15% of the total binding in all experiments. Each point was measured in duplicate, and each experiment was replicated at least four times. Calculation of affinity was performed by determining the IC50 using the curve-fitting program KaleidaGraph (Synergy Software). The Student’s t test was used to determine the statistical significance of differences in affinity of each chimera, point and combination point mutant receptors compared with its own wild type receptor, binding similar amounts of radioligand.
Measurement of inositol phosphates
The ability of peptide #4, the most selective for BRS-3, to activate the various mutant receptors and wild type receptor was examined and compared to the ability of the prototype peptide #5. Peptide #5 was used for these studies because it has an N-terminal Phe similar to peptide #4, whereas the other prototype peptide, i.e. peptide #1, has a Tyr in this position similar to peptides #2 and #3 (Table 1) and thus was used for the binding studies. Changes in total [3H]inositol phosphates ([3H]IP) was measured as described previously (Ryan, et al., 1998). Briefly, BRS-3*-, GRPR-, chimeric-, point- or combination point mutant-transfected Balb 3T3 cells were subcultured into 24-well plates (5.0 x 104 cells/well) in regular propagation media and then incubated for 24 h at 37oC in a 5% CO2 atmosphere. The cells were then incubated with 3 Ci/ml of myo- [2-3H] inositol in growth media supplemented with 2% FBS for an additional 24 hr. Before assay, the 24-well plates were washed by incubating for 30 min at 37oC with 1 ml/well of PBS (pH 7.0) containing 20 mM lithium chloride. The wash buffer was aspirated and replaced with 500 ml of IP assay buffer containing 135 mM sodium chloride, 20 mM HEPES (pH 7.4), 2 mM calcium chloride, 1.2 mM magnesium sulfate, 1 mM EGTA, 20 mM lithium chloride, 11.1 mM glucose, 0.05% BSA (w/v) and incubated with or without any of the peptides studied. After 60 min of incubation at 37oC, the experiments were terminated by the addition of 1 ml of ice cold 1% (v/v) hydrochloric acid in methanol. Total [3H]IP was isolated by anion exchange chromatography as described previously (Ryan, et al., 1998). Briefly, samples were loaded onto Dowex AG1-X8 anion exchange resin columns, washed with 5 ml of distilled water to remove free [3H]inositol, then washed with 2 ml of 5 mM disodium tetraborate/60 mM sodium formate solution to remove [3H]glycerophosphorylinositol. Two ml of 1 mM ammonium formate/100 mM formic acid solution were added to the columns to elute total [3H]IP. Each eluate was mixed with scintillation cocktail and measured for radioactivity in a scintillation counter.
Modeling of the hBRS-3-receptor
Molecular modeling was performed as described previously (Nakagawa, et al., 2005). Briefly, the primary sequence of the hBRS-3-receptor was input into the Swiss-PDB Viewer, DeepView and a suggested homology modeling template 1U19 (Okada, et al., 2004), a crystal structure of bovine rhodopsin at 2.2 Å resolution, downloaded from the PDB archives. The crystal structure of bovine rhodopsin (1U19) was loaded into Deep View, the sequence of hBRS-3 threaded onto the structure and the second extracellular domain (EC2) and the adjacent ends of the helical transmembrane domains (i.e. TM2 and TM3) aligned onto the structure of bovine rhodopsin. To accommodate the longer EC2 loop region of hBRS-3 relative to the shorter EC2 loop of 1U19, the central loop residues were excised. The model of hBRS-3 was imported into SYBYL 7.3 (Tripos International, St Louis, MO). The extraneous loop regions were removed and the ends of the helical transmembrane regions capped. The alpha carbon atoms of the transmembrane regions were immobilized and the remaining atoms relaxed through extensive energy minimization using the AMBER99 force field (Case, et al., 2005). Candidates for the three-dimensional structure of the EC2 loop were generated by the COMPOSER module of SYBYL which produced 25 EC2 model loop candidates. Each of the hBRS-3/EC2 models were optimized by energy minimization. The resulting structures were examined and fell into ~ 5 topological categories. These were further relaxed by molecular dynamics at reduced temperature (20ps, 100K) and finally reminimized. The putative binding sites of these EC2 loop models were visualized by the SiteID module in SYBYL (Tripos International, St Louis, MO). The models were solvated with water (solvation algorithm, 2 layers), and water molecules capable of defining a continuous cluster within 4Å of the protein were visualized as a surface. Amino acid residues within interaction distance (4Å) of the solvent accessible region were labeled. The models were examined and found to exhibit a wide range of possible conformations for the extracellular EC2 loop region, but with similar energies and the expected conformations for each of the loop amino acids from Ramachandran plots. One EC2 loop (#5) was determined to best fit the experimental data and is shown in the Figure 5. In this figure, the solvent-accessible interior of the extracellular region of the transmembrane receptor is indicated as a grey shape. This central portion of the receptor may accommodate and bind ligands. Solvent-accessible amino acid residues within interaction distance (4Å) of this interior space are shown as ball and stick models, with the residues identified in this work as important for binding colored orange.
Results
Wild-type BRS-3, BRS-3* and GRPR
To explore the molecular basis for the selectivity of three recently described synthetic Bn analogues (Peptides#2, #3, #4, Table 1) for the hBRS-3-receptor over other Bn receptors (GRPR, NMBR) (Mantey, et al., 2006;Mantey, et al., 2004;Mantey, et al., 2001;Boyle, et al., 2005), both loss-of-affinity and gain-of-affinity chimeric receptors as well as receptors with point mutations were made (Figures 1, 2, 3, Tables 2–3). These chimeric receptors were made using human BRS-3 and mouse GRPR, which has been used extensively in previous mutagenesis studies and allows comparison with previous results (Akeson, et al., 1997;Nakagawa, et al., 2005; Fathi, et al., 1993a; Tokita, et al., 2001b). Affinities and potencies for these peptides were compared to either [D-Tyr6, βAla11,Phe13,Nle14]Bn-(6–14) (Peptide#1, Table 1) or [D-Phe6, βAla11,Phe13,Nle14]Bn-(6–14) (Peptide#5, Table 1) which were used as the starting point for the development of the BRS-3 selective ligands, because they are the only known ligands to have high affinity for human and monkey BRS-3-receptors (Mantey, et al., 1997;Pradhan, et al., 1998;Sano, et al., 2004).
Figure 1.

Affinities of peptides#2–4 for BRS-3*, extracellular chimeric BRS-3*s, and wild-type GRPR expressed in CHOP cells (loss-of-affinity chimeras). The diagrams of the chimeric receptors formed are shown at the top. The chimeric BRS-3*s ([R127Q]BRS-3) were formed by replacing each of the extracellular domains of BRS-3 one at a time by the comparable GRPR extracellular domain. Results are expressed as the percentage of the saturable binding seen with no peptide#2-4 present that occurred when the indicated concentration of peptide#2-4 was present. Each point is the mean from at least 5 separate experiments, and in each experiment each point was measured in duplicate. EC2-, e3-, and e4-GRPR refer to the substitution of this extracellular domain of the GRPR for the comparable extracellular domain in BRS-3*. The arrows indicate large changes in affinity from BRS-3*.
Figure 2.

Affinities of peptides#2-4 for wild-type GRPR, extracellular chimeric GRPRs, and wild-type BRS-3 expressed in CHOP cells (gain-of-affinity chimeras). Diagrams of the chimeric receptors formed are shown at the top. The chimeric GRPRs were formed by replacing each of the extracellular domains of GRPR by the comparable domain of BRS-3 one at a time. Results are expressed as the percentage of the saturable binding seen with no peptide#2-4 present that occurred when the indicated concentration of peptide#2–4 was present. Each point is the mean from at least 5 separate experiments, and in each experiment each point was measured in duplicate. EC2-, e3-, and e4-BRS-3 refer to the substitution of this extracellular domain of BRS-3 for the comparable extracellular domain in GRPR. The arrows indicate large changes in affinity from the wild-type GRPR.
