

Abstract
The persistence of Porphyromonas gingivalis in the inflammatory environment of the periodontal pocket requires an ability to overcome oxidative stress. DNA damage is a major consequence of oxidative stress. Unlike the case for other organisms, our previous report suggests a role for a non-base excision repair mechanism for the removal of 8-oxo-7,8-dihydroguanine (8-oxo-G) in P. gingivalis. Because the uvrB gene is known to be important in nucleotide excision repair, the role of this gene in the repair of oxidative stress-induced DNA damage was investigated. A 3.1-kb fragment containing the uvrB gene was PCR amplified from the chromosomal DNA of P. gingivalis W83. This gene was insertionally inactivated using the ermF-ermAM antibiotic cassette and used to create a uvrB-deficient mutant by allelic exchange. When plated on brucella blood agar, the mutant strain, designated P. gingivalis FLL144, was similar in black pigmentation and beta-hemolysis to the parent strain. In addition, P. gingivalis FLL144 demonstrated no significant difference in growth rate, proteolytic activity, or sensitivity to hydrogen peroxide from that of the parent strain. However, in contrast to the wild type, P. gingivalis FLL144 was significantly sensitive to UV irradiation. The enzymatic removal of 8-oxo-G from duplex DNA was unaffected by the inactivation of the uvrB gene. DNA affinity fractionation identified unique proteins that preferentially bound to the oligonucleotide fragment carrying the 8-oxo-G lesion. Collectively, these results suggest that the repair of oxidative stress-induced DNA damage involving 8-oxo-G may occur by a still undescribed mechanism in P. gingivalis.
The importance of Porphyromonas gingivalis as an etiologic agent of adult periodontitis is well established (reviewed in reference 30). In addition to its anaerobic requirement, the association of P. gingivalis with inflammatory diseases implies that adaptability to oxidative stress is paramount for its survival. In an inflammatory microenvironment, reactive oxygen species (ROS) are important components (35). In the periodontal pocket, these oxygen metabolites are generated mostly from polymorphonuclear leukocyte and macrophage activities (10) or the occasional exposure of P. gingivalis to air. Two cellular systems that are coordinately regulated are known to function in bacteria to protect against oxidative stress (5, 32). In one system, antioxidant enzymes such as superoxide dismutase (SOD), catalase, peroxidase, and oxidase diminish or eliminate molecular oxygen and ROS before they can damage cellular components (4). As demonstrated in Escherichia coli, the other system involves the repair of the oxidatively damaged nucleic acids by endonucleases, using several mechanisms, including base excision repair (BER) and nucleotide excision repair (NER) (8, 28, 32). To date, we know little about the mechanism(s) of oxidative stress resistance in P. gingivalis. While this organism is oxygen tolerant and is missing catalase activity, it has been shown to express SOD activity (11). This SOD activity, however, is protective only for tolerance to atmospheric oxygen and is ineffective against hydrogen peroxide or exogenously generated ROS (33). Recently, the rbr, feoB2, dps, and ahpC genes were shown to provide oxidative stress protection against hydrogen peroxide (23, 27, 49, 52). Additionally, cell surface heme acquisition has been postulated to be a unique defense mechanism against ROS in P. gingivalis (47, 48). The storage of heme on the cell surface, which gives the organism its characteristic black pigmentation, can form μ-oxo dimers in the presence of ROS and can give rise to the catalytic degradation of hydrogen peroxide (47).
DNA is a major target of ROS (reviewed in reference 35). While oxidant-induced DNA damage generates over 20 different oxidatively altered bases (13), 8-oxo-7,8-dihydroguanine (8-oxo-G) is by far the major product (44). Unlike several other modified DNA bases, 8-oxo-G does not block replication. Instead, it can Watson-Crick base pair with cytosine as well as Hoogsteen base pair with adenine (21, 50). The polymerases can efficiently incorporate both cytosine and adenine across from 8-oxo-G. Mispairing with adenine often leads to GC-to-TA transversion mutations that can be deleterious to the cell (21). Because the average G+C content of the genome of P. gingivalis is 49% (37), a mechanism(s) to prevent or repair lesions resulting from guanine oxidation could be significant. This is further underscored by the observations that the salivary levels of 8-oxo-G and the presence of P. gingivalis and Tannerella forsythia in periodontitis patients were significantly higher than those in healthy subjects (42). In addition, the presence of 8-oxo-G was significantly correlated with P. gingivalis (42).
BER and NER are two known DNA repair mechanisms that are conserved in many organisms, including eukaryotes. Removal of 8-oxo-G appears to occur mostly by BER, which in E. coli involves the foramidopyrimidine glycosylase (Fpg) enzyme, encoded by the mutM gene (7, 8, 18). NER is unique due to its ability to repair a wide spectrum of DNA lesions. Proteins including UvrA, -B, -C, and -D are involved in the recognition of the lesion and the release of the patch of DNA, including the damaged base (6). While these DNA repair mechanisms have been described for many organisms, there is a gap in our understanding of the repair of oxidatively induced DNA damage in P. gingivalis.
Under oxidative stress conditions, we previously observed higher levels of 8-oxo-G in P. gingivalis FLL92, a nonpigmented isogenic mutant, than in the wild-type strain (27). Enzymatic removal of 8-oxo-G was catalyzed by a mechanism that did not include BER, as observed in E. coli. Compared to the parent strain, 8-oxo-G repair activity was also increased in P. gingivalis FLL92. Also, in comparison with other anaerobic periodontal pathogens, only P. gingivalis demonstrated a different pattern for the enzymatic removal of 8-oxo-G from that observed in E. coli. Because DNA cleavage occurred several bases away from the 8-oxo-G lesion, this raised the possibility of a NER-like repair mechanism, which is also known to repair similar lesions (27). In this study, we report that NER does not play a role in the repair of the 8-oxo-G lesion in P. gingivalis. Rather, a hypothetical protein of unknown function, via an as yet undescribed mechanism, may be involved in 8-oxo-G repair activity. Additionally, while P. gingivalis FLL144, a uvrB-defective mutant, was dramatically more sensitive than the wild type to UV, there was no difference in their sensitivities to hydrogen peroxide.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Strains and plasmids used in this experiment are listed in Table 1. P. gingivalis strains were grown in brain heart infusion (BHI) broth (Difco Laboratories, Detroit, MI) supplemented with hemin (5 μg/ml), vitamin K (0.5 μg/ml), and cysteine (0.1%). E. coli strains were grown in Luria-Bertani broth (LB) (40a). Unless otherwise stated, all cultures were incubated at 37°C. P. gingivalis strains were maintained in an anaerobic chamber (Coy Manufacturing, Ann Arbor, MI) in 10% H2, 10% CO2, and 80% N2. Growth rates for P. gingivalis and E. coli strains were determined spectrophotometrically (optical density at 600 nm [OD600]). Antibiotics were used at the following concentrations: clindamycin, 0.5 μg/ml; erythromycin, 300 μg/ml; and carbenicillin, 100 μg/ml.
TABLE 1.
Plasmids and bacterial strains used in this study
| Plasmid or strain | Phenotype, genotype, or description | Source or reference |
|---|---|---|
| Plasmids | ||
| pCR-XL-TOPO | Apr Kmr | Invitrogen |
| pVA2198 | SprermF-ermAM | 17 |
| Bacterial strains | ||
| Porphyromonas gingivalis strains | ||
| W83 | Wild type | 1 |
| FLL32 | recA defective | 2 |
| FLL92 | vimA defective | 1 |
| FLL144 | uvrB defective | This study |
| Escherichia coli strains | ||
| DH5α | F− φ80dlacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK− mK+) phoA supE44 thi-1 gyrA96 relA1 | Invitrogen |
| Top10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 ara-139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15Tn10(Tetr)] | Stratagene |
DNA isolation and analysis.
Plasmid and P. gingivalis chromosomal DNA preparations and analyses were performed as previously described (53). For large-scale preparation, plasmids were purified using a Qiagen (Santa Clarita, CA) plasmid maxi kit. DNA was digested with restriction enzymes as specified by the manufacturer (Roche, Indianapolis, IN). For DNA subcloning, the desired fragments were isolated from 1% agarose gels run in Tris-acetate-EDTA buffer and then purified using Amicon Ultrafree-DA (Millipore, Bellerica, MA). Southern blot alkaline transfer was performed according to the method of Roche Diagnostics Corporation (Indianapolis, IN). The PCR-amplified 3.0-kb uvrB and 2.1-kb ermF-ermAM genes were digoxigenin (DIG) labeled and used as probes in hybridization experiments. DNA labeling, hybridization, and detection were performed using a DIG High Prime labeling and detection starter kit II (Roche, Indianapolis, IN) according to the manufacturer's instructions.
