

Abstract
Cholera, a severe diarrheal disease, is caused by ingestion of the gram-negative bacterium Vibrio cholerae. Expression of V. cholerae virulence factors is highly regulated at the transcriptional and posttranscriptional levels by a complex network of proteins and small noncoding RNAs. The direct activator of transcription of most V. cholerae virulence genes is the ToxT protein. ToxT binds to a 13-bp sequence, the toxbox, located upstream of genes in its regulon. However, the organization of toxboxes relative to each other and to the core promoter elements at different genes varies dramatically. At different ToxT-activated genes a single toxbox may be necessary and sufficient for full activation, or pairs of toxboxes organized as either inverted or direct repeats may be required for full activation. Although all toxboxes are located at positions consistent with a class I promoter architecture, the locations of toxboxes relative to the transcription start site also vary from gene to gene. To further assess the ability of ToxT to activate transcription from different configurations relative to the core promoter elements, we constructed promoter-lacZ fusions having altered spacing both between toxbox pairs and between the promoter-proximal toxbox and the −35 box at five different ToxT-activated promoters. Our results suggest that that ToxT has remarkable flexibility in its positioning as a transcription activator and that different interactions between ToxT and RNA polymerase occur during transcription activation of promoters having different toxbox configurations.
The gram-negative bacterium Vibrio cholerae, the causative agent of the severe diarrheal disease cholera, continues to be a serious public health problem despite over 100 years of research on its pathogenesis. The current cholera pandemic, which began in 1961, continues unabated, and an estimated 5 million cases of cholera occur worldwide each year. Although over 200 known serogroups of V. cholerae have been identified, only the O1 and O139 serogroups are capable of causing pandemic cholera (34, 35).
Pandemic V. cholerae strains require two major virulence factors for colonization and pathogenesis. The first factor, cholera toxin, is an A1B5-type ADP-ribosylating toxin that is responsible for producing the voluminous watery diarrhea characteristic of cholera (8, 26). The genes encoding cholera toxin, ctxAB, are carried in the genome of a lysogenic bacteriophage, CTXΦ (40). The second major virulence factor is the toxin-coregulated pilus (TCP) (38), a type IV pilus that initiates microcolony formation and is required for intestinal colonization by V. cholerae (39). Genes encoding the TCP, which are in a long operon beginning with the tcpA pilin gene, are located in the Vibrio pathogenicity island (VPI) (22). In addition to its role as a colonization factor, the TCP is also the receptor for CTXΦ and thus allows horizontal transfer of ctxAB to nontoxigenic V. cholerae carrying the VPI (40). In addition to the TCP-encoding genes, several other genes thought to be involved in pathogenesis are located in the VPI (1). These genes encode the accessory colonization factors (acfA, acfB, acfC, and acfD) (6, 17, 18), a putative lipoprotein (tagA) (9), an aldehyde dehydrogenase (aldA) (32), and a putative methyl-accepting chemotaxis protein (tcpI) (10).
The expression of V. cholerae virulence genes is highly regulated. A cascade of both positive and negative transcription regulators is involved in initiating expression of all of the genes mentioned above (29). Furthermore, some gene products in the regulatory cascade are further controlled posttranscriptionally (30). Historically, the virulence regulon is referred to as the ToxR regulon. However, the direct transcriptional activator of ctxAB, the tcpA operon, the acfABCD genes, aldA, tagA, and tcpI, is a 32-kDa protein, ToxT. The combined actions of the transcription activators ToxR and TcpP are required for expression of ToxT (5,11-13, 23, 24).
ToxT is a member of the large AraC/XylS family of transcription regulators (14). The C-terminal 100 amino acids of ToxT form the AraC/XylS homology domain (7, 27) and likely bind to DNA, whereas the remaining 176 amino acids form an N-terminal domain that has no homology to any other protein based on BLAST and PSI-BLAST searches and protein threading programs. ToxT binds to a degenerate 13-bp DNA site, the toxbox, which is located upstream of all the genes in its regulon (41). The toxbox consists primarily of a T tract toward the 5′ end and A/T-rich sequence in the 3′ half. Mutagenesis of every base pair in the ToxT binding region upstream of tcpA indicated that as few as 5 bp and at most 7 bp in one 13-bp toxbox have sequence-specific roles in activation by ToxT (41). Furthermore, in addition to having sequence diversity, toxboxes also have diverse configurations relative to the core promoter elements (Fig. 1). Upstream of aldA, a single toxbox is required for transcription activation (42). Upstream of tcpA, two toxboxes configured as a direct repeat are required for transcription activation (41). Upstream of tagA, two toxboxes configured as an inverted repeat are required (42). Between acfA and acfD, two toxboxes configured as an inverted repeat are required for transcription activation of both genes (43). The acfA-acfD inverted repeat differs from the tagA inverted repeat both in the spacing between the toxboxes and in the spacing between the promoter-proximal toxbox and the −35 box (Fig. 1).