Figure 3.

Effect of various point mutations alone or in combination in the 2nd extracellular domain and adjacent transmembrane domains BRS-3* on affinity for peptides#3 and #4. (Top panel) Alignment of amino acids sequences in the second extracellular domain or adjacent transmembrane regions of GRPR and BRS-3. The boxes indicate divergent amino acids between these two receptors in these regions. Shown are the ten BRS-3* mutants made to explore the importance of ten amino acid differences for determining peptide#4 and peptide#3 selectivity. Arrows indicate that position of the point mutations made in BRS-3* by substituting into BRS-3* the divergent amino acid from the comparable position in GRPR. (Middle and Bottom panel) Affinities of peptide#4 and peptide#3 for BRS-3*, second extracellular domain or adjacent TM or point and combination point mutants of BRS-3* expressed in CHOP cells (BRS-3* loss-of-affinity point and combination point mutants). Results are expressed as the percentage of the saturable binding seen with no peptide#3 or 4 present that occurred when the indicated concentration of peptide#3 or 4 was present. Each point is the mean from at least 5 separate experiments, and in each experiment each point was measured in duplicate. The arrows indicate large changes in affinity of the BRS-3* by the mutations. Abbreviations: BRS-3*-[R127Q]BRS-3; [EC2-GRPR]BRS-3 refers to the substitution of the 2nd extracellular domain from GRPR into the comparable location in BRS-3.
Table 2.
Affinities of peptides #1–4 for wild-type BRS-3, BRS-3*, wild-type GRPR and extracellular chimeric BRS-3s and GRPRs
| IC50 (μM)
|
||||
|---|---|---|---|---|
| Peptide#4 | Peptide#3 | Peptide#2 | Peptide#1 | |
| BRS-3 | 0.49 ± 0.1 | 0.04 ± 0.01 | 0.0048 ± 0.0009 | 0.0027 ± 0.0001 |
| BRS-3* | 1.3 ± 0.1 | 0.08 ± 0.01 | 0.0033 ± 0.0009 | 0.0022 ± 0.0002 |
| GRPR | ≫100 | 2.8 ± 0.1 | 0.200 ± 0.004 | 0.0009 ± 0.0001 |
| Extracellular chimeras (loss-of-affinity) | ||||
| [EC2-GRPR]BRS-3* | 33.1 ± 1.21 | 0.34 ± 0.031 | 0.002 ± 0.001 | 0.0007 ± 0.0001 |
| [e3-GRPR]BRS-3* | 1.7 ± 0.1 | 0.85 ± 0.041 | 0.0120 ± 0.00011 | 0.0025 ± 0.0001 |
| [e4-GRPR]BRS-3* | 1.2 ± 0.1 | 0.06 ± 0.01 | 0.0009 ± 0.0001 | 0.0009 ± 0.0001 |
| Extracellular chimeras (gain-of-affinity) | ||||
| [EC2-BRS-3]GRPR | 6.9 ± 0.22 | 0.91 ± 0.072 | 0.200 ± 0.009 | 0.0007 ± 0.0001 |
| [e3-BRS-3]GRPR | ≫100 | ≫1002 | 0.021 ± 0.0012 | 0.0027 ± 0.0004 |
| [e4-BRS-3]GRPR | ≫100 | 3.0 ± 0.1 | 0.200 ± 0.007 | 0.0008 ± 0.0001 |
Abbreviations: BRS-3*, [R127Q]BRS-3; EC2, e3, e4 refer to the extracellular domain of the indicated receptor replacing the comparable domain of BRS-3* or GRPR as indicated.
p <0.0001 lower affinity than the wild type BRS-3* receptor and
p<0.001 higher affinity than the wild type GRPR receptor.
Table 3.
Affinities of Peptide#4, Peptide#3 and Peptide#1 for wild-type BRS-3, BRS-3*, wild-type GRPR and TM2, 2nd extracellular domain, TM3 point or combination point mutants in BRS-3*.
| IC50 (μM)
|
||||
|---|---|---|---|---|
| Peptide#4 | Peptide#3 | Peptide#1 | ||
| BRS-3* | 1.3 ± 0.1 | 0.08 ± 0.01 | 0.0022 ± 0.0001 | |
| GRPR | ≫100 | 2.8 ± 0.1 | 0.0009 ± 0.0001 | |
| EC2[GRPR]BRS-3* | 33.1 ± 1.31 | 0.34 ± 0.031 | 0.0007 ± 0.0001 | |
| TM2 point mutant in BRS-3* | ||||
| [L98V]BRS-3* | 1.8 ± 0.1 | 0.28 ± 0.011 | 0.0030 ± 0.0001 | |
| [V101A]BRS-3* | 3.2 ± 0.11 | 0.36 ± 0.011 | 0.0030 ± 0.0001 | |
| 2nd extracellular domain point mutant in BRS-3* | ||||
| [T106S]BRS-3* | 1.3 ± 0.1 | 0.16 ± 0.011 | 0.0022 ± 0.0001 | |
| [H107K]BRS-3* | 33.1 ± 1.21 | 0.35 ± 0.011 | 0.0020 ± 0.0001 | |
| [E111D]BRS-3* | 1.9 ± 0.1 | 0.06 ± 0.01 | 0.0019 ± 0.0001 | |
| [G112R]BRS-3* | 4.8 ± 0.11 | 0.04 ± 0.01 | 0.0028 ± 0.0004 | |
| TM3 point mutant in BRS-3* | ||||
| [V122L]BRS-3* | 1.8 ± 0.1 | 0.04 ± 0.01 | 0.0028 ± 0.0001 | |
| [L123I]BRS-3* | 1.9 ± 0.1 | 0.08 ± 0.01 | 0.0022 ± 0.0002 | |
| [S124P]BRS-3* | 1.1 ± 0.1 | 0.03 ± 0.01 | 0.0019 ± 0.0001 | |
| Point combination mutants in BRS-3* | ||||
| [V101A, H107K]BRS-3* | 34.7 ± 2.11 | 0.0018 ± 0.0001 | ||
| [H107K, G112R]BRS-3* | 36.3 ± 2.21 | 0.0021 ± 0.0001 | ||
| [V101A, G112R]BRS-3* | 15.1 ± 0.71,2 | 0.0053 ± 0.0001 | ||
| [V101A, H107K, G112R]BRS-3* | ≫1002,3 | 0.0022 ± 0.0001 | ||
| [L98V, V101A]BRS-3* | 0.36 ± 0.031 | 0.0016 ± 0.0001 | ||
| [T106S, H107K]BRS-3* | 0.42 ± 0.021 | 0.0019 ± 0.0001 | ||
| [V101A, T106S, H107]BRS-3* | 1.0 ± 0.11,4 | 0.0053 ± 0.0001 | ||
| [L98V, V101A, T106S, H107]BRS-3* | 0.93 ± 0.051,4 | 0.0039 ± 0.0001 | ||
Results are calculated from data in Figure 3 as described in METHODS.
Abbreviations: BRS-3*, [R127Q]BRS-3; point and point combination mutants refer to the BRS-3* receptor in which the indicated amino acid(s) of BRS-3* was replaced by the comparable amino acid of GRPR; TM, transmembrane domain.
p <0.05 compared to BRS-3*,
p <0.0001 compared to single mutant [V101A] or [G112R],
p <0.0001 compared to [H107K], [V101A,H107K] or [H107K, G112R] and
p <0.001 compared to [L98V, V101A] or [T106S, H107K].