PCR analysis of P. gingivalis chromosomal DNA.
Oligonucleotide primer design (Table 2) and general PCR amplification were done as previously described (2, 14, 41). Briefly, a 50-μl reaction mixture containing 1 μl of template DNA (0.5 μg), a 1 μM concentration of each primer, 25 μl high-fidelity PCR master enzyme mix (Roche, Indianapolis, IN), and distilled water was prepared. The PCR consisted of 25 cycles, with a temperature profile of 94°C for 30 s, 55°C for 1 min, and 72°C for 2 min. The final extension was performed at 72°C for 7 min. The PCR-amplified DNA was then identified by 1% agarose gel electrophoresis.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′) | Characteristics |
|---|---|---|
| P1 | GGA AAA CAC ACT CTT GCA GC | uvrB 3.0 forward |
| P2 | GCC GTT CCT ATC ATC TTT GC | uvrB 3.0 reverse |
| P3 | AAT TAG GCC TTA GTA ACG TGT AAC TTT | ermF 2.1 right |
| P4 | TAT TAG GCC TAT AGC TTC CGC TATT | ermF 2.1 left |
| P5 | ATG CGG TGG AGT ATT TCG TC | uvrB 1.2 forward |
| P6 | ATG TAG CCG GTG CTG ATA CC | uvrB 1.2 reverse |
| P7 | ATG CCC ATC CCT CTA TAC CTG | vimA forward |
| P8 | TAC CTG TTT TTG CTG ACC GG | vimA reverse |
Reverse transcriptase PCR (RT-PCR) analysis of DNase-treated RNA extracted from P. gingivalis.
Total RNA was extracted from P. gingivalis grown to mid-log phase (OD600 of 0.7) by use of a RiboPure kit (Ambion, Austin, TX). Reverse transcription and PCR amplification were performed with a Perkin-Elmer Cetus DNA thermal cycler (Perkin-Elmer Corporation, Norwalk, CT). The final products were analyzed by 1% agarose gel electrophoresis.
Cloning and mutagenesis of the P. gingivalis uvrB gene.
The 3.0-kb fragment carrying uvrB and flanking regions was PCR amplified from P. gingivalis W83 chromosomal DNA, using primers P1 and P2 (Table 2). This fragment was cloned into pCR2.1-TOPO plasmid vector (Invitrogen, Carlsbad, CA) and designated pFLL142. The ermF-ermAM antibiotic cassette was amplified from pVA2198 by use of Pfu Turbo polymerase (Stratagene, La Jolla, CA) and then ligated into the StuI restriction site of uvrB. The resultant recombinant plasmid, designated pFLL143, was used in the electroporation of P. gingivalis W83 as previously described (1, 16).
Gingipain activity assay.
P. gingivalis extracellular protein extracts were prepared as previously reported (45). The presence of Arg-X and Lys-X activity was determined using a microplate reader (Bio-Rad Laboratories, Hercules, CA) according to the methods of Potempa et al. (39).
Sensitivity to hydrogen peroxide and UV irradiation.
P. gingivalis strains were grown to early log phase (OD600 of 0.2) in BHI broth. Hydrogen peroxide at concentrations of 0.25 and 0.5 mM was then added to the cell cultures and further incubated for 20 h. The OD600 was then measured at 4-h intervals over a 24-h period. Cell cultures without hydrogen peroxide were used as controls. A UV sensitivity test was done as previously reported (1).
Oligonucleotide labeling and annealing procedures.
Oligonucleotide fragments (Table 3) used in this study were synthesized by Synthegen (Houston, TX). Labeling and annealing procedures were performed as previously described (27).
TABLE 3.
Oligonucleotides used in this study
| Oligonucleotide | Sequencea |
|---|---|
| O1 | 5′-GGCTATCGTGGCXGGCCACGACGG-3′ |
| 3′-CCGATAGCACCGCCCGGTGCTGCC-5′ | |
| O2 | 5′-GGCTATCGTGGCXGGCCACGACGG-3′ |
| 3′-CCGATAGCACCGACCGGTGCTGCC-5′ | |
| O3 | 5′-GACTACGTACTGTTACGGCTCCATCXCTACCGCATTCAGGCCAGATCTGC-3′ |
| 3′-CTGATGCATGACAATGCCGAGGTAGCGATGGCGTAAGTCCGGTCTAGACG-5′ | |
| O4 | 5′-GACTACGTACTGTTACGGCTCCATCGCTACCGCATTCAGGCCAGATCTGC-3′ |
| 3′-CTGATGCATGACAATGCCGAGGTAGCGATGGCGTAAGTCCGGTCTAGACG-5′ |
X, 8-oxo-G (in O1 and O3) or U (in O2). The positions of lesions are shown in bold.
Preparation of crude bacterial extracts.
Bacterial protein extracts were prepared as previously described (27). Briefly, P. gingivalis cultures were grown overnight in BHI broth. A 1/10 dilution of each bacterial strain was made in fresh BHI medium and grown to an OD600 of 0.6. E. coli was grown in a similar manner under aerobic conditions. The cell pellets were collected by centrifugation (9,000 × g for 10 min at 4°C), treated with protease inhibitors, resuspended in 5 ml of 50 mM Tris-HCl (pH 8.0)-1 mM EDTA lysis buffer, and subjected to eight freeze-thaw cycles. Cell debris was removed by centrifugation at 12,000 × g for 20 min at 4°C. The protein concentration of the supernatant was determined using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL).
Glycosylase assay.
Labeled and annealed oligonucleotides (2 pmol) were incubated at 37°C for 1 h with P. gingivalis or E. coli cell extract (2 μg) in a 1× enzyme buffer supplied with the uracil DNA glycosylase (Ung) or formamidopyrimidine-DNA glycosylase (Fpg) enzyme (Trevigen Inc., Gaithersburg, MD). An equal volume of loading buffer (98% formamide, 0.01 M EDTA, 1 mg/ml xylene cyanol, and 1 mg/ml bromophenol blue) was added to stop the reaction. Fifty picomoles of competitor oligonucleotide was added to each reaction mix and heated to 95°C for 5 min to denature the duplex, after which it was resolved by gel electrophoresis. As controls, Fpg and Ung control reactions were performed according to the methods of Liu et al. (31). Briefly, 2 pmol of specific oligonucleotide was incubated with 1 unit of the enzyme at 37°C for 1 h in reaction buffers provided by the manufacturers. Cleavage of abasic sites after glycosylase treatment with Ung was performed by adding 5 μl 0.1 M NaOH for 30 min at 37°C.
Gel electrophoresis and analysis of cleavage.
Reaction samples were loaded onto a 20% denaturing polyacrylamide gel (7 M urea) and run for 90 min at 500 V. The resulting bands, corresponding to the cleavage products and uncleaved substrate, were visualized using a Molecular Dynamics phosphorimager (Amersham Biosciences, Piscataway, NJ) and ImageQuant 5.0 software.
Pull-down assay.
DNA affinity fractionation was performed according to a modification of the method of Parham et al. (38). Briefly, phosphoramidite was used to covalently attach three biotin molecules to each oligonucleotide, using conventional coupling chemistry (21a). Streptavidin-coupled superparamagnetic polystyrene beads (2.8-μm diameter; Invitrogen, Carlsbad, CA) were coupled with the O3 or O4 oligonucleotide (Table 3). Oligonucleotide-attached beads were then incubated with 2 mg of the P. gingivalis FLL92 protein extract at 37°C for 10 min. The mixture was then placed on a Dynal MPC-S magnetic particle concentrator (Invitrogen, Carlsbad, CA) for 3 min. The supernatant was removed, and 10 μl 0.1% sodium dodecyl sulfate (SDS) was added to the immobilized beads, which were then incubated at 100°C for 7 min. The supernatant was removed and concentrated using an Ultrafree-MC 5,000 NMWL centrifugal filter device (Millipore, Bedford, MA). Aliquots containing 50 μg of protein were prepared for SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
Protein fractionation and digestion of extracted P. gingivalis FLL92 protein.