FIG. 1.

Promoter architecture and design of spacing mutations. The numbers indicate distances from the transcription start site (indicated by a bent arrow). The numbers for the acfA promoter are underlined to distinguish them from the numbers for the acfD promoter. The boxes indicate the positions of −35 and −10 promoter elements. The arrows below the thick black lines indicate the positions of toxboxes at each promoter. The open triangles indicate the locations of insertion or deletion mutations. “+/−p” indicates that insertions or deletions were made between the promoter-proximal toxbox and the −35 box, and “+/−t” indicates that insertions or deletions were made upstream of the toxbox (for aldA) or between the toxboxes.
Based on this diversity in DNA binding site configurations, we have proposed that ToxT binds to independent toxboxes as a monomer (41, 43). This hypothesis was tested by inserting 5 and 10 bp between either the acfA and acfD toxboxes or the tcpA toxboxes and assessing the occupancy of each toxbox using copper-phenanthroline (CP) footprinting. We found that ToxT bound to toxboxes regardless of their location relative to each other at promoters having either a direct repeat (tcpA) or an inverted repeat (acfA-acfD) configuration (41, 43). This strongly suggested that ToxT binds to individual toxboxes as an independent monomer. However, the insertions between toxboxes abrogated transcription activation by ToxT, suggesting that the spacing of toxboxes relative to each other is important for ToxT function. Furthermore, insertion of 5 or 10 bp between the promoter-proximal toxbox and the −35 box at the tcpA promoter also abrogated transcription activation by ToxT (unpublished data), suggesting that the spacing of the toxboxes relative to the promoter is important for activation. There is evidence from domain swapping experiments that ToxT N-terminal domains can associate with each other to form dimers or multimers (33, 37), but full-length ToxT was not observed to dimerize in such an assay (37). It is possible that ToxT monomers associate with each other at some promoters and that this association is important for transcription activation.
In this work, we further assessed the importance of toxbox spacing for ToxT-mediated transcription activation. We constructed promoter-lacZ fusions having altered spacing between toxboxes at four different dual toxbox promoters and between the promoter-proximal toxbox and the −35 box at five promoters. Our findings indicate that each promoter has an optimal spacing for activation by ToxT that is equal to or close to the wild-type spacing, although the optimal spacing is different for different promoters. We propose that at least three different types of interactions between ToxT and RNA polymerase (RNAP) occur in transcription activation depending on the ToxT binding site position and orientation relative to the promoter and that these interaction types differ in their sensitivity to spacing alterations.
MATERIALS AND METHODS
V. cholerae strains and plasmids.
The strains used experimentally in this work were classical V. cholerae strain O395 and derivatives of this strain. The strains were grown in LB medium at 37°C (overnight cultures) or in LB medium with a starting pH of 6.5 at 30°C (inducing conditions). The strains used for β-galactosidase assays were O395 and an O395 ΔtoxT derivative (VJ740) carrying the previously specified promoter fusion in plasmid pTL61T (3). Spacing mutations were created using the splicing-by-overlap-extension technique (15, 16), and all promoter constructs were cloned between the HindIII and XbaI sites of pTL61T (25). Plasmid sequences were confirmed by the University of Michigan DNA sequencing core. Antibiotics were used at the following concentrations: ampicillin, 100 μg/ml; and streptomycin, 100 μg/ml. V. cholerae was transformed with plasmid DNA by electroporation using a Bio-Rad MicroPulser.
DNA manipulation.
Plasmids were purified using a Promega Wizard Plus Miniprep kit. PCR was performed using Taq DNA polymerase (Denville Scientific) as specified by the manufacturer and an Eppendorf Mastercycler gradient. Restriction enzymes were purchased from New England Biolabs and were used as specified by the manufacturer.
β-Galactosidase assays.
Strains were cultured overnight in LB medium at 37°C and then subcultured using a 1/50 dilution in inducing medium and grown for 3 h at 30°C with vigorous aeration. The bacteria were then placed on ice, and 0.5 mg ml−1 chloramphenicol was added. β-Galactosidase assays were performed using the basic procedure of Miller as previously described (31).