Similar to reported in other studies (Mantey, et al., 1997;Pradhan, et al., 1998) peptide#1 showed a high affinity for the human BRS-3-receptor (IC50 0.0027 μM) and BRS-3*([R127Q]BRS-3) (IC50 0.0022 μM) as well as for GRPR (IC50 0.0009 μM) (Table 2) which allowed it to be radiolabeled and used as a universal ligand for all receptor mutation studies. Using 125I-peptide#1, related BRS-3 selective peptides (peptide#2, #3 and #4) were found to have 61-, 35- and 77-fold higher affinity for BRS-3* over GRPR (Fig. 1, Table 2). Even at a concentration of 30 μM, peptide#4 interacted only minimally with GRPR (Fig. 1, Table 2).
Extracellular Chimeric Receptors
Chimeric human BRS-3 and mouse GRPR’s were made by substituting extracellular domains to explore the BRS-3 selectivity of peptides#2–4. Three loss-of-affinity BRS-3 chimeric receptors were made by substituting the extracellular domains of GRPR for the comparable domains in BRS-3* (Fig. 1) and three potential gain-of-affinity GRPR chimeras were made by substituting the extracellular domains of BRS-3 for the comparable domain in GRPR (Fig. 2).
With the loss-of-affinity BRS-3* receptor chimeras, the substitution of the second extracellular domain (EC2) in the BRS-3* by the comparable domain of GRPR decreased affinity for peptide#4 by 26-fold (from 1.3 ± 0.1 to 33.1 ± 1.2 μM) (Fig. 1 and Table 2). In contrast, the substitution of the third (EC3) or fourth (EC4) extracellular domains of GRPR in BRS-3* did not alter the affinity of peptide#4 for BRS-3* (Fig. 1 and Table 2). In the case of peptide#3, not only did the replacement of the EC2 of BRS-3* with the comparable GRPR domain produce a decrease of 4.3-fold in affinity (from 0.08 ± 0.01 to 0.34 ± 0.03 μM) (Fig. 1 and Table 2), but also the substitution of the EC3 reduced the affinity for peptide#3 by 11-fold (from 0.08 ± 0.01 to 0.85 ± 0.07 μM) (Fig. 1 and Table 1). In contrast, with peptide#2, which closely resembles peptide#3 in structure (Table 1), only the substitution of the EC3 caused a loss-of-affinity of 3.6-fold (0.0033 ± 0.0001 to 0.0120 ± 0.001 μM) (Fig. 1 and Table 2), however the substitution of EC2 or EC4 had not effect (Table 2).
When the reverse study was performed by substitution in the GRPR the extracellular domains (EC) of BRS-3 to attempt to gain affinity, peptide#4 showed the greatest effect with the substitution of the EC2 of BRS-3 into GRPR, by increasing its affinity 14.5-fold (from >100 to 6.9 ± 0.2 μM) (Fig. 2 and Table 2). In contrast, the substitution of the EC3 and EC4 extracellular domains of BRS-3 with those from GRPR had no effect in peptide#4 affinity (Fig. 2, Table 2). With peptide#3 the substitution of the EC2 resulted in an increment in the affinity by 3.1-fold (from 2.8 ± 0.1 to 0.91 ± 0.07 μM) (Fig. 2 and Table 2). In contrast, substitution of the EC3 showed the most important effect with peptide#2 by increasing the affinity 9.5-fold (from 0.200 ± 0.004 to 0.021 ± 0.001 μM) (Fig. 2 and Table 2). The differences seen with peptides#2–4 with the loss-of-affinity and gain-of-affinity chimeric receptors was not due to a global alteration of receptor affinity, because each chimera receptor retained high affinity, for each peptide#1, #5 or one of the BRS-3 selective ligands (Fig. 2, 3 and Table 2).
These results show that the location of receptor domains most important for determining BRS-3 selectivity of these three peptides, have important differences, even though each of these peptides was derived from the studies of the some parent peptide (i.e. peptide#5). Specifically, differences in the EC2 domain of BRS-3 and GRPR are primarily responsible for peptide#4 selectivity, differences in the EC2 and EC3 domains for peptide#3’s selectivity, whereas for peptide#2’s selectivity For BRS-3 was partially dependent of differences in EC3, but in large part determined by differences outside the TM regions.
BRS-3 Second Extracellular Domain (EC2) and Adjacent Transmembrane (TM) region mutants (Loss-of-affinity Point and Combination Mutants)
Because the selectivity of the related peptides #3 and #4 were both influenced by differences in the EC2 of BRS-3 and GRPR, we wanted to determine whether the molecular basis for this was similar with these two peptides. To investigate further the molecular basis for the selectivity of peptide#4 and #3, we attempted to determinate which specific amino acid(s) are responsible for the high affinity of both peptides for BRS-3 by analyzing the amino acid(s) differences and similarities in the EC2 domain and adjacent transmembrane regions (TM) of these two receptors (Fig. 3, Top panel). The two receptors in this region (Fig. 3, Top panel) differed in ten amino acid(s), occurring at positions 98, 101, 106–107, 111–112, 122–124 and 127of BRS-3, which are comparable with positions 92, 95, 100–101,105–106, 116–118 and 121 of GRPR (Fig. 3, Top panel). To study the ten amino acid(s) differences, we first made ten BRS-3 losses of affinity point mutants by substituting in BRS-3 the comparable different amino acid from GRPR.
For peptide#4, the substitution of lysine instead of histidine at position 107 (H107K) in the EC2 of BRS-3* produced the greatest effect, decreasing the affinity by 26-fold (33.1 ± 1.2 μM) (Fig. 3, Middle panel and Table 3). This decrease in the affinity for peptide#4 was the same extent seen with the substitution of the whole EC2 domain in BRS-3 by the comparable domain of GRPR (33.1 ± 1.3 μM) (Table 3). Point mutation [V101A] (3.2±0.1 μM) in TM2 or [G112R] (4.8 ± 0.1 μM) in EC2 of BRS-3* (Fig. 3, Middle panel and Table 3) caused only a loss of 3–4-fold in the affinity of peptide#4 for BRS-3* (Fig. 3, Middle panel and Table 3). In contrast, point substitutions [L98V] in TM2, [T106S], [E111D] in EC2, or [V122L], [L123I] and [S124P] in TM3 of BRS-3* had no effect on the affinity of peptide#4 for BRS-3* (Fig. 3, Middle panel and Table 3).
With peptide#3, the largest decrease in affinity was seen with the substitution of 3[H107K] in EC2 of BRS-3* (Fig. 3 Middle panel, Table 3) which resulted in a 5-fold decrease (0.35 ± 0.01 μM) which was similar to the decrease caused by the substitution of [V101A] (0.36 ± 0.01 μM) in TM2 and the whole replacement of EC2 domain of BRS-3 (0.34 ± 0.03 μM) (Table 3). Point mutations [L98V] in TM2 or [T106S] in EC2 of BRS-3* resulted in small (2-fold) decreases in the affinity of peptide#3 for BRS-3 (Fig. 3 Middle panel, Table 3). In contrast, substitution in BRS-3 of either [E111D] or [G112R] in EC2, as well as either [V122L], [L123I] or [S124P] in TM3 of BRS-3*, did not affect the affinity of peptide#3 for the BRS-3* receptor (0.08 ± 0.01 μM) (Fig. 3, Middle panel and Table 3).