SDS-PAGE was performed with a 10% bis-Tris gel in 1× SDS-PAGE running buffer (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. The gels were run for 1.5 cm and then stained with SimplyBlue safe stain (Invitrogen, Carlsbad, CA) to visualize bands. After being destained briefly in water, the gel was cut into seven equally spaced slices (∼2 mm each) for trypsin digestion (see Fig. S1 in the supplemental material). As a control, a gel slice was cut from a blank region of the gel and processed in parallel with the sample. The excised gel pieces were dehydrated in acetonitrile and dried in a vacuum centrifuge for 30 min. The proteins were reduced in 20 μl of 20 mM dithiothreitol in 100 mM NH4HCO3 (sufficient to cover the gel pieces) for 1 h at 60°C. After cooling to room temperature, the dithiothreitol solution was replaced with an alkylating solution consisting of 20 μl of 200 mM iodoacetamide in 100 mM NH4HCO3. After 30 min of incubation at ambient temperature in the dark, the gel pieces were washed twice with 150 μl 100 mM NH4HCO3, finely minced with a flame-sealed polypropylene pipette tip, dehydrated by the addition of acetonitrile, and then dried in a vacuum centrifuge. The gel pieces were rehydrated and incubated overnight at 37°C in 20 μl digestion buffer containing 0.1 μg of mass spectrometry (MS)-grade trypsin (Promega, Madison, WI) in 50 mM acetic acid with equal parts of 100 mM NH4HCO3. The digestion reaction was stopped with 10 μl of 5% formic acid. The digest solution (aqueous extraction) was transferred to a clean 0.65-ml siliconized tube. To the gel pieces, 30 μl of 50% acetonitrile with 5% formic acid was added, and the mixture was vortexed for 30 min, centrifuged, and then sonicated for 5 min. This process was repeated, and both aqueous extractions were combined and concentrated to 10 μl in a vacuum centrifuge. Peptides were extracted and purified using standard C18 ZipTip technology following the manufacturer's directions (Millipore, Bedford, MA). The final volume of each preparation was 20 μl in 0.1% formic acid.
MS and data analysis.
The extracted peptides from each gel piece were analyzed using an LCQ Deca XP Plus system (Thermo Finnigan, San Jose, CA) with nano-electrospray technology (New Objectives, Woburn, MA). This consisted of reverse-phase C18 separation of peptides on a 10-cm by 75-μm capillary column with direct electrospray injection to the intake port of the LCQ system. MS and MS/MS analyses were accomplished with a four-part protocol that consisted of one full MS analysis (from 150 to 2,000 m/z) followed by three MS/MS events using data-dependent acquisition, where the most intense ion from a given full MS scan was subjected to collision-induced dissociation, followed by the second and third most intense ions (56). With the cycle repeating itself approximately every 2 s, the nanoflow buffer gradient was extended over 45 min, using a 0 to 60% acetonitrile gradient from buffer B (95% acetonitrile with 0.1% formic acid) developed against buffer A (2% acetonitrile with 0.1% formic acid) at a flow rate of 250 to 300 nl/min, with a final 5-min 80% bump of buffer B before reequilibration. Flow stream splitting (1:1,000) and a Scivex 10 port automated valve (Upchurch Scientific, Oak Harbor, WA), together with a Michrom nanotrap column (Michrom Bioresources, Auburn, CA), were used to move the 20-μl sample from the autosampler to the nanospray unit. The positive ion mode was employed, and the spray voltage and current were set at 2.2 kV and 5.0 μA, with a capillary voltage of 25 V. The spray temperature was set at 160°C for peptides. Data were collected with Xcalibur software (Thermo Electron) and screened with Bioworks 3.1. Peptide MS/MS spectra were processed by Turbo SEQUEST software (v.27) to produce unfiltered data and out files (24, 25) for each analysis, utilizing the latest version of the P. gingivalis Fasta database available from NCBI (January 2008). Proteome Software's SCAFFOLD 1.7. meta-analysis software, together with X! TANDEM (www.thegpm.org), was then used to statistically validate the peptide and protein findings of SEQUEST (60). Proteins were considered confidently identified when at least two different peptides were present at a probability of at least 95% and the protein probability was also 95% or higher. Individual peptide matches were then confirmed with the BLAST database at http://www.oralgen.lanl.gov.
RESULTS
Inactivation of the uvrB homolog in P. gingivalis W83.
A survey of the P. gingivalis genome (http://www.oralgen.lanl.gov) revealed the presence of a uvrB homolog which is 70% identical to E. coli uvrB. uvrB-defective isogenic mutants of P. gingivalis were created by allelic exchange mutagenesis. The recombinant plasmid pFLL143, carrying the ermF-ermAM cassette within the StuI restriction sites of the uvrB gene, was introduced into P. gingivalis W83 by electroporation. After 5 days of incubation on selective medium, several erythromycin-resistant colonies were observed, and eight were randomly chosen for further analysis. Similar to the wild type, all colonies plated on brucella blood agar were pigmented black and exhibited beta-hemolysis. Chromosomal DNAs from these colonies and the wild-type W83 strain were analyzed by PCR to confirm the inactivation of the uvrB gene. If the uvrB gene was inactivated by the ermF-ermAM cassette, a 5.1-kb fragment should be amplified from the erythromycin-resistant colonies by use of the P1 and P2 primers (Table 2). As shown in Fig. 1, a 5.1-kb band was observed for the erythromycin-resistant mutant, in contrast to a 3.1-kb band observed for the wild-type P. gingivalis strain W83. Furthermore, using primers P3 and P4, the ermF-ermAM cassette was amplified only from erythromycin-resistant P. gingivalis mutants (Fig. 1B). Chromosomal DNAs from three randomly chosen colonies and the wild-type W83 strain were digested with BamHI and subjected to Southern blot analysis. If uvrB was interrupted by the ermF-ermAM cassette, a 10-kb fragment should be observed, in contrast to an 8-kb fragment for W83 probed with DIG-labeled uvrB. In addition, hybridization performed with DIG-labeled ermF-ermAM (2.1 kb) should yield a 10-kb fragment for only the erythromycin-resistant mutants. The expected hybridizing fragment was observed for the mutants. Additionally, the ermF-ermAM probe did not hybridize to wild-type chromosomal DNA (see Fig. S2 in the supplemental material; also data not shown). One mutant, designated P. gingivalis FLL144, was randomly chosen for further study. The growth rate of FLL144 in BHI broth was similar to that of the wild-type P. gingivalis W83 strain. Collectively, these data indicated that uvrB in P. gingivalis FLL144 was inactivated with the ermF-ermAM cassette and that uvrB inactivation had no effect on black pigmentation, beta-hemolysis, and growth rate under normal experimental conditions.
FIG. 1.
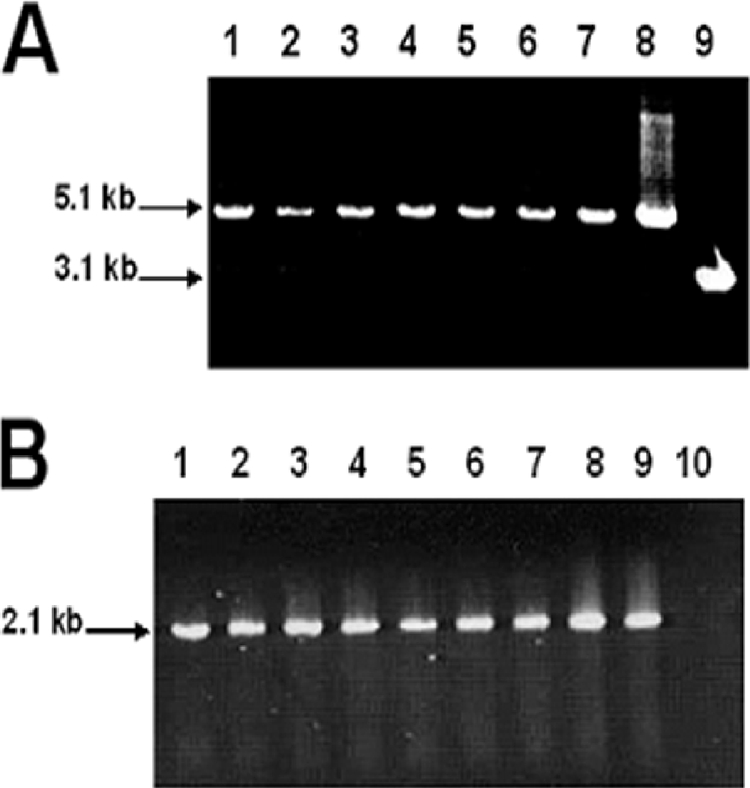
Inactivation of P. gingivalis uvrB by allelic mutagenesis. PCR amplification was used to determine the inactivation of the P. gingivalis uvrB gene in seven erythromycin-resistant P. gingivalis colonies. (A) Lanes 1 to 7, primers P1 and P2 (Table 2) were used to amplify the 5.1-kb uvrB::ermF-ermAM cassette; lane 8, pFLL143; lane 9, P. gingivalis wild-type W83 (Table 2). (B) Lanes 1 to 7, amplification of ermF-ermAM cassette from seven erythromycin-resistant colonies, using the P3 and P4 primers; lane 8, amplification of ermF-ermAM cassette from pFLL143; lane 9, pVA2198; lane 10, P. gingivalis W83.