CP footprinting.
CP footprinting was performed as previously described (41-43). Chemical cleavage was done in a gel after separation of free DNA and a bound ToxT-DNA complex by using an electrophoretic mobility shift assay. DNA was produced by PCR after one primer was end labeled with 32P using T4 polynucleotide kinase (New England Biolabs). Polyhistidine-tagged ToxT was purified as previously described (45). The ratio of ToxT to DNA used was adjusted such that approximately 50% of the labeled DNA formed a bound complex with ToxT.
RESULTS
Design of spacing variants.
Previous work indicated that insertion of 5 bp and insertion of 10 bp between the toxboxes, resulting in rotation of approximately one half turn and one full turn of the DNA helix, respectively, at the tcpA and acfA-acfD promoters abrogated transcription activation by ToxT, although the toxboxes were still occupied by ToxT in DNA footprinting experiments (41, 43). Insertion of 5 bp and insertion of 10 bp between the promoter-proximal toxbox and the −35 box at the tcpA promoter, resulting in rotation of approximately one half turn and one full turn of the DNA helix, respectively, between both toxboxes and the core promoter elements also abrogated activation by ToxT (unpublished data). Because there is natural variation in the positions of toxboxes at different promoters (Fig. 1), we constructed variant promoter regions in which the spacing either between toxboxes (+t or −t) or between the promoter-proximal toxbox and the −35 box (+p or −p) was altered in 1-bp increments by either insertion or deletion (Fig. 1). The resulting promoters were fused to lacZ in pTL61T (25) and then tested for activation by ToxT by comparing β-galactosidase levels in classical V. cholerae strains having wild-type toxT or a toxT deletion.
Effects of spacing mutations on aldA activation.
The aldA promoter is the only known promoter in which a single toxbox is necessary and sufficient for activation by ToxT (42), although ToxT may act as a repressor at other single toxboxes (J. H. Withey, unpublished data). Therefore, insertions upstream of the toxbox should have no effect on the level of activation conferred by ToxT. As shown in Fig. 2A, this was indeed the case. However, alteration of the spacing between the toxbox and the −35 box by even 1 bp abrogated activation of aldA transcription by ToxT. The aldA toxbox is in the orientation opposite that of toxboxes at similar locations in other promoters (at aldA the toxbox “points toward” the promoter [Fig. 1]), so these results suggest that a single toxbox in this orientation is extremely sensitive to spacing changes. aldA transcription is also relatively weakly activated by ToxT even with wild-type spacing of the toxbox and promoter (Fig. 2A).
FIG. 2.

Effects of promoter spacing mutations on β-galactosidase expression of promoter spacing mutants. The gray bars indicate the results for wild-type toxT V. cholerae strains (WT), and the open bars indicate the results for ΔtoxT strains. The number above each pair of bars indicates the difference in β-galactosidase expression between the wild-type toxT and ΔtoxT strains. The bars indicate the means of at least three separate experiments, and the error bars indicate standard deviations. (A) aldA-lacZ fusions. (B) tcpA-lacZ fusions. (C) acfA-lacZ fusions. (D) acfD-lacZ fusions. (E) tagA-lacZ fusions.
Effects of spacing mutations on tcpA activation.
Two toxboxes in a direct repeat configuration are required for activation of the tcpA promoter by ToxT (41). Transcription of tcpA is very strongly activated by ToxT, as might be expected given the crucial role of the TCP in pathogenesis (39). In contrast to what we observed for the aldA promoter, activation of tcpA transcription by ToxT was observed despite considerable changes in spacing between the proximal toxbox and the −35 box. Constructs having up to 3-bp insertions and a 1-bp deletion were significantly activated by ToxT (Fig. 2B). Insertion of 4 or 5 bp abrogated activation of tcpA by ToxT (data not shown). The difference in spacing between the tcpA +3p and tcpA −1p constructs would be rotation of approximately 137° and a distance of 1.32 nm given B-form DNA structure. Thus, altering the spacing between the tcpA toxboxes and the −35 box while retaining wild-type spacing between tcpA toxboxes was tolerated to a large degree.