To explore the possible impact of multiple amino acid substitutions, we next made mutant BRS-3*’s with combinations of these point mutations. With peptide#4, the triple mutant [V101A, H107K, G112R] in the BRS-3-receptor caused the largest effect (>100 μM) (Fig. 3, Bottom panel and Table 3) showing almost a complete loss-of-affinity, which is similar to its affinity for the native GRPR. Neither of the double combinations of point mutants which included the substitution [H107K] including [V101A, H107K] (34.7 ± 2.1 μM) or [H107K, G112R] (36.3 ± 2.2 μM) (Fig. 3, Bottom panel and Table 3) showed a greater decrease in the affinity than that caused by the substitution of [H107K] alone (33.1 ± 1.2 μM) (Table 2). However, an additive effect for the loss-of-affinity of peptide#4 for BRS-3* was produced by the double mutant [V101A, G112R] (15.1 ± 0.7 μM) (Fig. 3, Bottom panel and Table 3).
With peptide#3, neither the combination of two point mutants compared to either mutation alone (Table 3) in the EC2 domain which affected its affinity [T106S, H107K] (0.42 ± 0.02 μM) (Fig. 3, Bottom panel and Table 3) nor combination of the two TM2 point mutations that altered its affinity [L98V, V101A] (0.36 ± 0.03 μM) alone had an additive effect (Fig. 3, Bottom panel and Table 3). The triple mutant [V101A, T106S, H107K] had a marked effect decreasing the affinity by 12.5-fold (1.0 ± 0.1 μM) (Fig. 3, Bottom panel and Table 3) which was not different from [L98V, V101A, T106S, H107K] (0.93 ± 0.05 μM) (Fig. 3, Bottom panel and Table 3).
Stimulation of phospholipase C
To determinate the effect of these receptor mutations on activation of the GRPR or BRS-3-receptor, which are coupled to phospholipase C (Jensen, et al., 2007;Ryan, et al., 1998), we determined the ability of the most selective peptide, peptide#4 to stimulate [H3]IP formation in BRS-3*-, GRPR-, chimeric-, point- or combination mutants-transfected Balb 3T3 cells (Fig. 4, Table 4). Because peptide #4 has a Phe at the amino terminus instead of a Tyr, as seen in peptides #2 and #3 (Table 1), we compared the results from peptide #4 with those of [D-Phe6,βAla11,Phe13,Nle14]Bn-(6–14) (peptide #5) instead of the D-Tyr6 analogue (peptide #1) (Table 1). Peptide #5 caused an 1358±199 dpm increase in [H3]IP and the stimulation occurred in a concentration-dependent manner (Fig. 4 and Table 4) with a half-maximal effect in [H3]IP GRPR at 0.005 ± 0.001 μM and for BRS-3* at 0.012 ± 0.001 μM. Peptide #5 and #1 were equipotent (data not shown).
Figure 4.

Ability of peptides#4 and #5 to stimulate increases in [3H]IP formation in second extracellular domain or adjacent transmembrane regions point and combination point mutants of GRPR, and BRS-3*-R-transfected Balb-3T3 cells. The different nature and mutant receptors (5.0x104 cells/well) were loaded with myo-[2-3H]inositol as described in METHOS, washed, and incubated with the indicated concentrations of peptide#4 or #5 for 45 min at 37oC. Values are expressed as the percent of the total [3H]IP release stimulated by 1 μM peptide#5. Results are the means ±SEM from at least 5 experiments, and each point was determined in duplicate. The control and maximal value for BRS-3* were 1358±199 dpm and 3958±889 dpm (n=6).
Table 4.
Ability of peptides #4 and #5 to stimulate [3H]IP generation in Balb 3T3 cells stably transfected with BRS-3*, GRPR, various point mutants or chimeric receptors.
| [3H]IP
|
||||
|---|---|---|---|---|
| Fold increase | EC50 (μM)
|
|||
| Peptide#5 (1 μM) | Peptide#4 | Peptide#5 | ||
| BRS-3* | 2.0 ± 0.11 | 0.14 ± 0.01 | 0.012 ± 0.001 | |
| GRPR | 5.9 ± 0.71 | ≫10 | 0.005 ± 0.001 | |
| Extracellular chimeras (loss-of-affinity) | ||||
| [EC2-GRPR]BRS-3* | 1.2 ± 0.1 | - | - | |
| TM2 point mutant | ||||
| [V101A]BRS-3* | 2.0 ± 0.11 | 0.32 ± 0.022 | 0.011 ± 0.001 | |
| 2nd extracellular domain point mutant | ||||
| [H107K]BRS* | 1.1 ± 0.1 | - | - | |
| [G112R]BRS-3* | 3.0 ± 0.11 | 0.91 ± 0.032 | 0.013 ± 0.001 | |
| Point combination mutant | ||||
| [V101A, G112R]BRS-3* | 2.3 ± 0.21 | 1.48 ± 0.102 | 0.010 ± 0.001 | |
Results are calculated from data shown in Figure 4 as described in METHODS. Fold increase refers to the maximal stimulation seen with 1 μM peptide#5 expressed as a ratio (experimental/control). EC50 is the concentration of the indicated peptide causing half-maximal stimulation seen with 1 μM peptide#5. Results are the mean±SEM of 4 experiments.
Abbreviations: BRS-3*, [R127Q]BRS-3; point and combination mutants refer to the BRS-3* receptor in which the indicated amino acid(s) of BRS-3* was replaced by the comparable amino acid of GRPR.
p <0.05 compared to non stimulated cells and
p <0.003 or lower compared to BRS-3*.
Single point mutants [V101A] in TM2, or [G112R] in EC2 or the combination [V101A, G112R] in BRS-3* had no effect on the efficacy or potency of the nonselective agonist, peptide#5, for stimulating an increase in [H3]IP (Fig. 4 left and Table 4). In contrast, the point mutant [H107K] in the EC2 of BRS-3* or replacement of the EC2 of BRS-3* by that from GRPR, resulted in receptors, which were not able to stimulate of [H3]IP even at concentrations of agonist as high as 10 μM (Fig. 4 left and Table 4). Even though had both retained high binding affinity for peptide#1 (Table 2 and 3).
Peptide#4 showed high affinity, high potency and selectivity for activating the BRS-3* receptor (0.14 ± 0.01 μM) (Fig. 4 and Table 4), however, even at concentrations as high as 10 μM, it did not activate GRPR (Fig. 4 right and Table 4). A decrease in the potency for stimulation of [H3] IP was caused by point mutants [V101A] (0.32 ± 0.02 μM) and [G112R] (0.91 ± 0.03 μM) in BRS-3* (Fig. 4 right and Table 4) which is consistent with the binding results. However, the combination [V101A, G112R] did not have a greater effect on potency for stimulating [H3]IP (1.479 ± 0.096 μM) than [G112R] alone (Fig. 4 right and Table 4).