RT-PCR confirmation of uvrB inactivation in P. gingivalis FLL144.
To further confirm the inactivation of uvrB in P. gingivalis FLL144, total RNAs were isolated from the wild-type W83 and uvrB-defective mutant FLL144 strains grown to mid-log phase (OD600, 0.6 to 0.8). Specific intragenic oligonucleotide primers (P5 and P6) (Table 2) for amplification of uvrB were used in RT-PCR analysis. The P5 and P6 primers specific for uvrB should yield a 1.2-kb fragment only for the wild-type W83 strain. As shown in Fig. 2, a 1.2-kb fragment was amplified from the wild type by use of primers P5 and P6. As expected, there was no amplified fragment corresponding to uvrB in pFLL144. In addition, no amplification was observed for either the wild-type strain W83 or the uvrB-defective mutant strain FLL144 in the absence of RT. As a control, the vimA gene was observed in both P. gingivalis strains (Fig. 2, lanes 2 and 4). Taken together, these data confirm the inactivation of uvrB in P. gingivalis FLL144.
FIG. 2.
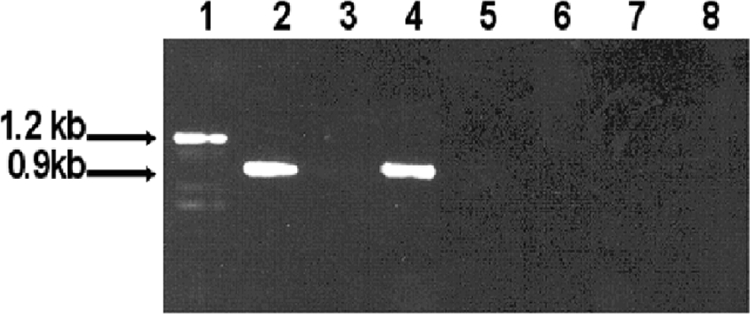
RT-PCR analysis of P. gingivalis W83 and P. gingivalis FLL144. DNase-treated total RNAs extracted from P. gingivalis strains W83 and FLL144 were subjected to RT-PCR. Lanes 1 and 3, primers P5 and P6 (Table 2) were used to amplify the 1.2-kb uvrB gene from P. gingivalis W83 and FLL144, respectively; lanes 2 and 4, primers P7 and P8 (Table 2) were used to amplify the 0.9-kb vimA gene from P. gingivalis W83 and FLL144, respectively; lanes 5 to 8, no-RT negative controls for uvrB and vimA from P. gingivalis W83 and FLL144. All lanes contained 10 μl of amplified mixture.
Gingipain activity is unaffected by inactivation of uvrB.
Because the gingipains are known to be associated with oxidative stress resistance (29), P. gingivalis W83 and FLL144 were assayed for gingipain activity, using α-benzoyl-dl-arginine p-nitroanilide (BAPNA) and z-lysine ρ-nitroanilide (ALNA). In late-exponential-growth-phase cultures, both arginine-X and lysine-X proteolytic activities were similar for both P. gingivalis strains, suggesting that the inactivation of uvrB does not significantly affect gingipain activity (Fig. 3A and B).
FIG. 3.
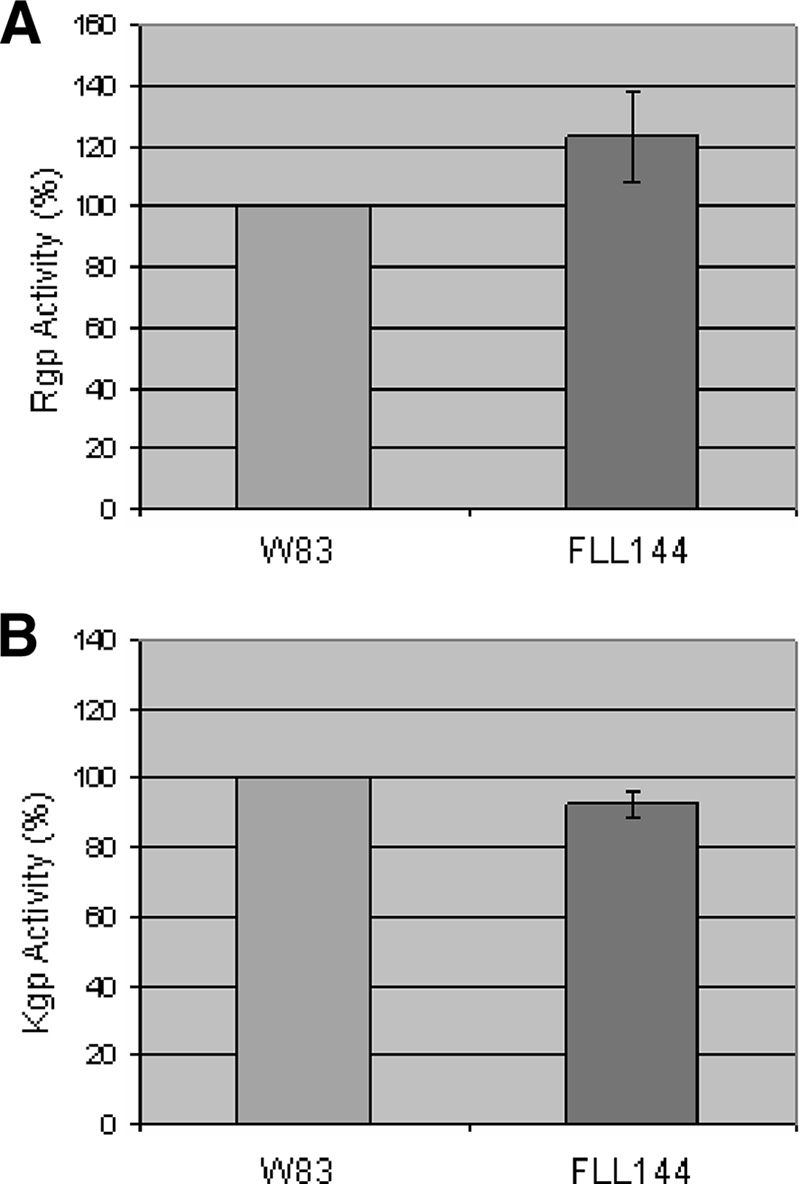
Proteolytic activity of P. gingivalis FLL144. P. gingivalis strains were grown to late log phase (OD600 of 1.2) in 50 ml of BHI broth supplemented with hemin and vitamin K. (A) A whole-cell culture was analyzed for Rgp (BAPNA) activity. (B) A whole-cell culture was analyzed for Kgp (ALNA) activity. The results shown are representative of three independent experiments performed in triplicate.
Inactivation of uvrB increases UV sensitivity of P. gingivalis.
