

Abstract
The Aspergillus nidulans NIMA kinase is essential for mitosis and is the founding member of the conserved NIMA-related kinase (Nek) family of protein kinases. To gain insight into NIMA function, a copy number suppression screen has been completed that defines three proteins termed MCNA, MCNB, and MCNC (multi-copy-number suppressor of nimA1 A, B, and C). All display a distinctive and dynamic cell cycle-specific distribution. MCNC has weak similarity to Saccharomyces cerevisiae Def1 within a shared CUE-like domain. MCNC, like Def1, is a cytoplasmic protein with slow mobility during sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and its deletion causes polarization defects and a small colony phenotype. MCNC enters nuclei during mitosis. In contrast, MCNB is a nuclear protein displaying increased nuclear levels as cells progress through interphase but is lost from nuclei at mitosis. MCNB is highly related to the Schizosaccharomyces pombe forkhead transcription factor Sep1 and is likely a transcriptional activator of nimA. Most surprisingly, MCNA, a protein restricted to the aspergilli and pathogenic systemic dimorphic fungi (the Eurotiomycetes), defines a nuclear body located near nucleoli at the nuclear periphery of G2 nuclei. During progression through mitosis, the MCNA body is excluded from nuclei. Cytoplasmic MCNA bodies then diminish during early stages of interphase, and single MCNA bodies are formed within nuclei as interphase progresses. Three sites of MCNA phosphorylation were mapped and mutated to implicate proline-directed phosphorylation in the equal segregation of MCNA during the cell cycle. The data indicate all three MCN proteins likely have cell cycle functions.
Genetic screens to isolate genes involved in cell cycle progression in Aspergillus nidulans (25) have identified numerous conserved proteins involved in several aspects of cell cycle progression. The most extensively studied of these is the mitotis-specific NIMA protein kinase, which is the founding member of the Nek family (NIMA-related kinases) of protein kinases (14, 28, 29). In A. nidulans NIMA is required for all cytological aspects of mitosis, and in its absence cells arrest in late G2, just before mitosis (4, 27). NIMA is subject to complex regulation to ensure that its activity is maximal during mitosis and reduced during exit from mitosis (11, 35-37, 41, 42, 53-56). These levels of control include cell cycle-specific modulation of NIMA mRNA levels, phosphorylation status, stability, protein kinase activity, and subcellular distribution.
Several approaches have been taken to identify proteins interacting with NIMA, including yeast two-hybrid analysis and extragenic suppressor screens. Two proteins that physically interact transiently with NIMA during the cell cycle, termed TINA (32) and TINC (9), were identified in two-hybrid screens. TINA is a protein that specifically locates to the spindle pole body (SPB) during mitosis, in a microtubule-dependent manner, and has been proposed to negatively regulate astral microtubule formation. A similar protein has been identified in Schizosaccharomyces pombe (Msd1p) (48) and, like TINA, this protein also localizes to SPBs only during mitosis. The S. pombe TINA-related protein has been proposed to play a role in anchoring microtubules to the SPB (48). Thus, these two proteins appear to play similar, and perhaps identical, functions at the mitotic SPB. Importantly, NIMA and Fin1, the S. pombe orthologue of NIMA, also locate to mitotic SPBs (12, 18, 19). TINC is a cytoplasmic protein that locates to nuclei during mitosis. The most notable characteristic of TINC is that expression of an N-terminally truncated version of the protein causes mitotic defects which prevent the process of nuclear envelope (NE) fission that normally generates two nuclei from one during exit from mitosis. TINC and NIMA have therefore been proposed to play a role in nuclear membrane fission (9), a process that is poorly understood.
Although NIMA is required for all cytological aspects of mitosis, it is not necessary for activation of CDK1 mitotic kinase activity (33). Insight into this surprising result has come from analysis of extragenic suppressors of the nimA1 temperature-sensitive allele of nimA (10, 51), which identified two nuclear pore complex (NPC) proteins (SONA and SONB) that themselves physically interact (11). Subsequent studies revealed that the NPC of A. nidulans undergoes massive reorganization during mitosis, during which peripheral nucleoporins (Nups) are removed from the NPC, whereas core components remain. The core components are thought to provide a conduit between the mitotic cytoplasm and nucleoplasm and act as a seed to which dispersed Nups return during mitotic exit to reestablish regulated nuclear transport during G1 (11, 31). Because NIMA is both required and sufficient (when ectopically expressed) to promote NPC disassembly, it is clear that one key role for NIMA during mitotic initiation is to promote NPC disassembly to allow tubulin (38) and mitotic regulators, such as CDK1-cylin B, access to nucleus-bound chromosomes (13).
To help further our understanding of the roles of NIMA in A. nidulans we have completed a copy number suppressor screen using temperature-sensitive alleles of nimA. The screen identified three new genes which are implicated in NIMA function. Importantly, all three proteins display distinctive changes in their subcellular distribution as cells progress through mitosis, strongly suggesting each potentially plays a role in cell cycle progression.
MATERIALS AND METHODS
General A. nidulans methodologies.
Standard growth and classical genetic methodologies for A. nidulans have been described previously (40). A strain list is provided in Table 1. Molecular genetic methods were as previously described and utilized fusion PCR for DNA constructions and strains with reduced nonhomologous recombination for gene targeting (26, 34, 47, 52).
TABLE 1.
A. nidulans strains used in this study
| Strain | Genotype | Derivation |
|---|---|---|
| LU28 | pyrG89 wA2 pyroA4 nicB2/nicA2 nimA1 pyr4-AMA1-mcnC | LPW3 transformant |
| LU29 | pyrG89 wA2 pyroA4 nicB2/nicA2 nimA1 pyr4-AMA1-mcnA | LPW3 transformant |
| LU30 | pyrG89 wA2 pyroA4 nicB2/nicA2 nimA1 pyr4-AMA1-mcnB | LPW3 transformant |
| LU31 | pyrG89 wA2 pyroA4 nicB2/nicA2 nimA1 pyr4-AMA1 | LPW3 transformant |
| LU32 | pyrG89 wA2 pyroA4 nicB2/nicA2 nimA1 pyr4-AMA1-nimA | LPW3 transformant |
| LU08 | pyrG89 wA2 pyroA4 nicB2/nicA2 nimA1 pyr4- alcA::mcnC | LPW3 transformant |
| LU07 | pyrG89 wA2 pyroA4 ΔmcnC::pyroA | GR5 transformant |
| LU390 | pyrG89 wA2 pyroA4 nimA1 ΔmcnC::pyroA | LU53 × LU07 |
| LU391 | pabaA1 pyroA4 nimA1 ΔmcnC::pyroA chaA1 | LU53 × LU07 |
| LU392 | pabaA1 pyroA4 nimA1 ΔmcnC::pyroA | LU53 × LU07 |
| LU393 | pabaA1 pyrG89 wA2 pyroA4 nimA1 ΔmcnC::pyroA | LU53 × LU07 |
| LU147 | pyrG89 argB2 pyroA4 An-mcnB-GFP::pyrGAf (sE151nirA141) | LU199 × UI224 |
| LU140 | (pyrG891) wA3 histone-H1-mRFP::pyrGAf An-mcnB-GFP::pyrGAf (sE151nirA141) | LU147 × LO1353 |
| LU146 | pyrG89 argB2 pyroA4 An-mcnA-GFP::pyrGAf (sE151 nirA141) | LU192 × UI224 |
| LU84 | (pyrG891) wA3 argB2 pyroA4 An-cgrA-mRFP::pyrGAf An-mcnA-GFP::pyrGAf | LU146 × LU165 |
| LU459 | (pyrG891) wA3 argB2 pyroA4 An-erg4-mRFP::pyrGAf An-mcnA-GFP::pyrGAf | LU146 × HA103 |
| LU102 | (pyrG891) argB2 An-bop1-chRFP::pyrGAf An-mcnA-GFP::pyrGAf (sE151nirA141) | LU45 × LU80 |
| LU460 | (pyrG891) wA3 argB2 An-fib-chRFP::pyrGAf An-mcnA-GFP::pyrGAf (nirA141) | LU192 × CDS410 |
| LU39 | (pyrG891) wA3 argB2 pyroA4 histone-H1-mRFP::pyrGAf An-mcnA-GFP::pyrGAf | LU146 × LO1353 |
| LU61 | pyrG89 argB2 ΔnkuAku70::argB pyroA4 nkuAAn-mcnC-Stag::pyrGAf sE15 nirA14 | SO451 transformant |
| LU256 | pyrG89 wA2 pyroA4 mcnA(3S→A)-GFP::pyGAf | ΔmcnA transformant |
| LU432 | pyrG89 mcnA(3S→A)-GFP::pyGAf An-fib-chRFP::pyrGAf (nirA141) | LU256 × CDS410a |
Copy number suppressor screen.