Changes in the spacing between the toxboxes at tcpA were less well tolerated. A constructs having a 1-bp insertion and a construct having a 1-bp deletion between the toxboxes were activated 13- and 26-fold, respectively, and a construct having a 2-bp deletion between the toxboxes was activated 8.3-fold. Any other changes in spacing abrogated activation by ToxT. Therefore, movement of the promoter-distal toxbox relative to the rest of the tcpA promoter caused a greater relative defect in activation by ToxT than movement of both toxboxes together.
Effects of spacing mutations on the acfA and acfD promoters.
The divergently transcribed acfA and acfD promoters have a central pair of toxboxes between them that are required for transcription activation of both genes by ToxT (43). However, the spacing of these toxboxes relative to the two core promoters differs (Fig. 1). The acfD promoter-proximal toxbox 5′ end is located at position −46, whereas the acfA promoter-proximal toxbox 5′ end is located at position −51, a difference of half a turn of the DNA helix. Given this natural difference, we expected to see significant flexibility in the toxbox-promoter spacing with which ToxT could effectively activate transcription of acfA and acfD. However, this was not the case. The two promoters behaved very similarly when base pairs were inserted between the proximal toxbox and the −35 box (Fig. 2C and 2D). A 1-bp insertion had no effect (acfD) or resulted in an even higher level of activation by ToxT (acfA). A 2-bp insertion decreased the level of activation of both promoters, and a 3-bp insertion abrogated activation by ToxT. A difference between the acfA and acfD promoters was observed with the deletion constructs. acfD was slightly activated by ToxT (3.5-fold) when 1 bp was deleted between the proximal toxbox and the −35 box, but further deletions abrogated activation. Deletions between the acfA-proximal toxbox and the −35 box resulted in high basal transcription levels with no activation conferred by ToxT. Instead, these constructs were slightly repressed by ToxT. acfA constructs having additional deletions (4 and 5 bp) were slightly activated by ToxT. It is notable that the acfA construct having a 5-bp deletion between the proximal toxbox and the −35 box had the same spacing as the wild-type acfD promoter, yet it was very poorly activated by ToxT. In the same vein, a construct having a 5-bp insertion between the acfD-proximal toxbox and the −35 box was not activated by ToxT (data not shown). This construct mimicked the spacing at the wild-type acfA promoter, yet this spacing was not functional for transcription activation of acfD by ToxT.
Changes in spacing between the acfA and acfD toxboxes were not as well tolerated by ToxT. Both acfA and acfD constructs having 1-bp insertions between the toxboxes were still activated by ToxT, albeit to a lesser degree for acfD. Any larger insertion abrogated activation. Deletion of one or both of the two base pairs between the acfA and acfD toxboxes resulted in no activation of either gene by ToxT. Thus, as observed for the tcpA promoter, changes in spacing between the promoter-proximal toxbox and the core promoter elements had less effect on activation by ToxT than changes in spacing between the toxboxes.
Effects of spacing mutations on the tagA promoter.
ToxT requires a pair of toxboxes in an inverted repeat configuration to activate tagA transcription (42). The spacing between the tagA inverted repeat toxboxes (8 bp) differs from the spacing between the acfA and acfD inverted repeat toxboxes (2 bp) (Fig. 1). As we observed for the tcpA promoter, ToxT was able to activate transcription of tagA constructs having a variety of spacing changes between the promoter-proximal toxbox and the −35 box (Fig. 2E). Constructs having 1- or 2-bp insertions or a 1-bp deletion were activated by ToxT at levels equal to or greater than the level observed with wild-type tagA spacing.
Constructs having insertions between the tagA toxboxes also exhibited a greater defect in activation by ToxT. β-Galactosidase expression from a construct having a 1-bp insertion and from a construct having a 1-bp deletion between the toxboxes was activated at relatively low levels by ToxT (4.4- and 3.4-fold, respectively). Further changes in spacing between the toxboxes abrogated activation of tagA by ToxT. This sensitivity to changes in spacing between the toxboxes exhibited by ToxT at tagA is similar to what we observed with the acfA and acfD promoters, suggesting that the inverted repeat toxbox configuration, regardless of the wild-type spacing between toxboxes, is not as tolerant of spacing changes as the direct repeat configuration at tcpA.
CP footprinting of ToxT on spacing variants.