The uvrB gene plays a significant role in NER in E. coli and other bacteria (46). Furthermore, it is documented that the NER mechanism functions primarily in the repair of UV-induced DNA damage (26, 36, 57). Therefore, the role of uvrB in the sensitivity to UV damage of P. gingivalis was examined. P. gingivalis W83, FLL144, and FLL32 (recA-defective isogenic mutant) were exposed to UV radiation at energies of 500 μJ and 1,000 μJ. P. gingivalis FLL32 was used as a positive control, as this mutant has been shown to demonstrate sensitivity to UV radiation (16). At 500 μJ, there was a 59% reduction in the number of wild-type W83 colonies, compared to 99% and 98% reductions in the numbers of uvrB-deficient mutant colonies and recA-defective isogenic mutant colonies, respectively (Fig. 4). Similarly, at 1,000 μJ, there was an 83% reduction in the number of wild-type W83 colonies, compared to 98% and 99% reductions in the numbers of uvrB-deficient mutant colonies and recA-defective isogenic mutant colonies, respectively. Taken together, these data show that under similar physiological conditions, the uvrB-defective mutant is markedly more sensitive to UV exposure than the wild-type W83 strain.
FIG. 4.

UV sensitivity of P. gingivalis is increased by inactivation of uvrB. P. gingivalis strains W83 and FLL144 were grown to mid-log phase (OD600 of 0.6), spread on BHI plates, and then subjected to irradiation at increasing doses (0 μJ, 500 μJ, and 1,000 μJ) of UV in a Stratalinker 2400 apparatus (Stratagene, La Jolla, CA). After 7 to 10 days of incubation, surviving colonies were counted and compared to those on untreated plates. The results are representative of three independent experiments. Error bars represent standard errors of the means.
Effect of UvrB on the sensitivity of P. gingivalis to hydrogen peroxide.
P. gingivalis W83 and the isogenic mutant P. gingivalis FLL144 were evaluated for sensitivity to hydrogen peroxide. Both the parent strain and P. gingivalis FLL144 showed similar profiles of sensitivity to both concentrations (0.25 and 0.5 mM) of hydrogen peroxide tested (Fig. 5A and B). Collectively, these data suggest that inactivation of the uvrB gene does not affect the sensitivity of P. gingivalis to hydrogen peroxide.
FIG. 5.

Sensitivity of P. gingivalis strains W83 and FLL144 to hydrogen peroxide. P. gingivalis was grown to early log phase (OD600 of 0.2) in BHI broth, 0.25 mM (A) or 0.5 mM (B) H2O2 was then added to the cell cultures, and the cultures were further incubated for 24 h. Cell cultures without H2O2 were used as controls. The results shown are representative of three independent experiments.
8-oxo-G repair activities are similar in both wild-type and mutant P. gingivalis strains.
The possibility of a NER-like repair mechanism is raised because the repair of the 8-oxo-G lesion involves DNA cleavage several bases away from the damaged base (27). P. gingivalis W83 and the isogenic mutant P. gingivalis FLL144 were assessed for enzymatic removal of 8-oxo-G. Bacterial extracts from the P. gingivalis isogenic strains grown in the presence or absence of hydrogen peroxide were used in glycosylase assays with a [γ-32P]ATP-5′-end-labeled 8-oxo(dG-dC)-containing oligonucleotide (24-mer) (Table 3). As shown in Fig. 6A, the Fpg enzyme generated a 12-mer fragment. Additionally, a cleavage product of 17 bases was seen for both P. gingivalis strains W83 and FLL144 (Fig. 6A, lanes 1 to 4). In a similar assay using a [γ-32P]ATP-5′-end-labeled 8-oxo(dG-dC)-containing 50-mer oligonucleotide, Fpg generated a 25-mer cleavage fragment. Interestingly, a 28-mer cleavage product was observed for P. gingivalis strains W83 and FLL144, even though the level of activity for the removal of 8-oxo-G under those conditions seemed to be different (Fig. 6B). As a control, the removal of uracil was also examined using the same extracts. As shown in Fig. 6C, the levels of activity for Ung were similar for both P. gingivalis strains. Taken together, these data suggest that the inactivation of uvrB in P. gingivalis W83 does not affect the removal of 8-oxo-G, regardless of the size of the oligonucleotide.
FIG. 6.
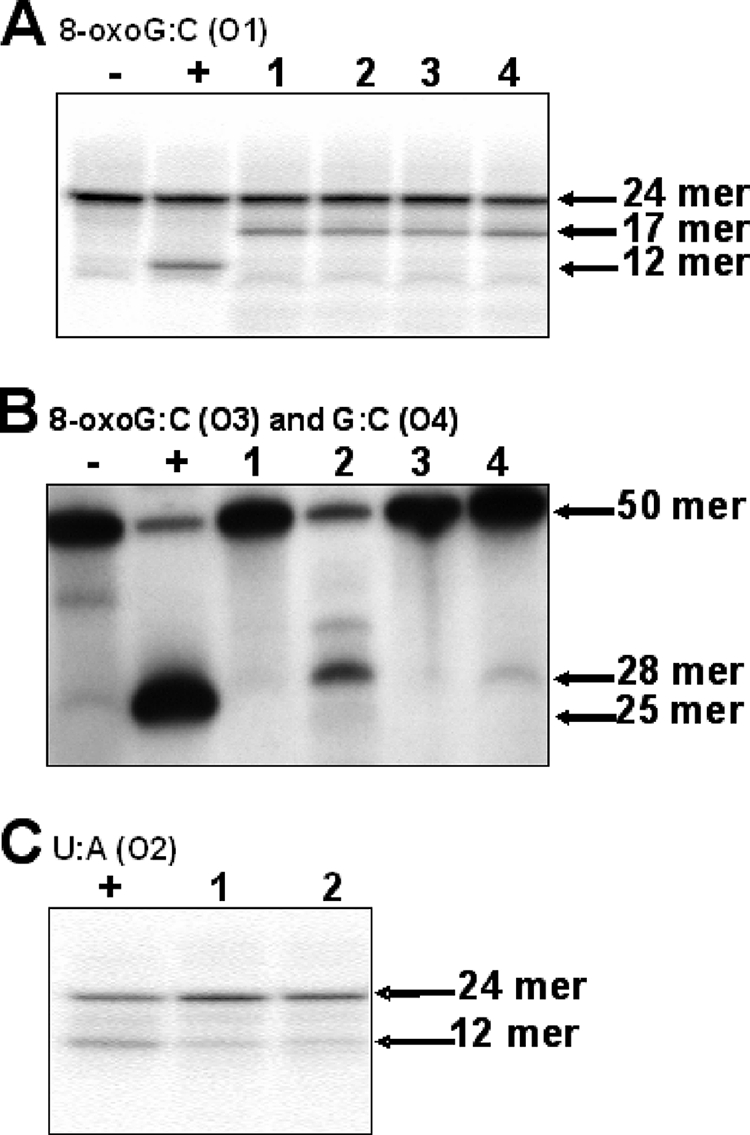
8-oxo-G and uracil removal activities of P. gingivalis strain W83 and FLL144 cell extracts. [γ-32P]ATP-5′-end-labeled oligonucleotide (O1) was incubated with P. gingivalis extracts for 1 h (Fpg) or 1 min (Ung), electrophoresed, and visualized. (A) Lane −, negative control containing O1; lane +, positive control containing O1 and Fpg enzyme; lane 1, O1 and treated P. gingivalis W83 extract; lane 2, O1 and treated P. gingivalis FLL144 extract; lane 3, O1 and P. gingivalis W83 extract; and lane 4, O1 and P. gingivalis FLL144 extract. (B) [γ-32P]ATP-5′-end-labeled oligonucleotides (O3 and O4) were incubated with P. gingivalis extracts for 20 min, electrophoresed, and visualized. Lane −, negative control containing O4 and Fpg enzyme; lane +, positive control containing O3 and Fpg enzyme; lane 1, O4 and treated P. gingivalis W83 extract; lane 2, O3 and treated P. gingivalis W83 extract; lane 3, O4 and treated P. gingivalis FLL144; and lane 4, O3 and treated P. gingivalis FLL144 extract. (C) Lane +, positive control containing O2 and Ung enzymes; lane 1, O2 and P. gingivalis W83 extract; and lane 2, O2 and P. gingivalis FLL144 extract.
Identification of proteins bound to 8-oxo-G-containing oligonucleotides by MS analysis.