Molecular modeling of the BRS3 receptor
To attempt to gain additional insight into why the different amino acids in the hBRS-3-receptor EC2 and adjacent transmembrane regions are important for high affinity and selectivity for peptide#4, the most selective agonist, and to a lesser extent for peptide#3, three-dimensional modeling of this region of the hBRS-3-receptor was performed (Fig. 5). Since the mutagenesis studies demonstrated the importance of residues V101, H107, G112 and R127 in the binding of peptides#3 and #4 to the hBRS-3-receptor, the environment of these crucial residues was investigated by molecular modeling. The primary sequence of hBRS-3 was entered into the Swiss-PDB Viewer, DeepView (Nakagawa, et al., 2005) and a search for suitable homology templates located a high resolution structure of bovine rhodopsin 1U19 (Okada, et al., 2004). Since the length of extracellular domain EC2 was greater in hBRS3 than in 1U19, it was excised. The model of the transmembrane region of the receptor was exported to SYBYL 7.3 molecular modeling suite (Tripos International, St Louis, MO) and the homology modeling COMPOSER module used to build candidates for the EC2 loop. This generated 25 potential loop conformations based on the crystal structure of homologous protein sequences. The alpha-carbons of the transmembrane regions were constrained and these loop models were optimized by energy minimization. Further optimization of the loops by constrained molecular dynamics followed by energy minimization produced loop models in which all amino acids were in favored positions in a Ramachandran plot. Inspection of these models showed that they fell into ~5 conformational categories. The potential binding sites in these models were probed using the SiteID module of SYBYL. Each model was solvated with water and clusters of water molecules within interaction distance of the protein interior were defined. This produced an estimate of the solvent accessible interior of the transmembrane receptor, and which amino acids were solvent exposed and thus capable of interaction with a potential ligand. These static models clearly illustrated the flexibility of the solvated EC2 region of the receptor and the conformational space available to the residues determined in this study to be important for binding; H107, E111 & G112. The remaining residues (L98, V101, L123, R127) were located on the relatively fixed transmembrane regions adjacent to EC2, and were oriented towards the solvent-accessible interior of the receptor. In a cell, these membrane-bound receptors are in motion in a fluid environment in which the conformation of the binding site may be influenced by interaction with a ligand. However, static modeling can give insight into the locations and accessibilities of the important amino acids within the binding site and potentially guide the development of new agonists.
Figure 5.
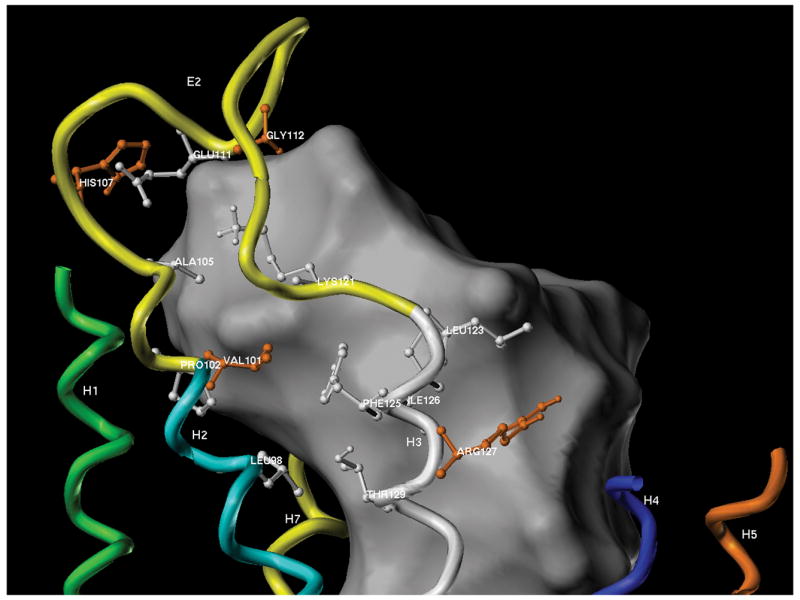
Three dimensions model of the putative binding pocket involving the EC2 region of the hBRS-3-receptor. This model was based on the studies of bovine rhodopsin as described in METHODS. The interior of the extracellular solvent-accessible region of the receptor is indicated as a white shape. This central portion of the transmembrane receptor may accommodate and bind ligands. Solvent-accessible amino acid residues within interaction distance (4Å) of this binding pocket are shown as ball and stick models, with the residues identified in this work as important for binding colored orange [Val101, His107, Gly112 and Arg127]. Residues L98, V101, H107, E111, G112, which differed between BRS-3 and GRPR (Fig. 3) are within 4Å of the binding pocket.
The EC2 loop model that most closely fitted the results of the mutagenesis studies is shown in Figure 5. In this figure, the transmembrane domains are shown as colored helices with the important extracellular EC2 loop (yellow) between TM2 (cyan) and TM3 (white). The model revealed the presence of a large solvent-accessible cavity within the transmembrane domain/extracellular domain interface, with potential interactions between a ligand and the interior-located transmembrane moieties and the flexible extracellular domains (grey area, Fig. 5). Seven of the ten amino acids, which differed in this region between BRS3 and GRPR and were examined in this study (L98, V101, H107, E111, G112, L123, R127) were found to be within 4Å of the proposed binding pocket. Alteration of four (V101, H107, G112, R127) of the seven amino acids studied within 4Å of the binding pocket caused the most significant decrease in affinity when present alone (3 to 26-fold), and the combination of all the members of this group caused a greater than additive decrease in affinity of peptide#4 for hBRS-3, showing almost a complete loss- of-affinity. These four important amino acids are highlighted in orange in Figure 5. All four of these important amino acids had their backbone residues orientated towards the interior of the receptor toward the binding pocket and thus would be available to interact with a ligand in the putative binding site. In contrast, the other three amino acids studied (T106S, V122, S124) which were not important for peptide#4 selectivity, are farther than 4Å of the binding pocket and oriented to the exterior of the receptor model. In the case of peptide#3 the situation is slightly different. Binding results showed the implication of four residues (L98, V101, T106, H107) in the affinity of peptide#3 for BRS-3. In our model three of these amino acids (L98, V101, H107) are situated in the hypothetical binding pocket, within 4Å, which could confirm their implication in the affinity of the selective compound.
Discussion
This study was undertaken to provide insight into the molecular basis for agonist high/affinityselectivity for human BRS-3(Mantey, et al., 2004;Mantey, et al., 2006;Boyle, et al., 2005;Mantey, et al., 2001). Nothing is known of the determinants of high affinity/selectivity for BRS-3. This has occurred because it is an orphan receptor, however is classified in the bombesin-receptor family, because of its high homology with the two mammalian members of this family(i.e. GRPR,NMBR)(Fathi, et al., 1993b;Jensen, et al., 2007). Furthermore, because of its low affinity for all natural-occurring Bn-related peptides(Mantey, et al., 1997;Jensen, et al., 2007), until the recent discovery of the synthetic Bn-analogue, [D-Phe6,βAla11,Phe13,Nle14]Bn-(6–14)(Mantey, et al., 1997), BRS-3’s pharmacology was largely unknown. Recent studies show [D-Phe6,βAla11,Phe13,Nle14]Bn-(6–14) a unique peptide, having high affinity/potency for almost all bombesin receptors. This synthetic Bn-analogue was used as the template to develop a number of selective human BRS-3 agonists(Mantey, et al., 2004;Mantey, et al., 2006;Boyle, et al., 2005;Mantey, et al., 2001), and this study explores the molecular determinants of their selectivity.
A number of our results show differences primarily in the extracellular(EC) domains, but also to a lesser extent in the upper transmembrane(TM) domains of bombesin receptors, play important roles in the selectivity of these peptide agonists for BRS-3. However, the extent of their importance and the specific receptor domains involved differ for the different BRS-3-preferring agonists. First, BRS-3 chimeric loss-of-affinity studies, demonstrated only substitution of EC2 of BRS-3 produced a markedly decrease in affinity of peptide#4, which was the most selective BRS-3 agonist. Second, with peptide#3 substitution of EC3, but also to a less extent EC2, decreased affinity. Third, with the selective BRS-3 ligand, peptide#2, only the substitution of EC3 altered affinity. Fourth, both the affinity of peptides #3 and #4 were affected by differences in the upper transmembrane regions, however the importance of differences in the upper TM2 and TM3 differed for these two peptides. Specifically, for peptide#4, but not peptide#3, differences in the upper TM3 [the presence of an arginine in the TM3 in BRS-3(ARG127) instead of a glutamine in the equivalent amino acid location in GRPR (GLU121)] was important for affinity. Fifth, these conclusions were supported by the reverse study examining gain-of-affinity GRPR chimeras constructed by inserting the extracellular domains of BRS-3 into GRPR. These results demonstrate that even though these three BRS-3-selective agonists were developed from the same template peptide,[D-Phe6,βAla11,Phe13,Nle14]Bn-(6–14), their molecular determinants of selectivity varied considerably.