Transformation and selection of transformants using AMA1-based genomic libraries were as previously described (10, 30), using the LPW3 nimA1 recipient strain (51). Complementing plasmids were isolated in Escherichia coli from the DNA of LPW3 transformants able to grow at 42°C. In order to distinguish suppressor plasmids from those containing wild-type nimA, restriction site mapping was employed. No suppressor plasmids could suppress the nimA5 loss-of-function allele, indicating that none are bypass suppressors. A combination of end sequence analysis and restriction site mapping defined three unique suppressors among the 14 identified. Subcloning experiments were used to define the genes able to suppress nimA1, showing 12 plasmids contained MCNC and 1 each contained MCNA and MCNB.
Isolation of full-length cDNAs by RACE-PCR.
To verify full-length cDNA clones for mcnA, mcnB, and mcnC, we utilized random amplification of cDNA ends-PCR (RACE-PCR). To generate an A. nidulans cDNA library, total RNA was prepared from 2 g of lyophilized R153 mycelia using the Ultraspec RNA isolation system (BIOTECX). Lyophilized mycelia were ground using a ceramic mortar and pestle pretreated in 2% H2O2 for 1 h to eliminate RNase activity. Ground mycelia were combined with 40 ml of Ultraspec RNA reagent and placed on ice for 5 min. Eight ml of chloroform was added and the material placed on ice for an additional 5 min. Cell debris was removed by centrifugation at 14,000 rpm for 15 min. RNA was precipitated using 2-propanol and resuspended in 1.5 ml diethyl pyrocarbonate-treated H2O. The RNA concentration (24 μg/μl) was determined by measuring the absorbance at 260 nm. Poly(A)+ mRNA was isolated using the PolyATtract mRNA isolation system (Promega). A 9.6-mg aliquot of total RNA was added to 4.46 ml of diethyl pyrocarbonate-treated H2O and placed in a 65°C water bath for 10 min. Ten μl of biotinylated oligo(dT) probe and 60 μl of 20× SSC (1× is 0.15 M NaCl plus 0.015 M sodium citrate) were added to the RNA and incubated at room temperature until cool. The entire annealing reaction mixture was incubated with streptavidin-paramagnetic particles, and mRNA was isolated by exposing particles to a magnet. mRNA was eluted in 500 μl of DEPC-treated H2O at a final concentration of 136 μg/ml. cDNA synthesis was performed using 1 μg poly(A)+ mRNA and cDNA synthesis reagents from the Marathon cDNA amplification kit (Clontech). Full-length mcnA, mcnB, and mcnC cDNAs were isolated using the touchdown PCR method. PCR was performed using an AP1 adaptor-ligated cDNA library, an adaptor primer (complementary to the adaptor sequence), and gene-specific primers for all three genes. The full-length cDNAs were cloned into the pCR 2 or pCR 2.1 vectors by using the TA cloning system (Invitrogen). cDNAs were completely sequenced.
ΔmcnA, ΔmcnB, and ΔmcnC phenotype testing.
The respective deleted transformants, ΔmcnA, ΔmcnB, and ΔmcnC, were tested for heterokaryon formation (34) by replica streaking on plates with and without selective pressure for the deletion marker. Deleted strains were tested for growth defects, with control wild-type strains R153 and JD100, under a variety of conditions. Strains were tested for their ability to undergo both self-crosses and outcrosses. Additionally, strains were spot inoculated onto YAGUU medium and tested under the following conditions: (i) temperatures of 20°C, 32°C, 37°C, and 42°C, (ii) osmotic stress with 1 M sucrose or 1 M sodium chloride, and (iii) drugs including nocodazole (0.1 μg/ml, 0.2 μg/ml, 0.3 μg/ml, 0.4 μg/ml, and 0.6 μg/ml), methyl methanesulfonate (0.01% and 0.02%), and hydroxyurea (4 mM, 6 mM, and 10 mM). All growth plates were incubated at 32°C except where indicated. The strains were also examined for cell cycle defects after 4′,6-diamidino-2-phenylindole (DAPI) staining. To examine polarization defects in ΔmcnC cells, 300 μl of inoculum containing 5 × 105 conidia/ml was placed on the surface of sterile coverslips and incubated at 32°C or 42°C to allow germination. Cells were fixed and DAPI stained using standard conditions. To quantify the polarization defect, DAPI staining was used to count the number of nuclei and the length of the germ tube was measured. These were then compared to measurements with control cells of strain JD100. At least 100 cells were characterized for each strain.
Affinity purifications using the S-Tag affinity peptide.
We have developed the S-Tag (21) as a clean and efficient single-step protein affinity purification system for A. nidulans. Details will be published elsewhere (H.-L. Liu et al., unpublished data). In brief, protein extracts derived from lyophilized mycelia, using HK buffer (33) supplemented with 300 mM NaCl, were incubated with S-protein agarose (catalog no. 69704-4; Novagen). After extensive washing, affinity-purified proteins were released using sodium dodecyl sulfate (SDS) sample buffer and separated by SDS-polylacrylamide gel electrophoresis (SDS-PAGE). Gels were either silver or Coomassie blue stained using standard methodologies. Mass spectrometry analysis was completed at the Ohio State University Campus Chemical Instrument Center using capillary liquid chromatography nanospray tandem mass spectrometry.
Western blot analysis.
Western blot analysis was performed to estimate protein expression levels and to verify proper integration of targeting constructs by confirming the protein size of chimeric proteins. Primary antibodies were anti-NIMA (E15III; 1:5,000 dilution), anti-green fluorescent protein (GFP; Living Colors; 1:5,000), anti-DsRed (Living Colors; 1:10,000), and anti-S peptide (Immunology Consultancy Laboratory; 1:5,000). Secondary antibodies used were enhanced chemiluminescence peroxidase-labeled antibodies from GE Healthcare. For MCNC-GFP Western blot analysis, 2 mg of total protein isolated in HK buffer (33) was immunoprecipitated using GFP antibody (Living Colors) and protein A/G beads. Purified MCNC-GFP was run on a 7% SDS-PAGE and subjected to Western blot analysis using standard methodologies.
In vitro λ-phosphatase assay.
After purification of protein via S-Tag affinity purification, beads were treated with 40 units of λ-phosphatase (New England Biolabs) with and without phosphatase inhibitors (1 mM Na-vanadate plus 50 mM Na-fluoride) after being washed in 2× phosphatase buffer, which included MnCl2 and protease inhibitors. The beads were mixed and incubated at 30°C for 30 min (mixing occasionally). Sample buffer was added and the supernatant collected and analyzed by SDS-PAGE.
Microscopy and image capture.
Fixed samples were examined using an E800 microscope (Nikon, Inc.) with DAPI, fluorescein isothiocyanate, or Texas red filters (Omega Optical, Inc.). Image capture was performed using an UltraPix digital camera (Life Science Resources, Ltd.). For live cell microscopy, strains were germinated in 3 ml of minimal medium in 35-mm glass-bottom petri dishes (MatTek Cultureware). Visualization was performed using a Nikon Eclipse TE300 inverted microscope (Nikon, Inc.) in conjunction with an Ultraview spinning disk confocal system (Perkin-Elmer) and an Orca ER digital camera (Hamamatsu). Image capture on both microscopes was performed using Ultraview image capture software (Perkin-Elmer).
Site-specific mutagenesis.
Site-directed mutagenesis was performed to generate the triple MCNA-3A mutant. The DNA sequence to be mutagenized was amplified from the GFP-tagged mcnA strain (LU256). The amplified DNA contained sequences for the mcnA gene fused to GFP and also the pyrGAF auxotrophic marker. The DNA was cloned into the pCR-Blunt II-TOPO vector using the Zero blunt TOPO PCR cloning kit (Invitrogen). The insert was subjected to three rounds of site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (catalog number 200518; Stratagene). Mutations were confirmed by sequencing, and the insert was released using BamHI and NotI and gel purified using a Qiagen gel purification kit. One μg of insert DNA was used to transform strain LU217, in which mcnA is deleted by the pyroA marker, selecting for pyrG+ colonies. Transformants were confirmed by spotting them on minimal medium (MM) plates with or without pyridoxine. Colonies that were pyrG-positive but pyroA-negative were considered positives.
RESULTS
nimA1 copy number suppressor screen.
Plasmids based on the AMA1 replicator of A. nidulans are maintained as extrachromosomal elements with an average copy number of 10 to 15 (1, 2). A. nidulans genomic libraries have been constructed in AMA1-containing vectors (30) and used to clone wild-type copies of conditional mutants by complementation. This approach also allows copy number suppressors of the original mutation to be cloned (10). We have completed a copy number suppressor screen by transformation of AMA1 libraries into a recipient nimA1 strain (41), looking for suppression of nimA1 temperature sensitivity. As described in Materials and Methods, in total three copy number suppressor genes were isolated and named mcnA (AN2692), mcnB (AN8858), and mcnC (AN2871) for multi-copy-number suppressors of nimA1.
MCNA and MCNB expression increases the level of NIMA protein.