Chromosomal DNA from P. gingivalis FLL92 exposed to hydrogen peroxide revealed higher levels of 8-oxo-G, in addition to increased repair activity in cell extracts, than those from the parent strain (27). Therefore, DNA affinity fractionation was performed to further identify a role for another putative unique protein(s) in the repair of 8-oxo-G lesions in P. gingivalis. An oligonucleotide fragment carrying the 8-oxo-G lesion (O3) was incubated with protein extracts from P. gingivalis FLL92 exposed to hydrogen peroxide. As a negative control, the protein extracts were also incubated with the normal oligonucleotide (O4) and with beads only. The extracted peptides were analyzed by MS. A total of >50,000 MS/MS spectra were acquired by searching against the P. gingivalis proteome with Bioworks Browser 3.1 SR1 ALPHA7 software (60). More than 500 peptides were identified, with 35 peptides representing unique spectra for the 16 proteins listed in Table 4 (4 to 23% coverage). These proteins were different from those that were attached to the control oligonucleotides without the 8-oxo-G lesion (see Table S1 in the supplemental material). Because the probability of protein identification from MS/MS spectra is a direct function of peptide identification, we tested for false identifications by using a decoy database consisting of both the forward (correct) and reversed (decoy) protein sequences (12, 24). Peptides that matched the reversed and/or forward sequence were considered false-positive results. Applying this criterion, the rate of false-positive identification was <1% (12, 15). There were no false-positive identifications for the 16 proteins listed in Table 4. We therefore concluded that any false-positive identifications determined did not have a significant impact on the analysis results. From these data, a conserved hypothetical protein, PG1037, was of particular interest. This protein seems to be encoded as part of an operon which is flanked by two genes, namely, PG1036 (uvrA), involved in DNA replication, recombination, and repair, and PG1038 (prcA), encoding an ATP-dependent DNA helicase (DNA helicase II) also involved in DNA replication, recombination, and repair. Taken together, these data suggest that a unique mechanism may be involved in the repair of 8-oxo-G lesions in P. gingivalis.
TABLE 4.
Proteins identified by MS analysis
| Gene ID | Gene name | Functional role | Other roles |
|---|---|---|---|
| PG0553 | Extracellular protease, putative | Protein fate; degradation of proteins, peptides, and glycopeptides | ATP synthesis coupled with proton transport |
| PG0514a | Preprotein translocase subunit (SecA) | Protein fate, protein and peptide secretion and trafficking | Nucleic acid-binding helicase activity, protein binding, ATP binding |
| PG0052a | Histidine kinase sensor protein | Uncategorized | Two-component sensor activity, protein histidine kinase activity, ATP binding, transferase activity |
| PG0295b | DNA processing protein subunit A (Smf family) | Cellular processes | DNA replication, recombination, and repair; intracellular trafficking and secretion |
| PG1576c | l-Aspartate oxidase (quinolinate synthetase B) | Biosynthesis of cofactors, prosthetic groups, and carriers; pyridine nucleotides | l-Aspartate oxidase activity, oxidoreductase activity |
| PG1487d | Hypothetical protein | Unknown, hypothetical | |
| PG1951 | Glutaminyl-tRNA synthetase | Translation, aminoacyl-tRNA synthetases | Aminoacyl-tRNA ligase activity, glutamate-tRNA ligase activity, glutamine-tRNA ligase activity, ATP binding |
| PG1657c | Methylmalonyl-coenzyme A mutase beta subunit | Fatty acid and phospholipid metabolism, degradation | Isomerase activity, intramolecular transferase activity, cobalamin binding, metal ion binding |
| PG0565d | Hypothetical protein | Unknown, hypothetical | |
| PG1341d | Hypothetical protein | Unknown, hypothetical | |
| PG2143c | YphC | Probable GTP binding protein | Monooxygenase activity, iron ion binding, GTP binding, heme binding |
| PG1037c,d | Conserved hypothetical protein | Unknown | Zinc ion binding |
| PG1985b,d | CRISPR-associated TM1792 family protein | Unknown, hypothetical | RAMP (repair-associated mysterious proteins) |
| PG0685a,c | ABC transport component, ATPase component | Transport and binding | Nucleotide binding, ATP binding, ATPase activity, nucleoside-triphosphatase activity |
| PG1937 | 50S ribosomal protein L4 | Translation, ribosomal proteins | |
| PG0690c | Cat2, a 4-hydroxybutyrate coenzyme A transferase | Catalytic activity | Acetyl-coenzyme A metabolic process |
Binding and transferase activity.
DNA replication, recombination, and repair.
Correlation with microarray data (not shown).
Hypothetical protein.
DISCUSSION
In this study, we examined the role of the P. gingivalis uvrB gene in the repair of oxidative stress-induced DNA damage. UvrB is a significant part of an important complex of proteins that function via NER in the repair of UV irradiation-induced DNA damage or bulky lesions sometimes induced by ROS (6, 22, 55). P. gingivalis has a single uvrB gene, which is highly homologous to the uvrB genes from other organisms, including Parabacteroides distasonis (72%) and Bacteroides fragilis (67%) (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The possibility of a NER mechanism, which is also known to repair similar lesions, was raised because enzymatic removal of 8-oxo-G in P. gingivalis occurred by DNA cleavage several bases away from the lesion (27). Similar to the case in other organisms (40, 51), uvrB in P. gingivalis appears to play the expected role, as the uvrB-defective mutant was more sensitive to UV irradiation than the wild-type strain. There was, however, no difference in sensitivity to oxidative stress in the uvrB-defective mutant (FLL144) compared to the wild type, suggesting that UvrB is not involved in oxidative stress resistance in P. gingivalis.
Repair of oxidative stress-induced DNA damage was similar for both the wild type and P. gingivalis FLL144, the uvrB-defective mutant. This further correlated with their similarity in sensitivity to hydrogen peroxide. Taken together, this may indicate that the uvrB-dependent NER system is not used in P. gingivalis to remove 8-oxo-G. Instead, a still undescribed mechanism(s) might be involved in this process. In bacteria, redundant mechanisms are observed to play a role in DNA repair (34, 54, 58). In a limited number of microorganisms, there is evidence of an additional excision repair pathway, alternative excision repair (AER) (3, 19, 59). Repair is catalyzed by a UV DNA damage endonuclease that binds a wide spectrum of DNA lesions. This protein nicks immediately 5′ of a lesion, leaving a 5′-phosphate and a 3′-OH. The remaining repair is enacted by enzymes that degrade the damaged displaced fragment, followed by DNA synthesis filling in the resulting gap and, finally, ligation. Because our studies have demonstrated a nick 3′ of the damaged base (27), it is unlikely (although it cannot be ruled out) that AER is functional under these experimental conditions in P. gingivalis exposed to hydrogen peroxide. In preliminary microarray studies (data not shown), we observed the upregulation of a putative exonuclease gene in cells exposed to hydrogen peroxide. This enzyme could be associated with a methyl-directed mismatch repair system that has also been demonstrated to play a role in the repair of 8-oxo-G (58). In E. coli, this system is initiated by the binding of MutS to the mismatch. MutL binds to MutS and MutH binds to a nearby d(GATC) recognition sequence (reviewed in reference 43). An incision is made by MutH 5′ of the d(GATC) sequence on the unmethylated strand, and the nicked strand is displaced by the combined activities of MutS, MutL, and MutU, a helicase. The displaced DNA strand is degraded by a specific exonuclease which is dependent on the position of the incision relative to the mismatch repair. The final repair involves a single-strand binding protein, DNA polymerase, and DNA ligase for filling in the gap left by the excision. In our preliminary studies, we observed the upregulation of mutS in P. gingivalis exposed to hydrogen peroxide. Because both oligonucleotides carrying the 8-oxo-G lesion had similar cleavage profiles and only one had a d(GATC) sequence, it is unclear if this mechanism may be functional in P. gingivalis for the repair of oxidative stress-induced DNA damage. In addition, there is no MutH homolog in the P. gingivalis genome (http://www.oralgen.lanl.gov). Further studies are in progress to investigate a role for the putative exonuclease and MutS in the repair of oxidative stress-induced DNA damage in P. gingivalis.