These results have similarities and differences from studies of selectivity of Bn peptides for the NMBR/GRPR and other peptide agonists with other G-protein-coupled receptors(GPCR). Compared to the NMBR and GRPR, our results are similar in that for selectivity of NMB or GRP, with both extracellular and transmembrane regions being important(Fathi, et al., 1993a;Sainz, et al., 1998;Akeson, et al., 1997; Tokita, et al., 2001b; Nakagawa, et al., 2005). Compared to other GPCRs, our results are similar to the peptide agonist, substance P with NK1 receptors(Li, et al., 1996) and CCK-8 with CCK-B receptors(Silvente-Poirot, et al., 1998), where both the extracellular and transmembrane domains are important for selectivity. In contrast, the affinity of CCK-8 for CCK-A receptor(Silvente-Poirot, et al., 1998) or secretin for secretin receptors(Holtmann, et al., 1995) is determined by the extracellular domains, whereas differences in the transmembrane regions play the critical role in the affinity of nociceptin for orphanin FQ receptors(Meng, et al., 1996).
In terms of the specific extracellular or TM-receptor domains important for ligand selectivity, our with BRS-3 selective agonists differ from the location of those determining selectivity of NMB [i.e. in EC3 and EC4(Sainz, et al., 1998)], or GRP[critical differences are in EC3(Tokita, et al., 2001b) and in a lesser degree in EC4(Akeson, et al., 1997) and EC2(Nakagawa, et al., 2005)]. Similar to our results for the most selective BRS-3 ligand, peptide#4, with the mouse p opioid receptor and rat AT1 receptor, the EC2 plays a critical role for high-affinity interaction with selective peptide DAMGO(Wang, et al., 1995) or angiotensin II(Hjorth, et al., 1994), respectively. Similarly with CCKBR both EC2/EC3 were important for the CCK affinity(Silvente-Poirot, et al., 1998). In contrast, only the TM3 region of the mouse PGD receptor was essential for PGD2 binding(Kobayashi, et al., 1997). These results demonstrate the marked variability in molecular determinants not only for different unrelated receptors, but also for closely-related receptors for peptide agonists.
Differences in EC2 were the most important for determining the selectivity of peptides#3/#4, the most selective BRS-3 ligands. Within EC2 there were 4 amino acid differences between BRS-3 and GRPR and 6 more differences in the adjacent upper TM’s(TM2/TM3). Site-directed mutagenesis demonstrated histidine107 of BRS-3, instead of lysine101 in GRPR, as the most important difference, and to a lesser extent, glycine112, instead of arginine106 in a comparable position of GRPR in the EC2 as responsible for these peptides’ BRS-3-selectivity. Histidine in other GPCRs plays an important role in determining high affinity interaction/selectivity of various peptide/nonpeptide agonists and/or antagonists(Fong, et al., 1993). Histidine in EC3 of CCKB receptors is necessary for high affinity for CCK(Silvente-Poirot, et al., 1998): in EC1 of AT1 receptors(Hjorth, et al., 1994)for angiotensin’s high affinity; in TM7 of CCKB receptors for high affinity/potency for CCK (Paillasse, et al., 2006) and in TM3 of corticotropin-releasing-factor receptors for the nonpeptide antagonist NBI-27914(Liaw, et al., 1997). The replacement of histidine197 in TM5 of neurokinin-1 receptors by lysine, results in a 500-fold affinity reduction of the nonpeptide antagonist, CP-96345(Fong, et al., 1993) suggesting histidine is involved in amino-aromatic interactions with the benzhydryl-moiety of CP-96345. It was proposed, the δ(+)amino group of histidine has electrostatic interactions with the benzhydryl δ(-)-π-electrons, however, the longer lysine side-chain creates unfavorable positioning for the ligand and decreases the affinity for CP-96345(Fong, et al., 1993). A benzhydryl moiety is also presented in peptide#4 raising the possibility that a similar interaction occurs between the imidazole of histidine107 of BRS-3 with the benzhydryl of peptide#4. This possibility is supported by the marked decrease in affinity for peptide#4 by replacement of histidine by lysine, as well as the much reduced effect of this substitution on the affinity of peptides #2 and #3 which lack benzhydryl moieties. In contrast, lysine101, instead of histidine in EC2 of GRPR is critical for the selectivity of GRP for GRPR over BRS-3 (Nakagawa, et al., 2005).
Five of the 10 other amino acids(i.e. [BRS-3,L98,V101,T106,G112,R127]) that differ between BRS-3 and GRPR in EC2/adjacent upper TM2 or TM3, also contributed modestly(<5-fold) to BRS-3’s selectivity for agonists#3 and #4. Arginine127 in BRS-3 instead of glutamine in the comparable position of GRPR (Q121) had a modest(3-fold) effect on the affinity of BRS-3-selective ligand peptide#4. This differed markedly from the effect of the reverse substitution of arginine for glutamine in this position in GRPR(Q121R)(Akeson, et al., 1997;Nakagawa, et al., 2005) which resulted in >10,000 decrease in affinity for GRP. The addition of a V101A substitution(TM2) to two differences EC2(H107K, G112R) in hBRS-3, which alone had modest effects, had a profound effect on peptide#4’s affinity, decreasing it to >100 μM. The ability of multiple mutations to potentiated each other’s effect, is reported for the selectivity of PHI for rat over human VPAC1 receptor(Couvineau, et al., 1996), GRP for GRPR over human BRS-3(Nakagawa, et al., 2005)and the peptide antagonist JMV594/JMV641 for GRPR over NMBR(Tokita, et al., 2001b). Possible explanations that could be proposed for the potentiating effects of multiple substitutions is that each amino acids have additive interactions with the ligand, that they are needed to maintain a basic binding site-conformation or cause a more global alteration in the BRS-3-receptor-conformation. The latter possibility is not supported by our results, because the various mutant receptors maintained high affinity for the synthetic Bn analogue[D-Phe6, βAla11,Phe13,Nle14]Bn(6–14). The first possibility is supported by our molecular modeling results because each of the three amino acids [V101A,H107K,G112] that together have a large influence on peptide#4 affinity, all were within 4Å distance of the likely binding pocket with their side-groups orientated inward, supporting the possibility of a direct interaction with the ligand.