Arrest in G2 under restrictive conditions for nimA1 (42°C) allows some accumulation of NIMA1 protein kinase activity, but this level is not sufficient to trigger entry into mitosis (51). We therefore considered the possibility that the copy number suppressors might act by increasing the expression levels of NIMA1, leading to suppression. To investigate this, nimA1 strains containing empty AMA1 vector (negative control), AMA1 plus nimA (positive control), and AMA1 plus mcnA, AMA1 plus mcnB, or AMA1 plus mcnC were shifted from permissive to restrictive temperature to impose the nimA1 G2 arrest. In these strains the nimA1 G2 arrest will be counteracted by wild-type AMA1-nimA and potentially by the copy number suppressors, allowing cells to enter mitosis. As NIMA is degraded if cells exit mitosis, and this degradation is prevented if cells are arrested in mitosis by activation of the spindle assembly checkpoint (54), we performed the experiments in the presence of the microtubule poison nocodazole. By depolymerizing microtubules and activating the spindle assembly checkpoint, NIMA was stabilized in those cells that passed the nimA1 G2 arrest point. By following this protocol we were able to compare the relative levels of NIMA in cells that arrest in G2 to those that were suppressed for the G2 arrest by expression of MCNA to -C or NIMA itself (Fig. 1). As revealed by Western blot analysis of cells arrested in G2/mitosis, expression of the copy number suppressors MCNA and MCNB markedly increased the level of NIMA protein. In fact both MCNA- and MCNB-expressing nimA1 cells arrested with comparable amounts of NIMA protein to that of the nimA1 plus NIMA positive control cells. In contrast, MCNC-expressing nimA1 cells had levels of NIMA protein comparable to the empty vector negative control cells. Ponceau staining of the transfer blot was used to confirm equal loading, and transfer of protein and nonspecific cross-reactive bands in the NIMA Western analysis also confirmed equal loading. These data indicate that MCNC is unlikely to suppress nimA1 through a mechanism involving increased expression of NIMA1 but that MCNA and MCNB could suppress nimA1 through this mechanism.
FIG. 1.
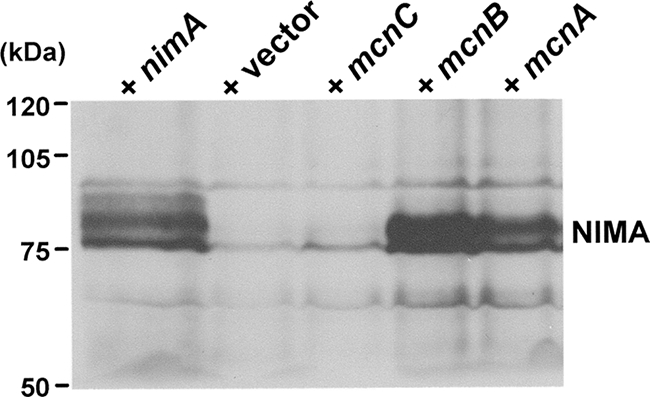
Effect of increased expression of MCN genes on NIMA1 protein levels. Western blot analysis of NIMA protein levels in a nimA1 strain containing AMA1 plasmids expressing no insert, wild-type nimA, or mcnC, mcnB, or mcnA (as indicated). Cells were incubated at the nimA1 restrictive temperature in the presence of nocodazole. Levels of the nonspecific band above NIMA acted as loading controls. The lane with empty vector represents basal levels of endogenous NIMA1 protein at the G2 arrest point.
MCNC is a cytoplasmic phospho-protein which enters nuclei during mitosis.
The mcnC gene (AN2871) contains two exons encoding an 870-amino-acid (aa) protein with a predicted molecular mass of 90.1 kDa which contains a potential CUE-like domain, suggesting a potential interaction with ubiquitinated proteins (20, 45). The mcnC gene and its structure are highly conserved within the eight sequenced aspergilli (http://www.broad.mit.edu/annotation/genome/aspergillus_group/GenomesIndex.html). Similar proteins, with sequence homology throughout the protein sequence, were detected in other filamentous ascomycetes. Limited similarity was also detected between MCNC, S. cerevisiae Def1, and Schizosaccharomyces pombe C354.10. Similarity to the yeast proteins was limited to the putative CUE domain (Fig. 2A). Proteins with similarity in this N-terminal portion of MCNC were detected within the basidiomycetes, but nonfungal organisms have no protein with significant similarity to MCNC.
FIG. 2.

MCNC protein has limited similarity to Def1 and locates to the cytoplasm. (A) Alignment of MCNC with proteins having a similar CUE-like domain (thick region of line) in their N-terminal regions, including S. cerevisiae Def1. An alignment of the CUE-like domain region within these proteins is shown. (B) MCNC-GFP is a cytoplasmic protein excluded from nuclei. Four single confocal slices at 0.2-μm intervals are shown, revealing other nonnuclear dark areas from which MCNC is excluded. The full z-series is presented as Movie S1 in the supplemental material. (C) Nuclei, as revealed by DAPI staining. (D) NLS-DsRED location, excluding MCNC-GFP. For panels C and D, the top panel shows nuclei, the middle shows MCNC-GFP, and the bottom shows the merged signals. (E) MCNC-GFP is not excluded from nuclei during mitosis but becomes excluded as cells exit mitosis. A time series of MCNC-GFP location is shown at 1-min intervals. A video is presented as Movie S2 in the supplemental material. Bar, ∼5 μm.
Endogenously C-terminally GFP-tagged MCNC resides throughout the cytoplasm (Fig. 2B), being excluded from nuclei as defined either by DAPI staining of DNA (Fig. 2C) or nuclear localization of an NLS-DsRed marker (46) (Fig. 2D). MCNC was not uniformly distributed within the cytoplasm but had an apparently structured distribution. This can be seen in the high-resolution through z series (Fig. 2B; see also Movie S1 in the supplemental material). It is likely that MCNC is excluded from vesicles and other membranous organelles within the cytoplasm, generating the darker-shaded areas observed throughout the cytoplasm. Live cell imaging revealed that MCNC-GFP entered nuclei during mitosis and was subsequently excluded from nuclei during exit from mitosis (Fig. 2E; see also Movie S2 in the supplemental material).
Western blot analysis of MCNC showed it to migrate during SDS-PAGE as a far larger protein than its predicted molecular mass (90 kDa). This was observed when using either GFP-tagged (Fig. 3A) or S-tagged versions of MCNC, both of which ran at nearly double their predicted molecular mass. We considered the possibility that the automated gene calling for mcnC might be inaccurate. However, 3′-RACE and sequence analysis confirmed the predicted stop codon. To confirm the start codon we expressed the predicted mcnC open reading frame (ORF) fused to GFP using the alcA-inducible promoter (49) and followed the molecular mass of the induced protein by Western blot analysis. Similar to the endogenously tagged protein, ectopically expressed MCNC-GFP ran at the higher-than-expected molecular mass (Fig. 3B). These data indicate that both the start and stop codons are defined correctly and that the anomalous migration of MCNC is either due to its intrinsic biophysical properties or posttranslational modifications. Interestingly, similar slow mobility during SDS-PAGE has been observed for S. cerevisiae Def1 (50).
FIG. 3.

MCNC migrates during SDS-PAGE as a larger protein than predicted. (A) Protein from an endogenously tagged MCNC-GFP strain was analyzed by Western blot analysis using anti-GFP antibodies. MCNC-GFP runs at approximately 200 kDa, while the predicted size for MCNC-GFP is 117 kDa. (B) Protein from a strain expressing tagged MCNC-GFP induced for 0, 1, and 2 h off the alcA promoter was analyzed by Western blot analysis using anti-GFP antibody. alcA::MCNC-GFP runs at the same size as endogenously tagged MCNC-GFP, indicating that the size discrepancies between the predicted and actual mobilities through SDS-PAGE are not due to incorrect gene annotation. For both blots additional lower-molecular-mass bands are present which are assumed to be proteolytic breakdown products.
We subjected S-tagged-purified MCNC to in vitro phosphatase reactions and detected slight band shifts consistent with MCNC being a phosphoprotein (data not shown). However, phosphatase-treated MCNC still migrated at a significantly higher molecular mass than predicted. Phosphorylation of MCNC does not therefore explain its aberrant mobility during SDS-PAGE. Preliminary mass spectroscopy analysis identified one potential site of MCNC serine phosphorylation within the sequence 434-AQPPQHSPVAPR-445 (data not shown).
Deletion of mcnC causes delayed growth polarization and exacerbates the temperature sensitivity caused by nimA1 or nimA7.
To generate a ΔmcnC null allele the coding region of mcnC was replaced using the pyroAAF nutritional marker. Deleted strains were tested for heterokaryon formation to see if mcnC is essential (34). This analysis revealed mcnC to be nonessential (data not shown). However, when compared to a control wild-type strain, ΔmcnC strains produced colonies at 32°C that were half the size of the control (Fig. 4A). This effect was more pronounced when the strains were grown at 42°C, suggesting the ΔmcnC allele causes slight temperature sensitivity (Fig. 4A).