8-oxo-G lesion-specific binding proteins may help to clarify a role for specific proteins in the repair process. Several specific proteins were observed to bind to the oligonucleotide carrying the 8-oxo-G lesion. Of specific interest is the novel hypothetical protein PG1037, which is part of the uvrA-pg1037-prcA operon. prcA encodes a putative ATP-dependent DNA helicase (DNA helicase II) which is also involved in DNA replication, recombination, and repair (http://www.oralgen.lanl.gov). Preliminary microarray analysis also demonstrated that this operon is upregulated in P. gingivalis exposed to hydrogen peroxide-induced oxidative stress (data not shown). In P. gingivalis, there are two UvrA paralogues that share 42% homology (http://www.oralgen.lanl.gov). It is likely that one UvrA paralogue may be associated with UvrB, while the other (PG1036) may be involved with PG1037. There is no homology between UvrB and PG1037. Taken together, the data show that it may be possible that the UvrA paralogues have distinct DNA repair activities which are dependent on the specific interacting protein pairs. Paralogues with distinct activities in DNA repair have been observed in other organisms (9). This is under further investigation in the laboratory. It is also likely that a protein complex is involved in the repair of the 8-oxo-G lesions. While we cannot rule out nonspecific protein-protein interactions, several of the 8-oxo-G lesion-specific binding proteins have putative functions that include ATP and metal ion binding and oxidoreductase activities. The presence of these proteins would be consistent with a DNA repair activity that would most likely be ATP and metal ion dependent (20). Also, this would further support our previous observations that BER, an ATP-independent mechanism, is not involved in the repair of 8-oxo-G lesions in P. gingivalis (27). Further clarification of the role of these proteins is being investigated.
In conclusion, we have evaluated a P. gingivalis uvrB-defective mutant for its response to oxidative stress and its ability to repair one of the more common lesions, 8-oxo-G, induced under those conditions. While DNA repair is one of the most highly conserved biological processes, it is likely that a novel mechanism may be utilized for the repair of 8-oxo-G in P. gingivalis. Further studies are needed to delineate a role for the 8-oxo-G lesion-specific binding proteins in DNA repair in P. gingivalis. Also, the determination of a role for these proteins in the pathogenicity of this organism and other anaerobes may have implications for novel therapeutic strategies.
Supplementary Material
Footnotes
Published ahead of print on 10 October 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Abaibou, H., Z. Chen, G. J. Olango, Y. Liu, J. Edwards, and H. M. Fletcher. 2001. vimA gene downstream of recA is involved in virulence modulation in Porphyromonas gingivalis W83. Infect. Immun. 69325-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abaibou, H., Q. Ma, G. J. Olango, J. Potempa, J. Travis, and H. M. Fletcher. 2000. Unaltered expression of the major protease genes in a non-virulent recA-defective mutant of Porphyromonas gingivalis W83. Oral Microbiol. Immunol. 1540-47. [DOI] [PubMed] [Google Scholar]
- 3.Alleva, J. L., S. Zuo, J. Hurwitz, and P. W. Doetsch. 2000. In vitro reconstitution of the Schizosaccharomyces pombe alternative excision repair pathway. Biochemistry 392659-2666. [DOI] [PubMed] [Google Scholar]
- 4.Amano, A., T. Ishimoto, H. Tamagawa, and S. Shizukuishi. 1992. Role of superoxide dismutase in resistance of Porphyromonas gingivalis to killing by polymorphonuclear leukocytes. Infect. Immun. 60712-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baillon, M. L., A. H. van Vliet, J. M. Ketley, C. Constantinidou, and C. W. Penn. 1999. An iron-regulated alkyl hydroperoxide reductase (AhpC) confers aerotolerance and oxidative stress resistance to the microaerophilic pathogen Campylobacter jejuni. J. Bacteriol. 1814798-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batty, D. P., and R. D. Wood. 2000. Damage recognition in nucleotide excision repair of DNA. Gene 241193-204. [DOI] [PubMed] [Google Scholar]
- 7.Boiteux, S., T. R. O'Connor, and J. Laval. 1987. Formamidopyrimidine-DNA glycosylase of Escherichia coli: cloning and sequencing of the fpg structural gene and overproduction of the protein. EMBO J. 63177-3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boiteux, S., and J. P. Radicella. 1999. Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress. Biochimie 8159-67. [DOI] [PubMed] [Google Scholar]
- 9.Carpenter, E. P., A. Corbett, H. Thomson, J. Adacha, K. Jensen, J. Bergeron, I. Kasampalidis, R. Exley, M. Winterbotham, C. Tang, G. S. Baldwin, and P. Freemont. 2007. AP endonuclease paralogues with distinct activities in DNA repair and bacterial pathogenesis. EMBO J. 261363-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapple, I. L. 1996. Role of free radicals and antioxidants in the pathogenesis of the inflammatory periodontal diseases. Clin. Mol. Pathol. 49M247-M255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi, J. I., N. Takahashi, T. Kato, and H. K. Kuramitsu. 1991. Isolation, expression, and nucleotide sequence of the sod gene from Porphyromonas gingivalis. Infect. Immun. 591564-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Godoy, L. M., J. V. Olsen, G. A. de Souza, G. Li, P. Mortensen, and M. Mann. 2006. Status of complete proteome analysis by mass spectrometry: SILAC labeled yeast as a model system. Genome Biol. 7R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demple, B., and L. Harrison. 1994. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 63915-948. [DOI] [PubMed] [Google Scholar]
- 14.Devereux, J., P. Haeberli, and O. Smithies. 1984. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 12387-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng, J., D. Q. Naiman, and B. Cooper. 2007. Probability-based pattern recognition and statistical framework for randomization: modeling tandem mass spectrum/peptide sequence false match frequencies. Bioinformatics 232210-2217. [DOI] [PubMed] [Google Scholar]
- 16.Fletcher, H. M., R. M. Morgan, and F. L. Macrina. 1997. Nucleotide sequence of the Porphyromonas gingivalis W83 recA homolog and construction of a recA-deficient mutant. Infect. Immun. 654592-4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fletcher, H. M., H. A. Schenkein, R. M. Morgan, K. A. Bailey, C. R. Berry, and F. L. Macrina. 1995. Virulence of a Porphyromonas gingivalis W83 mutant defective in the prtH gene. Infect. Immun. 631521-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fowler, R. G., S. J. White, C. Koyama, S. C. Moore, R. L. Dunn, and R. M. Schaaper. 2003. Interactions among the Escherichia coli mutT, mutM, and mutY damage prevention pathways. DNA Repair (Amsterdam) 2159-173. [DOI] [PubMed] [Google Scholar]
- 19.Freyer, G. A., S. Davey, J. V. Ferrer, A. M. Martin, D. Beach, and P. W. Doetsch. 1995. An alternative eukaryotic DNA excision repair pathway. Mol. Cell. Biol. 154572-4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedberg, E. C., G. C. Walker, W. Siede, R. D. Wood, R. A. Schultz, and T. Ellenberger. 2006. DNA repair and mutagenesis. ASM Press, Washington, DC.