To examine the ToxT occupancy of toxboxes with changes in the spacing between them, CP footprinting was performed. Previously, we found that 5- or 10-bp insertions between direct repeat or inverted repeat toxboxes did not affect the footprinting pattern of ToxT, although these insertions abrogated transcription activation by ToxT (41, 43). As shown in Fig. 3, we footprinted ToxT on the acfD +5t, acfD −2t, tcpA +4t, and tagA +3t fusion constructs. All of these constructs exhibited defects in ToxT activation (Fig. 2). The CP footprinting data obtained for the acfD +5t, tcpA +4t, and tagA +3t constructs indicate that in each case ToxT occupied both toxboxes regardless of their position relative to each other, as we previously observed with the acfD and tcpA promoters (41, 43). These results suggest that rotating the toxboxes by insertional mutation does not disrupt ToxT binding and also suggest that there is independent binding of ToxT to individual toxboxes. The CP footprinting data obtained for the acfD −2t construct revealed a different pattern of binding (Fig. 3). The acfD-distal toxbox was clearly protected by ToxT, whereas the acfD-proximal toxbox was more weakly protected by ToxT. Because the acfD −2t construct eliminated the only two base pairs between the toxboxes, it is likely that ToxT monomers impeded binding of each other on this construct through steric hindrance. In this case the observed defect in ToxT-activated transcription was likely due to the absence of ToxT bound to the acfD-proximal promoter.
FIG. 3.
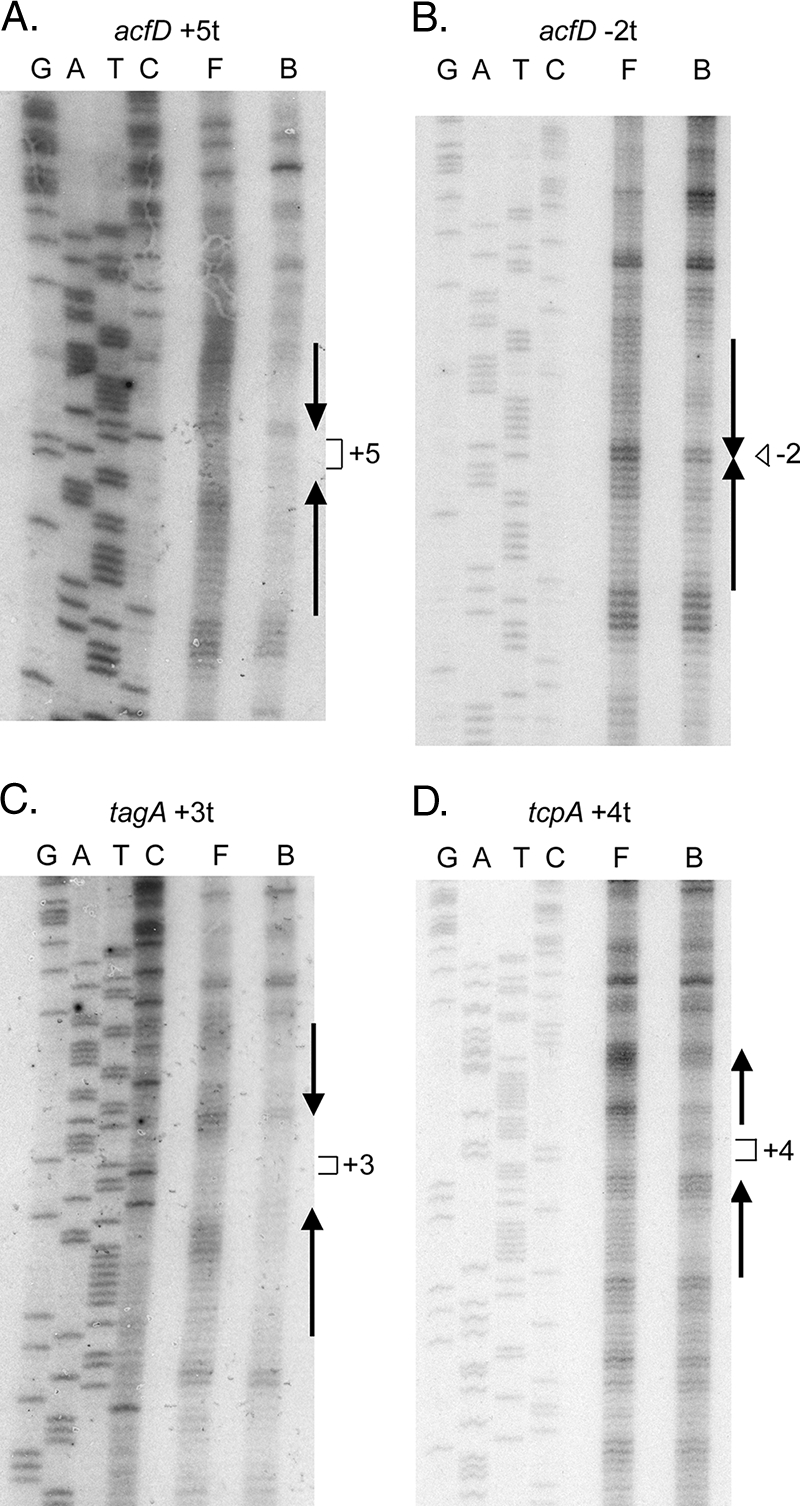
CP footprinting of ToxT on spacing variants. Experiments were performed using histidine-tagged ToxT and purified end-labeled DNA as described in Materials and Methods. Lanes G, A, T, and C are the sequencing reaction lanes used for size markers, lane F contained free DNA, and lane B contained DNA complexed with ToxT. The numbers indicate the positions of inserted or deleted DNA sequence. (A) CP footprinting for the acfD +5t construct. (B) CP footprinting for the acfD −2t construct. (C) CP footprinting for the tagA +3t construct. (D) CP footprinting for the tcpA +4t construct.