FIG. 4.

Deletion of mcnC causes a small colony phenotype and polarization defects. (A) The colony size of an ΔmcnC strain, along with a wild-type and nimA1 strain, at both 32°C and 42°C, is shown, demonstrating the ΔmcnC small colony size defect. (B) Bright field and DAPI images of ΔmcnC and wild-type strains grown for 8 h at 42°C. Panels a and b show wild-type (c) and (d) ΔmcnC cells. Bar, ∼5 μm. (C) The relationship between number of nuclei per cell and polarization of growth is graphed for wild-type and ΔmcnC spores grown as for panel B. In each case the number of cells, with the indicated number of nuclei, is characterized further as either nonpolarized or polarized. (D) Colony growth phenotypes of strains with the indicated mutations after 2 days growth at 37°C are shown.
Wild-type and ΔmcnC cells were grown at 42°C, and their nuclei were stained with DAPI to define potential effects of lack of mcnC on mitotic progression (Fig. 4B). As mcnC is a copy number suppressor of nimA1, it would be predicted to play a positive role to promote mitosis. Thus, in the ΔmcnC cells we had expected that progression into mitosis might be defective, leading to fewer nuclei per cell when compared to the wild type. However, this was not observed. In fact the most noticeable defect observed in ΔmcnC cells was their inability to establish polarized growth in a normal manner. Typically, when spores break dormancy and germinate they initiate biosynthetic activities and undergo a process of swelling and limited isotropic growth. After passage through the first mitosis, isotropic growth ceases and a single site of continued growth is established, producing highly polarized germlings, as seen for the wild-type cell in Fig. 4B, panels a and b. In ΔmcnC cells the transition from isotropic growth to polarized growth is defective, leading to larger and rounder cells forming before polarized growth occurs (Fig. 4B, panels c and d). With continued incubation ΔmcnC cells do establish polarized growth but form smaller colonies than the wild type (Fig. 4A).
To quantify the defects in polarized growth, spores of wild-type and ΔmcnC strains were grown for 9 hours, at which point the majority of wild-type cells had initiated polarized growth (Fig. 4C). Cells were fixed and stained with DAPI, and the number of nuclei per cell and establishment of polarization were assessed. All cells that had undergone at least one mitosis in the wild-type strain had established polarized growth, whereas over half the ΔmcnC cells that had completed mitosis failed to initiate polarized growth (Fig. 4C). In fact over 40% of ΔmcnC cells that had completed two mitotic divisions had not yet initiated polarized growth. Notably, the defect in polarized growth was not due to defects in transition through the cell cycle, as the average number of nuclei per cell was virtually identical between the two strains. Thus, ΔmcnC spores appear to have a defect in the transition from isotropic to polarized growth, which is independent of nuclear division.
As MCNC is predicted to play a positive role in NIMA function, we anticipated that the growth defects caused by temperature-sensitive loss-of-function nimA alleles might be exacerbated in combination with ΔmcnC. To test this idea, double nimA1 ΔmcnC and nimA7 ΔmcnC mutant strains were generated and tested for temperature sensitivity. It is apparent the temperature sensitivity of both nimA1 (Fig. 4D) and nimA7 (data not shown) is increased when mcnC function is missing. These findings are consistent with MCNC playing a positive role in the function of NIMA.
MCNB is a nuclear forkhead transcription factor implicated in nimA expression.
The mcnB gene (AN8858) contains a single exon encoding a protein of 718 aa with a predicted molecular mass of 78.7 kDa. MCNB contains a conserved forkhead domain, also known as a winged helix domain, found in several transcription factors, including some involved in cell cycle-specific gene expression, such as S. pombe Sep1 (57) (see Discussion, below). MCNB is highly conserved within the eight sequenced aspergilli. Similar proteins, with sequence homology throughout, can be detected in other filamentous ascomycetes but, outside this group of organisms, high similarity is restricted to the forkhead domain (aa 184 to 317) and can be detected from yeasts to vertebrates, including humans. Deletion of mcnB did not cause any detectable growth defects (data not shown).
To determine if MCNB displayed any cell cycle-specific changes in abundance and/or subcellular localization, it was endogenously tagged with GFP and viewed using confocal microscopy. MCNB-GFP was observed in asynchronous cell cultures and found to locate primarily within nuclei. Notably, however, there was considerable variation in the level of MCNB-GFP between nuclei of different cells, and 30 to 40% of nuclei appeared largely negative for MCNB-GFP. The variable amount of MCNB-GFP within nuclei suggests it is likely regulated through the cell cycle. To follow this possibility a strain was developed containing both MCNB-GFP and histone H1-red fluorescent protein (RFP), enabling us to follow the mitotic cycle and distribution of MCNB (Fig. 5). The level of MCNB-GFP within nuclei increased through the cell cycle, reaching a maximum level in late G2. Upon entry into mitosis, as defined by chromatin condensation, MCNB-GFP signal was lost from nuclei (Fig. 5B, 1′ 25″). Upon completion of DNA segregation and entry into G1, MCNB-GFP did not immediately reaccumulate in nuclei but did accumulate later in the cell cycle. Thus, the nuclear accumulation of MCNB is under cell cycle regulation such that it reaches a maximum in G2 and a minimum during mitosis. This dynamic accumulation might reflect a role for MCNB in cell cycle-specific expression of NIMA and/or other proteins required for entry into mitosis.
FIG. 5.

The nuclear levels of MCNB varies through the cell cycle. (A) Spores germinated to the 1-2 nuclear stage endogenously expressing MCNB-GFP. MCNB-GFP shows a nuclear location that varies between cells. (B) A time series from live cell confocal imaging of a strain expressing endogenous histone H1-RFP and MCNB-GFP. At mitosis chromatin condenses (1′25”) and MCNB-GFP leaves nuclei and does not immediately return to nuclei after completion of mitosis. Bar, ∼5 μm.
MCNA defines a novel nuclear body with a unique pattern of postmitotic segregation.
The mcnA gene (AN2692) contains two exons encoding a protein of 506 aa with a predicted molecular mass of 56.6 kDa. Both the gene structure and protein sequence of mcnA are conserved in all eight of the currently sequenced aspergilli. Highly related proteins are also encoded in the genomes of Coccidioides immitis RS and Histoplasma capsulatum (sequences available at NCBI), and MCNA-like proteins are present in two other Histoplasma strains (G217A and G186A) as well as Blastomyces dermatitidis (C. Rappleye, personal communication) and Penicillium marneffei (A. Andrianopoulos, personal communication). All three isolates of Paracoccidioides brasiliensis similarly contain a protein with high similarity to MCNC (http://www.broad.mit.edu/annotation/genome/paracoccidioides_brasiliensis/MultiHome.html). Thus, the aspergilli and the systemic dimorphic fungi (the Eurotiomycetes) all encode highly related MCNA proteins, whereas other sequenced fungi do not. Deletion of mcnA did not cause detectable growth defects (data not shown).
To determine if MCNA displayed cell cycle-specific changes in subcellular localization it was endogenously tagged with GFP, and the tagged strains were viewed after staining DNA with DAPI to reveal the location of nuclei. We initially observed MCNA-GFP in conidia, and each spore contained a single distinct focus of MCNA-GFP fluorescence (Fig. 6A). In germinated spores MCNA-GFP was also found to locate within nuclei, primarily as single discrete foci (Fig. 6B to E), which we have termed MCNA bodies.
FIG. 6.

MCNA locates to a subnuclear body, the MCNB body. (A) Spores from a strain endogenously expressing MCNA-GFP contain single foci of MCNA. The upper panel is with differential interference contrast and the lower panel is with GFP. (B) Nuclear location of the MCNA body in relationship to the nucleolus. A single nucleus is shown that expressed MCNA-GFP, DAPI-stained nucleus, and ANA1-stained nucleolus. (C) A two-nucleus germling, showing MCNA-GFP within DAPI-stained nuclei. (D) The location of MCNA-GFP (green) in relationship to the nucleolus (red), defined by CGRA, fibrillarin, and Bop1 endogenously tagged with red fluorescent protein. (E) The endoplasmic reticulum, which is contiguous with the nuclear envelope, visualized using red fluorescent protein-tagged Erg24 covisualized with MCNA-GFP, indicating MCNA bodies locate near the nuclear periphery. Bars, ∼5 μm (A and C to E) and ∼3 μm (B).
During observation of many nuclei it appeared that nuclear MCNA bodies locate more toward the nuclear periphery than the nuclear interior. To investigate this possibility more directly, a strain was developed in which the NE could be visualized using Erg24-chRED (a marker for the endoplasmic reticulum which is contiguous with the outer NE) and MCNA bodies using MCNA-GFP. Confocal imaging of this strain revealed that MCNA bodies do locate near the periphery of nuclei in the vicinity of the NE (Fig. 6E).