BRS-3-receptor activation stimulates tyrosine kinases and phospholipase C and D(Ryan, et al., 1998;Jensen, et al., 2007). However, almost nothing is known of the molecular determinants for its activation(Jensen, et al., 2007). Our results support the conclusion that histidine107 in EC2 of BRS-3, is essential for receptor-activation by synthetic agonist ligands independent of any effect on receptor-affinity. First, substitution of histidine107 by the GRPR-equivalent residue lysine did not alter the basal activity, but completely inhibited agonist activation. Second, lack of agonist activation was not due to failure to bind ligand, because the mutant receptor(H107K]BRS-3) retained high affinity for the agonist peptides#1/#5. These results demonstrate histidine107 is essential for BRS-3 activation by agonists, but not for high affinity binding. Histidine107 in BRS-3’s EC2 is the first extracellular amino acid in the bombesin-receptor family shown to be essential for receptor activation. Previously, amino acids essential for agonist activation and/or G-protein-coupling were described in transmembrane regions of GRPR[P98 in TM2(Donohue, et al., 1999), F270,N281,R304 in TM6(Lin, et al., 2000;Akeson, et al., 1997)], and in the 2nd [R139,I143,V144,M147](Benya, et al., 1994) and 4th intracellular[A264](Benya, et al., 1994) domains of GRPR. The role of extracellular domains in the biological activity of G protein-coupled receptors by peptide agonists is generally poorly understood, but numerous studies for other peptide/nonpeptide agonists report the importance of intracellular receptor/TM domains critical for their receptor’s activation(Kobilka, 2007). Similar to our results with histidine107, a few studies in other GPCRs reported with other peptide agonists, amino acids in EC domains were important for G-protein coupling and/or signaling, such as in EC2 for CCK-A receptor(Dong, et al., 2007) and VIP activation of hVPAC1 receptor(Knudsen, et al., 2000). These results suggest that with some peptide ligands, such as the synthetic BRS-3 selective ligands, amino acids in EC domains play a more prominent role in activation than was generally reported for nonpeptide agonists(Kobilka, 2007).Whether similar results will be found for the natural ligands of BRS-3 is at present unclear because the natural ligand is unknown.
Acknowledgments
This work was partial supported by intramural funds of the NIDDK and NCI, NIH and the Tulane University Peptide Research Fund
Abbreviations
- AT II
angiotensin II
- Bn
bombesin
- BRS-3
bombesin receptor subtype 3
- fBB4
bombesin receptor subtype 4
- BK
bradykinin
- BSA
bovine serum albumin fraction V
- CCK
cholecystokinin, CCKR, cholecystokinin receptor
- DMEM
Dulbecco’s minimum essential medium
- EC
extracellular
- FBS
fetal bovine serum
- GPCR
G protein-coupled receptor
- GRP
gastrin-releasing peptide
- GRPR
gastrin-releasing peptide receptor
- IP
inositol-phosphates
- NMB
neuromedin B
- NMBR
neuromedin B receptor
- PBS
phosphate-buffered saline
- PHI
peptide histidine isoleucine amide
- TM
transmembrane
- VPAC1R
vasoactive intestinal polypeptide receptor subtype 1
- VIP
vasoactive intestinal peptide
Footnotes
Recommended section: Gastrointestinal, hepatic, pulmonary, and renal
References
- Akeson M, Sainz E, Mantey SA, Jensen RT, Battey JF. Identification of four amino acids in the gastrin-releasing peptide C receptor that are required for high affinity agonist binding. J Biol Chem. 1997;272:17405–17409. doi: 10.1074/jbc.272.28.17405. [DOI] [PubMed] [Google Scholar]
- Benya RV, Akeson M, Mrozinski J, Jensen RT, Battey JF. Internalization of the gastrin-releasing peptide receptor is mediated by phospholipase C-dependent and -independent processes. Mol Pharmacol. 1994;46:495–501. [PubMed] [Google Scholar]
- Boyle RG, Humphries J, Mitchell T, Showell GA, Apaya R, Iijima H, Shimada H, Arai T, Ueno H, Usui Y, Sakaki T, Wada E, Wada K. The design of a new potent and selective ligand for the orphan bombesin receptor subtype 3 (BRS3) J Pept Sci. 2005;11:136–141. doi: 10.1002/psc.599. [DOI] [PubMed] [Google Scholar]
- Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvineau A, Rouyer-Fessard C, Maoret JJ, Gaudin P, Nicole P, Laburthe M. Vasoactive intestinal peptide (VIP)1 receptor. Three nonadjacent amino acids are responsible for species selectivity with respect to recognition of peptide histidine isoleucinemide. J Biol Chem. 1996;271:12795–12800. doi: 10.1074/jbc.271.22.12795. [DOI] [PubMed] [Google Scholar]
- Dong M, Ding XQ, Thomas SE, Gao F, Lam PC, Abagyan R, Miller LJ. Role of lysine187 within the second extracellular loop of the type A cholecystokinin receptor in agonist-induced activation. Use of complementary charge-reversal mutagenesis to define a functionally important interdomain interaction. Biochemistry. 2007;46:4522–4531. doi: 10.1021/bi0622468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue RJ, Sainz E, Akeson M, Kroog GS, Mantey SA, Battey JF, Jensen RT, Northrup JK. An aspartate residue at the extracellular boundary of TMII, and an arginine residue at TMVII of the gastrin-releasing peptide receptor interact to facilitate heterotrimeric G protein coupling. Biochemistry. 1999;38:9366–9372. doi: 10.1021/bi990544h. [DOI] [PubMed] [Google Scholar]
- Fathi Z, Benya RV, Shapira H, Jensen RT, Battey JF. The fifth transmembrane segment of the neuromedin B receptor is critical for high affinity neuromedin B binding. J Biol Chem. 1993a;268(20):14622–14626. [PubMed] [Google Scholar]
- Fathi Z, Corjay MH, Shapira H, Wada E, Benya R, Jensen R, Viallet J, Sausville EA, Battey JF. BRS-3: novel bombesin receptor subtype selectively expressed in testis and lung carcinoma cells. J Biol Chem. 1993b;268(8):5979–5984. [PubMed] [Google Scholar]
- Fong TM, Cascieri MA, Yu H, Bansal A, Swain C, Strader CD. Amino-aromatic interaction between histidine 197 of the neurokinin-1 receptor and CP 96345. Nature. 1993;362:350–353. doi: 10.1038/362350a0. [DOI] [PubMed] [Google Scholar]
- Hjorth SA, Schambye HT, Greenlee WJ, Schwartz TW. Identification of peptide binding residues in the extracellular domains of the AT1 receptor. J Biol Chem. 1994;269:30953–30959. [PubMed] [Google Scholar]
- Holtmann MH, Hadac EM, Miller LJ. Critical contributions of amino-terminal extracellular domains in agonist binding and activation of secretin and vasoactive intestinal polypeptide receptors. Studies of chimeric receptors. J Biol Chem. 1995;270:14394–14398. doi: 10.1074/jbc.270.24.14394. [DOI] [PubMed] [Google Scholar]
- Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. The bombesin Receptors: Nomenclature, distribution, pharmacology, signaling and functions in normal and disease states. Pharmacol Rev. 2007 doi: 10.1124/pr.107.07108. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RT, Moody TW. Bombesin-related peptides and neurotensin: effects on cancer growth/proliferation and cellular signaling in cancer. In: Kastin AJ, editor. Handbook of Biologically active peptides. Elsevier; Amsterdam: 2006. pp. 429–434. [Google Scholar]