DISCUSSION
The experiments described in this paper examined the ability of ToxT to activate transcription of V. cholerae virulence gene promoters having altered spacing between the toxboxes and the core promoter elements. Our results indicate that different toxbox configurations have different spacing requirements for ToxT activity. This finding suggests that the interactions between ToxT and RNAP involved in transcription activation differ depending on the toxbox configuration. All known ToxT-activated promoters have a class I architecture, suggesting that ToxT interacts with the α C-terminal domain (α-CTD) of RNAP (2). Overexpression of a mutant α subunit lacking the CTD resulted in a loss of tcpA activation by ToxT, strongly suggesting that this interaction is essential for transcription activation (19).
The promoter-proximal toxboxes at the acfD, tcpA and tagA promoters are located at similar positions and in the same orientation relative to the core promoter, and the differences between these promoters are the orientation of the promoter-distal toxbox and the spacing between the toxboxes (Fig. 1). ToxT was able to activate constructs from all three promoters having significant changes in spacing between the promoter-proximal toxbox and the −35 box. One explanation for this similarity is that the interactions between the ToxT monomer bound to the promoter-proximal toxbox and the RNAP α-CTD are similar at these three promoters. A caveat is that insertions or deletions between the core promoter and the proximal toxbox alter the spacing of both toxboxes relative to the promoter, so such changes could potentially affect interactions between both ToxT monomers bound to the two toxboxes and both α-CTD subunits. Further similarity between the acfD, tcpA, and tagA promoters was found in previous work with the ToxT antagonist virstatin (20), which interacts with the ToxT N-terminal domain and may inhibit interaction between monomers (37). These three promoters were found to be the promoters most sensitive to treatment with virstatin.
Altering the spacing between the toxboxes had different effects on the inverted repeat and direct repeat promoters. Transcription activation by ToxT from the inverted repeat toxbox pairs at acfD and tagA was severely reduced by a 1-bp insertion or deletion between the toxboxes, and a 2-bp insertion or deletion resulted in a complete loss of activation by ToxT. At the acfA inverted repeat toxbox pair, ToxT was able to activate a promoter having a 1-bp insertion between the toxboxes, but any other changes resulted in a loss of activation. Thus, the inverted repeat promoters are very sensitive to altered spacing between toxboxes. If ToxT monomers dimerize and this interaction is required for transcription activation, insertions between toxboxes could interfere; these changes in spacing could also interfere with interactions between ToxT bound to the distal toxbox and the RNAP α-CTD. Changes in spacing between the direct repeat toxbox pair at tcpA had less severe effects. ToxT activated transcription from constructs having a 1-bp insertion and a 1-bp deletion between the toxboxes 13- and 26-fold, respectively, and activated a construct having a 2-bp deletion 8.3-fold. This difference in sensitivity to changes in spacing between the inverted repeat and direct repeat promoters suggests that the interactions between ToxT bound to the promoter-distal toxbox and RNAP are similar at the acfA, acfD, and tagA promoters and different at the tcpA promoter, as we have previously proposed (41).
The promoter that has the lowest amplitude of activation by ToxT, aldA, was also the promoter most sensitive to any spacing changes. The aldA promoter is the only known promoter at which ToxT requires a single binding site for activation, and any change in spacing between this toxbox and the −35 box eliminated activation by ToxT. Spacing changes upstream of this toxbox had no effect, confirming that ToxT requires only this single site for activation of aldA. In addition to the presence of a single toxbox, the orientation of the aldA toxbox is opposite that of other promoter-proximal toxboxes. This suggests that the interactions between ToxT and RNAP involved in activation of aldA are different than those at other promoters having a toxbox at a similar position.