In addition to locating near the nuclear periphery, MCNA bodies locate near the DAPI shadow representing the nucleolus (Fig. 6C). To test for a potential association of MCNA with the nucleolus, the relationship between MCNA-GFP and the nucleolar antigens detected by the ANA1 antibody was defined. ANA1 is an autoimmune antibody that recognizes the human fibrillarin nucleolar protein and can be used as a marker for the nucleolus during immunofluorescence (23). These studies (Fig. 6B) indicate that MCNA bodies are associated with nucleoli. However, they are not located within the body of the nucleolus, as defined by ANA1 staining; rather, the MCNA bodies locate next to the nucleolus at the nuclear periphery (Fig. 6B). To further define the relationship between the nucleolus and MCNA bodies, we endogenously tagged CgrA, an orthologue of the S. cerevisiae nucleolar protein Cgr1p as defined for A. fumigatus (5, 6), as well as predicted orthologues of the fibrillarin and Bop1 nucleolar proteins. The relationship between MCNA bodies and these nucleolar proteins were the same as defined using the ANA1 antibody (Fig. 6D), indicating the MCNA body locates adjacent to the nucleolus but near the NE throughout much of the cell cycle. However, within an asynchronous growing population of cells, MCNA bodies were not exclusively located within nuclei and varied in size and number, suggestive of cell cycle regulation.
During live cell imaging, MCNA-GFP displayed a dynamic and unique pattern of segregation (Fig. 7A). Individual MCNA bodies diminished in intensity as two new MCNA foci appeared and increased in intensity. At the end of the process two new nuclear MCNA bodies were produced. Thus, the pattern, in general, for generation of new MCNA bodies was one going to three, which then resolved to two bodies. However, it was not uncommon to detect additional MCNA foci (Fig. 7B). This indicates that fragmentation of the parental bodies might occur or that new transient bodies can be generated. The process of generating two new MCNA bodies was quite variable but normally took between ∼30 and 40 min to complete.
FIG. 7.

Mitotic segregation of MCNA bodies. (A) Endogenously GFP-tagged MCNA in a two-nucleus germling is shown during its segregation. Note that three MCNA foci appear (25′) as an intermediary to segregation of MCNA-GFP equally to daughter nuclei and that additional foci also appear (seven foci at 35′, for example). (B) Segregation of MCNA bodies (arrowhead) occurs considerably after DNA segregation and occurs via a cytoplasmic intermediate (14′). Bars, ∼5 μm.
To relate the process of MCNA body segregation with mitosis, MCNA-GFP was observed with monomeric RFP-tagged histone H1. During DNA condensation, a single MCNA body remains associated with the nucleus (Fig. 7B, 0′). After anaphase the MCNA body is excluded from the nucleus and appears in the cytoplasm (Fig. 7B). MCNA protein then begins to accumulate in daughter nuclei in the vicinity of DNA (Fig. 7B, 14′). The nuclear signal increases and concentrates to the typical location of interphase MCNA bodies (Fig. 7B, 24′). The single cytoplasmic MCNA bodies sometimes appear to fragment, forming variable numbers of MCNA cytoplasmic foci (Fig. 7B, 24′). It is only after ∼35 min of the first appearance in daughter nuclei that MCNA bodies are no longer apparent in the cytoplasm. Therefore, MCNA is nuclear during G2. It is expelled to the cytoplasm during mitosis and locates in both the cytoplasm and nuclei during the early stages of G1. As the cell cycle progresses MCNA becomes exclusively nuclear.
We have recently shown that mitotic nucleolar segregation also involves a cytoplasmic intermediate (L. Ukil et al., submitted for publication). The nucleolus is expelled from nuclei during anaphase-telophase via a double pinch of the NE. The double pinch forms two daughter nuclei and a central nuclear remnant that contains nucleolar proteins, but not the nucleolar organizing regions which are segregated with DNA to daughter nuclei. The nucleolus undergoes stepwise disassembly, and released nucleolar proteins are transported into daughter nuclei in which new nucleoli then reform (Ukil et al., submitted). Although there are similarities between the pattern of segregation of MCNA bodies and nucleoli, it does not appear they undergo identical modes, or timing, of mitotic segregation. To directly address these differences, we visualized the localization of MCNA in comparison with the nucleolar protein fibrillarin (Fig. 8). At 4 min in this time course three Fib-chRFP structures are evident. The larger central structure is the parental nucleolus, while the smaller structures represent the reforming daughter nucleoli. At this time MCNA is in a distinct structure (Fig. 8, 4 min). This series was collected at 1-min intervals, and the full time course is shown in Movie S3 in the supplemental material. Segregation of Fib-RFP is complete at 6 min (see Movie S3 in the supplemental material). At this time the MCNA body is still intact and in the cytoplasm. A further 9 min passes before MCNA starts to undergo disassembly and appear in daughter nuclei. Its segregation then takes another 16 min to complete (Fig. 8; see also Movie S3 in the supplemental material). Thus, although the segregation of nucleolar proteins and MCNA follows a similar pattern, segregation of MCNA initiates at a later time during interphase and takes considerably longer to complete its segregation to daughter nuclei. Additional differences include the fragmentation of MCNA bodies in the cytoplasm and the extensive mobility of parental MCNA bodies throughout the cytoplasm (data not shown). Neither of these characteristics has been observed for the cytoplasmic nucleolus.
FIG. 8.

Segregation of MCNA bodies occurs through a similar mechanism to nucleolar segregation but is completed later in the cell cycle. Nuclear division of the nucleolus following Fib-RFP and MCNA bodies, shown using MCNA-GFP. At 4 min Fib-RFP is at a mid-stage of segregation, displaying a central cytoplasmic parental nucleolus flanked on either side by newly forming daughter nucleoli. By 8 min the nucleolus has completed its segregation to daughter nuclei but the MCNA body remains intact within the cytoplasm. At 16 min the parental MCNA body begins to diminish as new MCNA bodies appear in daughter nuclei next to the nucleoli. As the cell cycle continues the parental MCNA body disappears and new MCNA bodies are formed in daughter nuclei. Bar, ∼5 μm.
MCNA is a phosphoprotein, and its unique pattern of segregation is regulated by proline-directed phosphorylation.
Single-step affinity purification, using endogenously S-tagged MCNA, was used to purify MCNA to homogeneity. During SDS-PAGE, purified MCNA-S-tag proteins ran as several bands (Fig. 9A), which were found to be different species of MCNA protein based upon parallel Western blot analysis using anti-S-peptide antibodies (data not shown). A similar pattern of MCNA was also detected when Western blot analysis was completed on protein from MCNA-GFP strains using anti-GFP antibodies (Fig. 9B). Therefore, MCNA exists as several molecular weight species and does not apparently copurify with significant amounts of interacting partners.
FIG. 9.

MCNA phosphorylation is involved in its equal segregation. (A) Endogenously S-tagged MCNA was affinity purified and treated with λ-phosphatase with and without phosphatase inhibitors (1 mM Na-vanadate plus 50 mM Na-fluoride) and separated by SDS-PAGE. The gel was stained with silver. (B) MCNA-GFP and mutant MCNA-3A-GFP, with three phosphorylation sites mutated to alanine, were analyzed by Western blotting using GFP antibody. Note the appearance of two bands for MCNA-3A-GFP, indicating both that the mobility shifts seen for MCNA-GFP requires the three S sites mutated and that at least one additional site of phosphorylation exists. (C) MCNA-3A-GFP visualized in asynchronously grown cells fixed with DNA stained by DAPI. Note the unequal amounts of MCNA-3A-GFP in individual nuclei compared to wild-type MCNA-GFP cells (Fig. 6 to 8). Bar, ∼5 μm.
To see if the molecular weight shifts observed for MCNA were caused by phosphorylation, in vitro phosphatase reactions were completed on purified MCNA-S-tag. Dephosphorylation of MCNA caused proteins to collapse to a single molecular weight species (Fig. 9A). Addition of phosphatase inhibitors prevented the molecular weight collapse. These results, in combination with mass spectroscopy analysis as described below, confirm that the different protein bands purified are all MCNA and that MCNA is a phosphoprotein, likely phosphorylated at multiple sites.
To identify the phosphorylation sites within MCNA, purified protein was analyzed using capillary liquid chromatography-nanospray tandem mass spectrometry. During this analysis 56% sequence coverage was obtained and three sites of phosphorylation detected. Modification by phosphorylation was detected at serine 330, serine 365, and serine 442. All three phosphorylated sites conform to a minimal proline-directed kinase consensus “SP” site suggestive of potential regulation of MCNA by CDK1-mediated phosphorylation.