- 21.Grollman, A. P., and M. Moriya. 1993. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 9246-249. [DOI] [PubMed] [Google Scholar]
- 21a.Hamamoto, S., and H. Takaku. 1986. New approach to the synthesis of deoxyribonucleoside phosphoramidite derivatives. Chem. Lett. 19861401-1406. [Google Scholar]
- 22.Hanada, K., M. Iwasaki, S. Ihashi, and H. Ikeda. 2000. UvrA and UvrB suppress illegitimate recombination: synergistic action with RecQ helicase. Proc. Natl. Acad. Sci. USA 975989-5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He, J., H. Miyazaki, C. Anaya, F. Yu, W. A. Yeudall, and J. P. Lewis. 2006. Role of Porphyromonas gingivalis FeoB2 in metal uptake and oxidative stress protection. Infect. Immun. 744214-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higdon, R., J. M. Hogan, B. G. Van, and E. Kolker. 2005. Randomized sequence databases for tandem mass spectrometry peptide and protein identification. OMICS 9364-379. [DOI] [PubMed] [Google Scholar]
- 25.Higdon, R., and E. Kolker. 2007. A predictive model for identifying proteins by a single peptide match. Bioinformatics 23277-280. [DOI] [PubMed] [Google Scholar]
- 26.Jeggo, P. A. 1998. DNA breakage and repair. Adv. Genet. 38185-218. [DOI] [PubMed] [Google Scholar]
- 27.Johnson, N. A., R. McKenzie, L. McLean, L. C. Sowers, and H. M. Fletcher. 2004. 8-Oxo-7,8-dihydroguanine is removed by a nucleotide excision repair-like mechanism in Porphyromonas gingivalis W83. J. Bacteriol. 1867697-7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konola, J. T., K. E. Sargent, and J. B. Gow. 2000. Efficient repair of hydrogen peroxide-induced DNA damage by Escherichia coli requires SOS induction of RecA and RuvA proteins. Mutat. Res. 459187-194. [DOI] [PubMed] [Google Scholar]
- 29.Kuramitsu, H. K. 1998. Proteases of Porphyromonas gingivalis: what don't they do? Oral Microbiol. Immunol. 13263-270. [DOI] [PubMed] [Google Scholar]
- 30.Lamont, R. J., and H. F. Jenkinson. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 621244-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu, P., A. Burdzy, and L. C. Sowers. 2003. Repair of the mutagenic DNA oxidation product, 5-formyluracil. DNA Repair (Amsterdam) 2199-210. [DOI] [PubMed] [Google Scholar]
- 32.Lu, A. L., X. Li, Y. Gu, P. M. Wright, and D. Y. Chang. 2001. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem. Biophys. 35141-170. [DOI] [PubMed] [Google Scholar]
- 33.Lynch, M. C., and H. K. Kuramitsu. 1999. Role of superoxide dismutase activity in the physiology of Porphyromonas gingivalis. Infect. Immun. 673367-3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martins-Pinheiro, M., R. S. Galhardo, C. Lage, K. M. Lima-Bessa, K. A. Aires, and C. F. Menck. 2004. Different patterns of evolution for duplicated DNA repair genes in bacteria of the Xanthomonadales group. BMC Evol. Biol. 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller, R. A., and B. E. Britigan. 1997. Role of oxidants in microbial pathophysiology. Clin. Microbiol. Rev. 101-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Modrich, P., and R. Lahue. 1996. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 65101-133. [DOI] [PubMed] [Google Scholar]
- 37.Nelson, K. E., R. D. Fleischmann, R. T. DeBoy, I. T. Paulsen, D. E. Fouts, J. A. Eisen, S. C. Daugherty, R. J. Dodson, A. S. Durkin, M. Gwinn, D. H. Haft, J. F. Kolonay, W. C. Nelson, T. Mason, L. Tallon, J. Gray, D. Granger, H. Tettelin, H. Dong, J. L. Galvin, M. J. Duncan, F. E. Dewhirst, and C. M. Fraser. 2003. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J. Bacteriol. 1855591-5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parham, N. J., F. J. Picard, R. Peytavi, M. Gagnon, G. Seyrig, P. A. Gagne, M. Boissinot, and M. G. Bergeron. 2007. Specific magnetic bead based capture of genomic DNA from clinical samples: application to the detection of group B streptococci in vaginal/anal swabs. Clin. Chem. 531570-1576. [DOI] [PubMed] [Google Scholar]
- 39.Potempa, J., J. Mikolajczyk-Pawlinska, D. Brassell, D. Nelson, I. B. Thogersen, J. J. Enghild, and J. Travis. 1998. Comparative properties of two cysteine proteinases (gingipains R), the products of two related but individual genes of Porphyromonas gingivalis. J. Biol. Chem. 27321648-21657. [DOI] [PubMed] [Google Scholar]
- 40.Rivera, E., L. Vila, and J. Barbe. 1996. The uvrB gene of Pseudomonas aeruginosa is not DNA damage inducible. J. Bacteriol. 1785550-5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40a.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 41.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 745463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawamoto, Y., N. Sugano, H. Tanaka, and K. Ito. 2005. Detection of periodontopathic bacteria and an oxidative stress marker in saliva from periodontitis patients. Oral Microbiol. Immunol. 20216-220. [DOI] [PubMed] [Google Scholar]
- 43.Schofield, M. J., and P. Hsieh. 2003. DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 57579-608. [DOI] [PubMed] [Google Scholar]
- 44.Sekiguchi, M., and T. Tsuzuki. 2002. Oxidative nucleotide damage: consequences and prevention. Oncogene 218895-8904. [DOI] [PubMed] [Google Scholar]
- 45.Sheets, S. M., J. Potempa, J. Travis, H. M. Fletcher, and C. A. Casiano. 2006. Gingipains from Porphyromonas gingivalis W83 synergistically disrupt endothelial cell adhesion and can induce caspase-independent apoptosis. Infect. Immun. 745667-5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skorvaga, M., K. Theis, B. S. Mandavilli, C. Kisker, and B. Van Houten. 2002. The beta-hairpin motif of UvrB is essential for DNA binding, damage processing, and UvrC-mediated incisions. J. Biol. Chem. 2771553-1559. [DOI] [PubMed] [Google Scholar]
- 47.Smalley, J. W., A. J. Birss, and J. Silver. 2000. The periodontal pathogen Porphyromonas gingivalis harnesses the chemistry of the mu-oxo bishaem of iron protoporphyrin IX to protect against hydrogen peroxide. FEMS Microbiol. Lett. 183159-164. [DOI] [PubMed] [Google Scholar]
- 48.Smalley, J. W., J. Silver, P. J. Marsh, and A. J. Birss. 1998. The periodontopathogen Porphyromonas gingivalis binds iron protoporphyrin IX in the mu-oxo dimeric form: an oxidative buffer and possible pathogenic mechanism. Biochem. J. 331681-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sztukowska, M., M. Bugno, J. Potempa, J. Travis, and D. M. Kurtz, Jr. 2002. Role of rubrerythrin in the oxidative stress response of Porphyromonas gingivalis. Mol. Microbiol. 44479-488. [DOI] [PubMed] [Google Scholar]
- 50.Tchou, J., M. L. Michaels, J. H. Miller, and A. P. Grollman. 1993. Function of the zinc finger in Escherichia coli Fpg protein. J. Biol. Chem. 26826738-26744. [PubMed] [Google Scholar]
- 51.Thompson, S. A., R. L. Latch, and J. M. Blaser. 1998. Molecular characterization of the Helicobacter pylori uvrB gene. Gene 209113-122. [DOI] [PubMed] [Google Scholar]
- 52.Ueshima, J., M. Shoji, D. B. Ratnayake, K. Abe, S. Yoshida, K. Yamamoto, and K. Nakayama. 2003. Purification, gene cloning, gene expression, and mutants of Dps from the obligate anaerobe Porphyromonas gingivalis. Infect. Immun. 711170-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vanterpool, E., F. Roy, and H. M. Fletcher. 2004. The vimE gene downstream of vimA is independently expressed and is involved in modulating proteolytic activity in Porphyromonas gingivalis W83. Infect. Immun. 725555-5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viswanathan, M., V. Burdett, C. Baitinger, P. Modrich, and S. T. Lovett. 2001. Redundant exonuclease involvement in Escherichia coli methyl-directed mismatch repair. J. Biol. Chem. 27631053-31058. [DOI] [PubMed] [Google Scholar]
- 55.Wang, Y. 2008. Bulky DNA lesions induced by reactive oxygen species. Chem. Res. Toxicol. 21276-281. [DOI] [PubMed] [Google Scholar]
- 56.Wienkoop, S., and W. Weckwerth. 2006. Relative and absolute quantitative shotgun proteomics: targeting low-abundance proteins in Arabidopsis thaliana. J. Exp. Bot. 571529-1535. [DOI] [PubMed] [Google Scholar]
- 57.Wood, R. D. 1996. DNA repair in eukaryotes. Annu. Rev. Biochem. 65135-167. [DOI] [PubMed] [Google Scholar]
- 58.Wyrzykowski, J., and M. R. Volkert. 2003. The Escherichia coli methyl-directed mismatch repair system repairs base pairs containing oxidative lesions. J. Bacteriol. 1851701-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yasui, A., and S. J. McCready. 1998. Alternative repair pathways for UV-induced DNA damage. Bioessays 20291-297. [DOI] [PubMed] [Google Scholar]
- 60.Zhang, L., Y. Lun, D. Yan, L. Yu, W. Ma, B. Du, and X. Zhu. 2007. Proteomic analysis of macrophages: a new way to identify novel cell-surface antigens. J. Immunol. Methods 32180-85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.