Based on the results described above, we propose that ToxT interacts with RNAP in at least three different ways based on the position and orientation of the toxboxes and that these interactions have different spacing requirements (Fig. 4). A type 1 interaction occurs between the RNAP α-CTD and ToxT at a promoter-proximal toxbox oriented so that it “points away” from the promoter. This is the case for tcpA, tagA, and acfD. The type 1 interaction is relatively spacing insensitive and may involve an interaction between α-CTD and ToxT that is not localized to the DNA, as has been observed with some other AraC family members (4, 36). The type 1 interaction is very virstatin sensitive. A type 2 interaction between α-CTD and ToxT occurs at toxboxes oriented so that they “point toward” the promoter. This is the case for the inverted repeat toxbox promoters acfA, acfD, and tagA, as well as the single toxbox promoter aldA. The type 2 interaction is very spacing sensitive, as a difference of even 1 bp from the wild-type spacing abrogates transcription activation at some promoters. However, the acfA and aldA promoters were the promoters least sensitive to virstatin treatment (37), suggesting that the type 2 interaction is virstatin insensitive. A type 3 interaction occurs between α-CTD and ToxT at a distal toxbox oriented so that it “points away” from the promoter. This occurs at the tcpA promoter and has intermediate spacing sensitivity, as insertion or deletion of 1 bp is tolerated relatively well but larger changes are not well tolerated. In terms of activation by ToxT, the tcpA promoter, having type 1 and type 3 interactions, is most highly activated, the three promoters having type 1 and type 2 interactions are activated at intermediate levels, and the aldA promoter having only a type 2 interaction is weakly activated.
FIG. 4.

Model showing three types of interactions between ToxT and RNAP in transcription activation at promoters having different toxbox configurations. The open ellipses represent ToxT, the arrows represent toxboxes, and the shaded ellipses represent the RNAP α-CTDs. −35 and −10 boxes are represented by open rectangles, and the transcription start site is indicated by a bent arrow.
One promoter that does not fit as well into this simple scheme is acfA. acfA has a promoter-proximal toxbox that is located approximately 5 bp further upstream than is typical for other ToxT-activated promoters, and spacing alteration affects it differently than it affects the other promoters. Surprisingly, changing the spacing between the proximal acfA toxbox and the −35 box to resemble the spacing for more typical promoters did not result in a construct that was activated well by ToxT. This was despite the fact that the same toxbox pair is used by ToxT to activate acfD transcription with the more typical spacing. Previously, we observed that mutations in the toxbox pair between acfA and acfD had very similar effects on transcription activation of both promoters by ToxT (43). The results described here suggest that the DNA sequences of the individual toxboxes may play a further role in activation. Mutagenesis of every base pair within the tcpA toxboxes identified 7 of 13 bp that were important for activation of this promoter (41), but there may be additional sequence requirements for toxboxes at promoters having different configurations, such as acfA.
Much previous work in many laboratories on spacing requirements of class I promoters has focused on the effects of inserting or deleting a half or full turn of the DNA helix, although a few studies similar to this study have been described previously. The most relevant example is the MarA protein, another member of the AraC/XylS family that binds to DNA as a monomer. MarA is able to activate transcription from both class I and class II promoters depending on the location and orientation of its binding sites (27). Studies on the spacing between a MarA binding site and the −35 box required for transcription activation of a class I promoter found that the wild-type spacing (16 bp) is optimal and that there was a gradual decrease in activation amplitude as base pairs were either inserted or deleted to alter the spacing (28). Interestingly, the observed spacing range with which MarA was able to activate transcription is similar to the range for the ToxT-activated promoters described here as having a type 1 interaction with RNAP α-CTD. SoxS, a transcription activator closely related to MarA that recognizes the same DNA binding site, was found to require one particular orientation of its binding site when located close to the promoter and the opposite orientation of its binding site when located further upstream (44). This finding clearly differs from our observation that ToxT can use binding sites in either orientation at similar locations relative to the core promoter. A study of Escherichia coli CytR, a repressor in the LacI family, showed that CytR was able to function as a repressor when its individual binding sites were separated by between 1 and 16 bp in 2-bp increments (21). CytR binds to DNA as a dimer, and differences in the affinity of CytR for its binding sites correlated with the degree of repression observed with various spacings. We have previously observed that ToxT binds to DNA as a monomer and that rotating its binding sites by even a half turn of the DNA did not affect their occupancy by ToxT (41, 43).