To test the functional significance of the three phosphorylation sites, a gene replacement construct was generated in which all three sites were mutated to encode nonphosphorylatable alanine. This construct was introduced into a strain in which the coding region of mcnA was replaced using the pyroA nutritional marker. After homologous integration of the replacement construct, an endogenous GFP-tagged version of MCNA was generated as the sole copy of MNCA and in which the three serine phosphorylation sites were converted to alanine residues (MCNA-3A-GFP). In this manner we were able to define the contribution of these sites to the phosphorylation and distribution pattern of MCNA.
Western blot analysis was performed to check if mutating the three serine sites resulted in the abolishment of the mobility shifts caused by phosphorylation of MCNA. As can be seen (Fig. 9B) MCNA-3A did not run as several species but instead migrated as two bands. This indicates that the sites mutated are involved in the phosphorylation of MCNA and that there is at least one other unmapped phosphorylation site.
Live cell imaging of MCNA-3A-GFP was completed to determine if lack of phosphorylation caused by the 3A mutation had any effect on the location or segregation pattern of MCNA bodies. The most noticeable defect defined was the unequal segregation of mutant MCNA-3A-GFP (Fig. 9C). Wild-type MCNA-GFP is distributed to each nucleus in an even manner such that all nuclei receive a relatively equal amount of MNCA at the end of the segregation process (Fig. 6, 7, and 8). However, the amount of MCNA-3A-GFP per nucleus was variable (Fig. 9C) when compared to the equal distribution of the wild-type protein. The unequal segregation of MCNA-3A to daughter nuclei suggests that phosphorylation of MCNA plays an important role in its segregation during the cell cycle.
DISCUSSION
Three new proteins, MCNA, MCNB, and MCNC, have been identified by using a copy number suppression screen of the mitosis-specific NIMA protein kinase temperature-sensitive nimA1 allele. Increased expression of each suppressor gene, off of a multi-copy-number plasmid, was able to suppress the temperature sensitivity of the nimA1 allele but not the nimA5 allele. The nimA5 mutation is in the protein kinase domain and causes loss of kinase activity at the restrictive temperature. The suppressors are thus unable to bypass NIMA kinase function, but they are able to suppress the temperature sensitivity of nimA1, a mutation in the C-terminal regulatory domain of NIMA.
MCNC locates to the cytoplasm and is required for normal growth.
Expression of MCNC did not overtly affect the level of NIMA1 protein, suggesting that it causes suppression of nimA1 by a mechanism not involving increased NIMA1 expression. This function potentially involves ubiquitination, as MCNC contains a sequence motif similar to the CUE domain (45) which in other proteins is known to physically interact with monoubiquitinated proteins. The closest related protein to MCNC in S. cerevisiae is Def1 (50). Similarity is restricted to the CUE-like domain, but MCNC and Def1 also have similar biophysical properties, as both migrate during SDS-PAGE as far-larger proteins than their predicted sizes. In addition, deletion of either causes a small colony growth phenotype (8, 50). Def1 was isolated as a chromatin-associated binding partner of Rad26 and shown to be involved in the degradation of RNA polymerase II in response to DNA damage (43, 50). This might not be the sole function of Def1, as it was also isolated in association with the Rrm3 DNA helicase and shown to have a role in telomere length maintenance (8). Whether MCNC and Def1 are functionally related proteins remains to be determined. MCNC locates in the cytoplasm during interphase and enters nuclei during mitosis. Upon completion of mitosis MCNC no longer located within nuclei. This cell cycle-specific distribution is similar to that described for other cytoplasmic proteins, such as RanGAP (11), and is a result of the opening of NPCs at mitosis, which allows diffusion of cytoplasmic proteins into nuclei at this stage of the cell cycle (11, 13, 31). Deletion of mcnC enhanced the temperature sensitivity of the nimA1 and nimA7 alleles, consistent with MCNC playing a positive role in NIMA function. However, somewhat surprisingly, deletion of mcnC did not appear to affect cell cycle progression but instead caused a delay in the ability of germinating cells to establish polarized growth. In this regard it is interesting that a minor proportion of NIMA-GFP has been observed in the vicinity of the growing tip of germlings, and we have recently detected NIMA-GFP at the sites of septum formation (C. DeSouza, K.-F. Shen, and S. Osmani, unpublished data). Thus, NIMA may play roles to link cell morphogenesis with cell cycle progression in ways that could involve MCNC.
MCNB encodes an S. pombe Sep1-related forkhead transcription factor driving expression of nimA.
We have previously shown that the nimA1 allele, unlike nimA5 or nimA7, retains some kinase activity at the restrictive temperature (51). We therefore considered the possibility that the copy number suppressors might function to increase the expression of NIMA1. This mode of suppression was found to be a potential for MCNB and MCNA, because their increased expression caused a marked increase in the level of NIMA1 protein.
Insight into the possible function of MCNB was revealed by its strong sequence homology to forkhead transcription factors, which are known to be involved in cell cycle-specific gene expression. In fact, the S. pombe orthologue of MCNB is Sep1, which is involved in the cell cycle-specific transcription of Fin1, the S. pombe NIMA orthologue (18, 22), as well as other genes whose expression is highest in the M and G1 phases of the cell cycle (7, 57). Sep1 plays a positive role in the expression of these genes (39). We therefore suggest that MCNB is a cell cycle-specific transcription factor that positively influences nimA expression. This function would explain why increased expression of MCNB causes increased protein levels of NIMA1. The cell cycle distribution of MCNB fits well with this proposed function. For instance, there is a correlation between the nuclear accumulation of MCNB and the expression of NIMA, with both being maximal in late G2 and minimal in the early stages of interphase. Similarly, like S. pombe Sep1, MCNB is not an essential protein (44). However, there is one notable difference between the biologies of Sep1 and MCNB. The levels of nuclear MCNB are regulated through the cell cycle such that its nuclear levels are maximal in G2, but MCNB is not concentrated in nuclei during mitosis. On the other hand, Sep1 has been reported to constitutively locate to nuclei at a constant level, including during mitosis (57). The significance of this fundamental difference is at present unclear. Perhaps part of this difference is related to the fact that the partially disassembled mitotic NPCs of A. nidulans allow free diffusion in and out of nuclei, whereas NPCs in S. pombe remain intact during mitosis. The opening of the mitotic NPCs in A. nidulans likely has marked negative effects on transcription (see below). Interestingly, however, in S. pombe there is a cluster of genes, including Fin1, Cdc15, Spo12, Cdc19, Mid1, Sid2, Ppb1, and Plo1, which are transcriptionally activated during mitosis and into G1 (3). Several of these proteins are involved in septation, and septa begin to form during mitosis in S. pombe. It is thus logical for genes involved in septation to be activated during mitosis. In contrast, in A. nidulans septa are not always formed after each mitotic division and, when they are formed, the process takes place considerably after completion of mitosis, not in mitosis or early G1 (24). A. nidulans does not therefore need a mitotic wave of transcription to promote septation as S. pombe does. Consistent with this, we have recently found that the largest subunit of RNA polymerase II is released from nuclei during A. nidulans mitosis (S. Son and S. Osmani, unpublished data). This likely has the effect of stopping polymerase II-dependent transcription during A. nidulans mitosis, as occurs during mitosis in higher eukaryotes. These two reasons, lack of A. nidulans mitotic transcription and no need for septation gene expression during mitosis or early G1, may explain why Sep1 and MCNB have different nuclear accumulation profiles. Future experiments will define if MCNB is a direct regulator of nimA or other G2-regulated cell cycle-specific genes.
MCNA defines a new subnuclear body.
The other nimA1 suppressor that was able to promote increased levels of NIMA1 was MCNA. The sequence of MCNA does not provide insight into its function because no conserved domains are present and orthologues have not been studied. However, MCNA is a remarkable protein for several reasons. For example, MCNA has a highly specific distribution during evolution and is conserved in a very restricted group of filamentous ascomycetes, including the aspergilli and members of the pathogenic dimorphic filamentous fungi, such as Coccidioides and Histoplasma (within the Eurotiomycetes) (15). Orthologues within these fungi have high similarity to MCNA (at least 72% identity and 82% similarity within the aspergilli, 53% identity and 71% similarity in Histoplasma species, and 47% identity and 66% similarity in the Coccidioides), but no similar proteins could be detected in the genomes of other sequenced filamentous fungi, including other members of the ascomycetes. The high degree of conservation of MCNA within the Eurotiomycetes therefore suggests it might play a role specific to this group of organisms.