- Knudsen SM, Tams JW, Fahrenkrug J. Role of second extracellular loop in the function of human vasoactive intestinal polypeptide/pituitary adenylate cyclase activating polypeptide receptor 1 (hVPAC1R) J Mol Neurosci. 2000;14:137–146. doi: 10.1385/JMN:14:3:137. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Kiriyama M, Hirata T, Hirata M, Ushikubi F, Narumiya S. Identification of domains conferring ligand binding specificity to the prostanoid receptor. Studies on chimeric prostacyclin/prostaglandin D receptors. J Biol Chem. 1997;272:15154–15160. doi: 10.1074/jbc.272.24.15154. [DOI] [PubMed] [Google Scholar]
- Kobilka BK. G protein coupled receptor structure and activation. Biochim Biophys Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Hsu P, Sachais BS, Krause JE, Leeman SE, Boyd ND. Identification of the site in the substance P (NK-1) receptor for modulation of peptide binding by sulfhydryl reagents. J Biol Chem. 1996;271:1950–1956. doi: 10.1074/jbc.271.4.1950. [DOI] [PubMed] [Google Scholar]
- Liaw CW, Grigoriadis DE, Lorang MT, De Souza EB, Maki RA. Localization of agonist- and antagonist-binding domains of human corticotropin-releasing factor receptors. Mol Endocrinol. 1997;11:2048–2053. doi: 10.1210/mend.11.13.0034. [DOI] [PubMed] [Google Scholar]
- Lin Y, Jian X, Lin Z, Kroog GS, Mantey S, Jensen RT, Battey J, Northup J. Two amino acids in the sixth transmembrane segment of the mouse gastrin-releasing peptide receptor are important for receptor activation. JPET. 2000;294:1053–1062. [PubMed] [Google Scholar]
- Mantey SA, Coy DH, Entsuah LK, Jensen RT. Development of bombesin analogs with conformationally restricted amino acid substitutions with enhanced selectivity for the orphan receptor human bombesin receptor subtype 3. J Pharmacol Exp Ther. 2004;310:1161–1170. doi: 10.1124/jpet.104.066761. [DOI] [PubMed] [Google Scholar]
- Mantey SA, Coy DH, Pradhan TK, Igarashi H, Rizo IM, Shen L, Hou W, Hocart SJ, Jensen RT. Rational design of a peptide agonist that interacts selectively with the orphan receptor, bombesin receptor subtype 3. J Biol Chem. 2001;276:9219–9229. doi: 10.1074/jbc.M008737200. [DOI] [PubMed] [Google Scholar]
- Mantey SA, Gonzalez N, Schumann M, Pradhan TK, Shen L, Coy DH, Jensen RT. Identification of bombesin receptor subtype-specific ligands: effect of N-methyl scanning, truncation, substitution, and evaluation of putative reported selective ligands. J Pharmacol Exp Ther. 2006;319:980–989. doi: 10.1124/jpet.106.107011. [DOI] [PubMed] [Google Scholar]
- Mantey SA, Weber HC, Sainz E, Akeson M, Ryan RR, Pradhan TK, Searles RP, Spindel ER, Battey JF, Coy DH, Jensen RT. Discovery of a high affinity radioligand for the human orphan receptor, bombesin receptor subtype 3, which demonstrates it has a unique pharmacology compared to other mammalian bombesin receptors. J Biol Chem. 1997;272(41):26062–26071. doi: 10.1074/jbc.272.41.26062. [DOI] [PubMed] [Google Scholar]
- Meng F, Taylor LP, Hoversten MT, Ueda Y, Ardati A, Reinscheid RK, Monsma FJ, Watson SJ, Civelli O, Akil H. Moving from the orphanin FQ receptor to an opioid receptor using four point mutations. J Biol Chem. 1996;271:32016–32020. doi: 10.1074/jbc.271.50.32016. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Hocart SJ, Schumann M, Tapia JA, Mantey SA, Coy DH, Tokita K, Katsuno T, Jensen RT. Identification of key amino acids in the gastrin-releasing peptide receptor (GRPR) responsible for high affinity binding of gastrin-releasing peptide (GRP) Biochem Pharmacol. 2005;69:579–593. doi: 10.1016/j.bcp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Nakamichi Y, Wada E, Aoki K, Ohara-Imaizumi M, Kikuta T, Nishiwaki C, Matsushima S, Watanabe T, Wada K, Nagamatsu S. Functions of pancreatic beta cells and adipocytes in bombesin receptor subtype-3-deficient mice. Biochem Biophys Res Commun. 2004;318:698–703. doi: 10.1016/j.bbrc.2004.04.081. [DOI] [PubMed] [Google Scholar]
- Ohki-Hamazaki H, Watase K, Yamamoto K, Ogura H, Yamano M, Yamada K, Maeno H, Imaki J, Kikuyama S, Wada E, Wada K. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature. 1997;390(6656):165–169. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]
- Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- Paillasse MR, Deraeve C, de MP, Mhamdi L, Favre G, Poirot M, Silvente-Poirot S. Insights into the cholecystokinin 2 receptor binding site and processes of activation. Mol Pharmacol. 2006;70:1935–1945. doi: 10.1124/mol.106.029967. [DOI] [PubMed] [Google Scholar]
- Porcher C, Juhem A, Peinnequin A, Bonaz B. Bombesin receptor subtype-3 is expressed by the enteric nervous system and by interstitial cells of Cajal in the rat gastrointestinal tract. Cell Tissue Res. 2005;320:21–31. doi: 10.1007/s00441-004-1032-1. [DOI] [PubMed] [Google Scholar]
- Pradhan TK, Katsuno T, Taylor JE, Kim SH, Ryan RR, Mantey SA, Donohue PJ, Weber HC, Sainz E, Battey JF, Coy DH, Jensen RT. Identification of a unique ligand which has high affinity for all four bombesin receptor subtypes. Eur J Pharmacol. 1998;343:275–287. doi: 10.1016/s0014-2999(97)01527-6. [DOI] [PubMed] [Google Scholar]
- Ryan RR, Weber HC, Hou W, Sainz E, Mantey SA, Battey JF, Coy DH, Jensen RT. Ability of various bombesin receptor agonists and antagonists to alter intracellular signaling of the human orphan receptor BRS-3. J Biol Chem. 1998;273:13613–13624. doi: 10.1074/jbc.273.22.13613. [DOI] [PubMed] [Google Scholar]
- Sainz E, Akeson M, Mantey SA, Jensen RT, Battey JF. Four amino acid residues are critical for high affinity binding of neuromedin B to the neuromedin B receptor. J Biol Chem. 1998;273:15927–15932. doi: 10.1074/jbc.273.26.15927. [DOI] [PubMed] [Google Scholar]
- Sano H, Feighner SD, Hreniuk DL, Iwaasa H, Sailer AW, Pan J, Reitman ML, Kanatani A, Howard AD, Tan CP. Characterization of the bombesin-like peptide receptor family in primates. Genomics. 2004;84:139–146. doi: 10.1016/j.ygeno.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Schulz S, Rocken C, Schulz S. Immunohistochemical detection of bombesin receptor subtypes GRP-R and BRS-3 in human tumors using novel antipeptide antibodies. Virchows Arch. 2006;449:421–427. doi: 10.1007/s00428-006-0265-7. [DOI] [PubMed] [Google Scholar]
- Silvente-Poirot S, Escrieut C, Wank SA. Role of the extracellular domains of the cholecystokinin receptor in agonist binding. Mol Pharmacol. 1998;54:364–371. doi: 10.1124/mol.54.2.364. [DOI] [PubMed] [Google Scholar]
- Tokita K, Hocart SJ, Katsuno T, Mantey SA, Coy DH, Jensen RT. Tyrosine 220 in the fifth transmembrane domain of the neuromedin B receptor is critical for the high selectivity of the peptoid antagonist PD168368. J Biol Chem. 2001a;276:495–504. doi: 10.1074/jbc.M006059200. [DOI] [PubMed] [Google Scholar]
- Tokita K, Katsuno T, Hocart SJ, Coy DH, Llinares M, Martinez J, Jensen RT. Molecular basis for selectivity of high affinity peptide antagonists for the gastrin-releasing peptide receptor. J Biol Chem. 2001b;276:36652–36663. doi: 10.1074/jbc.M104566200. [DOI] [PubMed] [Google Scholar]
- Wang WW, Shahrestanifar M, Jin J, Howells RD. Studies on mu and delta opioid receptor selectivity utilizing chimeric and site-mutagenized receptors. Proc Natl Acad Sci (USA) 1995;92:12436–12440. doi: 10.1073/pnas.92.26.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]