Another intriguing result regarding MCNA concerns its subcellular distribution, as it defines a novel subnuclear body we have termed the MCNA body. The MCNA body has a very specific location within nuclei, residing next to the NE near the nucleolus. However, this is not a fixed position, because during mitosis the MCNA body is expelled from the nucleus into the cytoplasm. Recent work has shown that this behavior is shared between the MCNA body and the nucleolus (Ukil et al., submitted). Mitotic nucleolar expulsion occurs through a process involving a double pinch of the NE around the nucleolus during telophase. This generates two daughter nuclei and a central structure containing the nucleolus, which undergoes disassembly in the cytoplasm during exit from mitosis. The nucleolar proteins released from the nucleolus are imported into daughter nuclei in early G1 to reform nucleoli. We propose the MCNA body is similarly expelled from the nucleus via the double NE pinch mechanism. It then also disappears from the cytoplasm, and we suggest the MCNA protein is disassembled from the cytoplasmic foci and reimported to daughter nuclei in the same way nucleolar proteins are. However, MCNA does not accumulate in daughter nuclei until much later in the cell cycle, considerably after daughter nucleoli have been formed. MCNA does appear in all nuclei at a later stage of interphase such that during G2 each nucleus again contains a single MCNA body of similar size with no cytoplasmic signal apparent. Therefore, although MCNA bodies locate adjacent to nucleoli and they are both expelled from the nucleus during mitosis, they reappear in nuclei at different stages of the cell cycle. This suggests that the early stages of their segregation are mechanistically similar and regulated in a similar fashion but that their reassembly in daughter nuclei is regulated differently.
Insight into the regulation of MCNA segregation was uncovered by analysis of MCNA phosphorylation sites identified using mass spectroscopy. This analysis mapped three SP sites of phosphorylation which were mutated to alanine and found to play a role in the phosphorylation of MCNA and its equal segregation to daughter nuclei. Although there is likely one more MCNA site of phosphorylation, the data implicate Cdk1-cyclin B, or another proline-directed kinase, in the regulation of equal mitotic segregation of MCNA.
Although fascinating, these data do not readily explain how ectopic expression of MCNA causes increased accumulation of NIMA1, nor do they shed obvious light on the function of MCNA or the MCNA body. Because MCNA locates to the cytoplasm during the early stages of the cell cycle, but to the nucleus during later periods of the cell cycle, this suggests its function is regulated by being sequestered alternatively between these two subcellular compartments. To our knowledge, the only subnuclear body that behaves somewhat similarly to MCNA bodies is the TAM body of S. cerevisiae (16). During interphase, components of the TAM body locate to the nucleolus but, during mitosis, also locate to the cytoplasm in a single discrete spot located in the daughter cell. Thus, components of the TAM body behave somewhat similarly to MCNA bodies. TAM bodies have been proposed to function in the cytoplasmic degradation of cell cycle-specific mRNAs, such as the cyclin CLB2, which is known to be degraded in daughter nuclei during exit from mitosis (17). Our current data do not support any direct connection between TAM bodies and MCNA bodies. However, given the fact that nimA mRNA levels are under cell cycle-specific control (35), it is intriguing that a subnuclear body that behaves somewhat similarly to MCNA bodies plays a role in degradation of cell cycle-specific mRNAs. One possible mechanism of nimA1 suppression we are considering is that extra MCNA could cause defects in the turnover of nimA mRNA, allowing accumulation of NIMA1 protein and suppression of nimA1.
Clearly, more experimentation is required. Equally clear is the fascinating cell biology of the MCNA body and the possibility that this newly discovered nuclear structure might exist uniquely in a very important group of fungi which includes members we exploit in the food and biotechnology industries and other members that colonize us and are thus of significant medical importance.
Supplementary Material
Acknowledgments
We thank Sunghun Son and all members of the Osmani lab for their support and input to this work, particularly Colin De Souza for his insights and comments on the manuscript. We thank Greg May for AMA1 libraries. Thanks are also given to Chad Rappleye and Alex Andrianopoulos for their thoughts on the highly specific conservation of MCNA.
The work was supported by a grant from the NIH (GM042564) to S.A.O.
Footnotes
Published ahead of print on 17 October 2008.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Aleksenko, A., and A. J. Clutterbuck. 1996. The plasmid replicator AMA1 in Aspergillus nidulans is an inverted duplication of a low-copy-number dispersed genomic repeat. Mol. Microbiol. 19565-574. [DOI] [PubMed] [Google Scholar]
- 2.Aleksenko, A., and A. J. Clutterbuck. 1997. Autonomous plasmid replication in Aspergillus nidulans: AMA1 and MATE elements. Fungal Genet. Biol. 21373-387. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, M., S. S. Ng, V. Marchesi, F. H. MacIver, F. E. Stevens, T. Riddell, D. M. Glover, I. M. Hagan, and C. J. McInerny. 2002. Plo1+ regulates gene transcription at the M-G1 interval during the fission yeast mitotic cell cycle. EMBO J. 215745-5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergen, L. G., A. Upshall, and N. R. Morris. 1984. S-phase, G2, and nuclear division mutants of Aspergillus nidulans. J. Bacteriol. 159114-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhabhra, R., M. D. Miley, E. Mylonakis, D. Boettner, J. Fortwendel, J. C. Panepinto, M. Postow, J. C. Rhodes, and D. S. Askew. 2004. Disruption of the Aspergillus fumigatus gene encoding nucleolar protein CgrA impairs thermotolerant growth and reduces virulence. Infect. Immun. 724731-4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhabhra, R., W. Zhao, J. C. Rhodes, and D. S. Askew. 2006. Nucleolar localization of Aspergillus fumigatus CgrA is temperature-dependent. Fungal Genet. Biol. 431-7. [DOI] [PubMed] [Google Scholar]
- 7.Buck, V., S. S. Ng, A. B. Ruiz-Garcia, K. Papadopoulou, S. Bhatti, J. M. Samuel, M. Anderson, J. B. Millar, and C. J. McInerny. 2004. Fkh2p and Sep1p regulate mitotic gene transcription in fission yeast. J. Cell Sci. 1175623-5632. [DOI] [PubMed] [Google Scholar]
- 8.Chen, Y. B., C. P. Yang, R. X. Li, R. Zeng, and J. Q. Zhou. 2005. Def1p is involved in telomere maintenance in budding yeast. J. Biol. Chem. 28024784-24791. [DOI] [PubMed] [Google Scholar]
- 9.Davies, J. R., A. H. Osmani, C. P. De Souza, C. Bachewich, and S. A. Osmani. 2004. Potential link between the NIMA mitotic kinase and nuclear membrane fission during mitotic exit in Aspergillus nidulans. Eukaryot. Cell 31433-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Souza, C. P., K. P. Horn, K. Masker, and S. A. Osmani. 2003. The SONB(NUP98) nucleoporin interacts with the NIMA kinase in Aspergillus nidulans. Genetics 1651071-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Souza, C. P., A. H. Osmani, S. B. Hashmi, and S. A. Osmani. 2004. Partial nuclear pore complex disassembly during closed mitosis in Aspergillus nidulans. Curr. Biol. 141973-1984. [DOI] [PubMed] [Google Scholar]
- 12.De Souza, C. P., A. H. Osmani, L. P. Wu, J. L. Spotts, and S. A. Osmani. 2000. Mitotic histone H3 phosphorylation by the NIMA kinase in Aspergillus nidulans. Cell 102293-302. [DOI] [PubMed] [Google Scholar]
- 13.De Souza, C. P., and S. A. Osmani. 2007. Mitosis, not just open or closed. Eukaryot. Cell 61521-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fry, A. M., and E. A. Nigg. 1995. The NIMA kinase joins forces with Cdc2. Curr. Biol. 51122-1125. [DOI] [PubMed] [Google Scholar]
- 15.Geiser, D. M., C. Gueidan, J. Miadlikowska, F. Lutzoni, F. Kauff, V. Hofstetter, E. Fraker, C. L. Schoch, L. Tibell, W. A. Untereiner, and A. Aptroot. 2006. Eurotiomycetes: Eurotiomycetidae and Chaetothyriomycetidae. Mycologia 981053-1064. [DOI] [PubMed] [Google Scholar]
- 16.Gill, T., J. Aulds, and M. E. Schmitt. 2006. A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae. J. Cell Biol. 17335-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gill, T., T. Cai, J. Aulds, S. Wierzbicki, and M. E. Schmitt. 2004. RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: novel method of mRNA degradation. Mol. Cell. Biol. 24945-953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grallert, A., and I. M. Hagan. 2002. Schizosaccharomyces pombe NIMA-related kinase, Fin1, regulates spindle formation and an affinity of Polo for the SPB. EMBO J. 213096-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grallert, A., A. Krapp, S. Bagley, V. Simanis, and I. M. Hagan. 2004. Recruitment of NIMA kinase shows that maturation of the S. pombe spindle-pole body occurs over consecutive cell cycles and reveals a role for NIMA in modulating SIN activity. Genes Dev. 181007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hicke, L., H. L. Schubert, and C. P. Hill. 2005. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 6610-621. [DOI] [PubMed] [Google Scholar]
- 21.Kim, J. S., and R. T. Raines. 1993. Ribonuclease S-peptide as a carrier in fusion proteins. Protein Sci. 2348-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krien, M. J., R. R. West, U. P. John, K. Koniaras, J. R. McIntosh, and M. J. O'Connell. 2002. The fission yeast NIMA kinase Fin1p is required for spindle function and nuclear envelope integrity. EMBO J. 211713-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhn, G., M. Jarzabek-Chorzelska, M. Blaszczyk, T. P. Chorzelski, and S. Jablonska. 1990. The fibrillarin (Scl-34) autoantibody in systemic scleroderma. Dermatol. Monatsschr. 17619-26. (In German.) [PubMed] [Google Scholar]
- 24.Momany, M., and I. Taylor. 2000. Landmarks in the early duplication cycles of Aspergillus fumigatus and Aspergillus nidulans: polarity, germ tube emergence and septation. Microbiology 1463279-3284. [DOI] [PubMed] [Google Scholar]
- 25.Morris, N. R. 1976. Mitotic mutants of Aspergillus nidulans. Genet. Res. (Cambridge) 26237-254. [DOI] [PubMed] [Google Scholar]
- 26.Nayak, T., E. Szewczyk, C. E. Oakley, A. Osmani, L. Ukil, S. L. Murray, M. J. Hynes, S. A. Osmani, and B. R. Oakley. 2006. A versatile and efficient gene targeting system for Aspergillus nidulans. Genetics 1721557-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oakley, B. R., and N. R. Morris. 1983. A mutation in Aspergillus nidulans that blocks the transition from interphase to prophase. J. Cell Biol. 961155-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Connell, M. J., M. J. Krien, and T. Hunter. 2003. Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol. 13221-228. [DOI] [PubMed] [Google Scholar]
- 29.O'Regan, L., J. Blot, and A. M. Fry. 2007. Mitotic regulation by NIMA-related kinases. Cell Div. 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osherov, N., and G. May. 2000. Conidial germination in Aspergillus nidulans requires RAS signaling and protein synthesis. Genetics 155647-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osmani, A. H., J. Davies, H. L. Liu, and S. A. Osmani. 2006. Systematic deletion and mitotic localization of the nuclear pore complex proteins of Aspergillus nidulans. Mol. Biol. Cell 174946-4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osmani, A. H., J. Davies, C. E. Oakley, B. R. Oakley, and S. A. Osmani. 2003. TINA interacts with the NIMA kinase in Aspergillus nidulans and negatively regulates astral microtubules during metaphase arrest. Mol. Biol. Cell 143169-3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osmani, A. H., S. L. McGuire, and S. A. Osmani. 1991. Parallel activation of the NIMA and p34cdc2 cell cycle-regulated protein kinases is required to initiate mitosis in A. nidulans. Cell 67283-291. [DOI] [PubMed] [Google Scholar]
- 34.Osmani, A. H., B. R. Oakley, and S. A. Osmani. 2006. Identification and analysis of essential Aspergillus nidulans genes using the heterokaryon rescue technique. Nat. Protoc. 12517-2526. [DOI] [PubMed] [Google Scholar]
- 35.Osmani, S. A., G. S. May, and N. R. Morris. 1987. Regulation of the mRNA levels of nimA, a gene required for the G2-M transition in Aspergillus nidulans. J. Cell Biol. 1041495-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osmani, S. A., and X. S. Ye. 1996. Cell cycle regulation in Aspergillus by two protein kinases. Biochem. J. 317633-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osmani, S. A., and X. S. Ye. 1997. Targets of checkpoints controlling mitosis. Trends Cell Biol. 7283-288. [DOI] [PubMed] [Google Scholar]
- 38.Ovechkina, Y., P. Maddox, C. E. Oakley, X. Xiang, S. A. Osmani, E. D. Salmon, and B. R. Oakley. 2003. Spindle formation in Aspergillus is coupled to tubulin movement into the nucleus. Mol. Biol. Cell 142192-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papadopoulou, K., S. S. Ng, H. Ohkura, M. Geymonat, S. G. Sedgwick, and C. J. McInerny. 2008. Regulation of gene expression during M-G1-phase in fission yeast through Plo1p and forkhead transcription factors. J. Cell Sci. 12138-47. [DOI] [PubMed] [Google Scholar]
- 40.Pontecorvo, G., J. A. Roper, L. M. Hemmons, K. D. MacDonald, and A. W. Bufton. 1953. The genetics of Aspergillus nidulans, p. 141-238. In M. Demerec (ed.), Advances in genetics, vol. 5. Academic Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 41.Pu, R. T., Gang Xu, L. Wu, J. Vierula, K. O'Donnell, X. Ye, and S. A. Osmani. 1995. Isolation of a functional homolog of the cell cycle specific NIMA protein kinase and functional analysis of conserved residues. J. Biol. Chem. 27118110-18116. [DOI] [PubMed] [Google Scholar]
- 42.Pu, R. T., and S. A. Osmani. 1995. Mitotic destruction of the cell cycle regulated NIMA protein kinase of Aspergillus nidulans is required for mitotic exit. EMBO J. 14995-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reid, J., and J. Q. Svejstrup. 2004. DNA damage-induced Def1-RNA polymerase II interaction and Def1 requirement for polymerase ubiquitylation in vitro. J. Biol. Chem. 27929875-29878. [DOI] [PubMed] [Google Scholar]
- 44.Ribar, B., A. Grallert, E. Olah, and Z. Szallasi. 1999. Deletion of the sep1+ forkhead transcription factor homologue is not lethal but causes hyphal growth in Schizosaccharomyces pombe. Biochem. Biophys. Res. Commun. 263465-474. [DOI] [PubMed] [Google Scholar]
- 45.Shih, S. C., G. Prag, S. A. Francis, M. A. Sutanto, J. H. Hurley, and L. Hicke. 2003. A ubiquitin-binding motif required for intramolecular monoubiquitylation, the CUE domain. EMBO J. 221273-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suelmann, R., N. Sievers, and R. Fischer. 1997. Nuclear traffic in fungal hyphae: in vivo study of nuclear migration and positioning in Aspergillus nidulans. Mol. Microbiol. 25757-769. [DOI] [PubMed] [Google Scholar]
- 47.Szewczyk, E., T. Nayak, C. E. Oakley, H. Edgerton, Y. Xiong, N. Taheri-Talesh, S. A. Osmani, and B. R. Oakley. 2006. Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 13111-3120. [DOI] [PubMed] [Google Scholar]
- 48.Toya, M., M. Sato, U. Haselmann, K. Asakawa, D. Brunner, C. Antony, and T. Toda. 2007. Gamma-tubulin complex-mediated anchoring of spindle microtubules to spindle-pole bodies requires Msd1 in fission yeast. Nat. Cell Biol. 9646-653. [DOI] [PubMed] [Google Scholar]
- 49.Waring, R. B., G. S. May, and N. R. Morris. 1989. Characterization of an inducible expression system in Aspergillus nidulans using alcA and tubulin-coding genes. Gene 79119-130. [DOI] [PubMed] [Google Scholar]
- 50.Woudstra, E. C., C. Gilbert, J. Fellows, L. Jansen, J. Brouwer, H. Erdjument-Bromage, P. Tempst, and J. Q. Svejstrup. 2002. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature 415929-933. [DOI] [PubMed] [Google Scholar]
- 51.Wu, L., S. A. Osmani, and P. M. Mirabito. 1998. A role for NIMA in the nuclear localization of cyclin B in Aspergillus nidulans. J. Cell Biol. 1411575-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang, L., L. Ukil, A. Osmani, F. Nahm, J. Davies, C. P. De Souza, X. Dou, A. Perez-Balaguer, and S. A. Osmani. 2004. Rapid production of gene replacement constructs and generation of a green fluorescent protein-tagged centromeric marker in Aspergillus nidulans. Eukaryot. Cell 31359-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye, X. S., R. R. Fincher, A. Tang, K. O'Donnell, and S. A. Osmani. 1996. Two S-phase checkpoint systems, one involving the function of both BIME and Tyr15 phosphorylation of p34cdc2, inhibit NIMA and prevent premature mitosis. EMBO J. 153599-3610. [PMC free article] [PubMed] [Google Scholar]
- 54.Ye, X. S., R. R. Fincher, A. Tang, A. H. Osmani, and S. A. Osmani. 1998. Regulation of the anaphase-promoting complex/cyclosome by BIMA(APC3) and proteolysis of NIMA. Mol. Biol. Cell 93019-3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye, X. S., S. L. Lee, T. D. Wolkow, S. L. McGuire, J. E. Hamer, G. C. Wood, and S. A. Osmani. 1999. Interaction between developmental and cell cycle regulators is required for morphogenesis in Aspergillus nidulans. EMBO J. 186994-7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye, X. S., G. Xu, P. T. Pu, R. R. Fincher, S. L. McGuire, A. H. Osmani, and S. A. Osmani. 1995. The NIMA protein kinase is hyperphosphorylated and activated downstream of p34cdc2/cyclin B: coordination of two mitosis promoting kinases. EMBO J. 14986-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zilahi, E., E. Salimova, V. Simanis, and M. Sipiczki. 2000. The S. pombe sep1 gene encodes a nuclear protein that is required for periodic expression of the cdc15 gene. FEBS Lett. 481105-108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.