In summary, we analyzed the promoter spacing requirements for V. cholerae ToxT to activate transcription of virulence genes. Our results indicate that different interactions are likely to occur between ToxT and RNAP depending on the position and orientation of ToxT binding sites relative to each other and relative to the −35 box and that these ToxT-RNAP interactions differ in their spacing flexibility.
Acknowledgments
We thank the members of the Withey and Neely laboratories at Wayne State University School of Medicine for helpful discussions.
This work was supported by Public Health Service grant 1K22AI071011 from the National Institute of Allergy and Infectious Diseases (to J.H.W.) and by startup funds from Wayne State University.
Footnotes
Published ahead of print on 10 October 2008.
REFERENCES
- 1.Brown, R. C., and R. K. Taylor. 1995. Organization of tcp, acf, and toxT genes within a ToxT-dependent operon. Mol. Microbiol. 16425-439. [DOI] [PubMed] [Google Scholar]
- 2.Busby, S., and R. H. Ebright. 1994. Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell 79743-746. [DOI] [PubMed] [Google Scholar]
- 3.Champion, G. A., M. N. Neely, M. A. Brennan, and V. J. DiRita. 1997. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol. Microbiol. 23323-331. [DOI] [PubMed] [Google Scholar]
- 4.Dangi, B., A. M. Gronenborn, J. L. Rosner, and R. G. Martin. 2004. Versatility of the carboxy-terminal domain of the alpha subunit of RNA polymerase in transcriptional activation: use of the DNA contact site as a protein contact site for MarA. Mol. Microbiol. 5445-59. [DOI] [PubMed] [Google Scholar]
- 5.DiRita, V. J., C. Parsot, G. Jander, and J. J. Mekalanos. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 885403-5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Everiss, K. D., K. J. Hughes, M. E. Kovach, and K. M. Peterson. 1994. The Vibrio cholerae acfB colonization determinant encodes an inner membrane protein that is related to a family of signal-transducing proteins. Infect. Immun. 623289-3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallegos, M. T., R. Schleif, A. Bairoch, K. Hofmann, and J. L. Ramos. 1997. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61393-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill, D. M. 1976. The arrangement of subunits in cholera toxin. Biochemistry 151242-1248. [DOI] [PubMed] [Google Scholar]
- 9.Harkey, C. W., K. D. Everiss, and K. M. Peterson. 1995. Isolation and characterization of a Vibrio cholerae gene (tagA) that encodes a ToxR-regulated lipoprotein. Gene 15381-84. [DOI] [PubMed] [Google Scholar]
- 10.Harkey, C. W., K. D. Everiss, and K. M. Peterson. 1994. The Vibrio cholerae toxin-coregulated-pilus gene tcpI encodes a homolog of methyl-accepting chemotaxis proteins. Infect. Immun. 622669-2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hase, C. C., and J. J. Mekalanos. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 95730-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higgins, D. E., and V. J. DiRita. 1994. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol. Microbiol. 1417-29. [DOI] [PubMed] [Google Scholar]
- 13.Higgins, D. E., and V. J. DiRita. 1996. Genetic analysis of the interaction between Vibrio cholerae transcription activator ToxR and toxT promoter DNA. J. Bacteriol. 1781080-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins, D. E., E. Nazareno, and V. J. DiRita. 1992. The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J. Bacteriol. 1746974-6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horton, R. M., S. N. Ho, J. K. Pullen, H. D. Hunt, Z. Cai, and L. R. Pease. 1993. Gene splicing by overlap extension. Methods Enzymol. 217270-279. [DOI] [PubMed] [Google Scholar]
- 16.Horton, R. M., H. D. Hunt, S. N. Ho, J. K. Pullen, and L. R. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 7761-68. [DOI] [PubMed] [Google Scholar]
- 17.Hughes, K. J., K. D. Everiss, M. E. Kovach, and K. M. Peterson. 1995. Isolation and characterization of the Vibrio cholerae acfA gene, required for efficient intestinal colonization. Gene 15659-61. [DOI] [PubMed] [Google Scholar]
- 18.Hughes, K. J., K. D. Everiss, M. E. Kovach, and K. M. Peterson. 1994. Sequence analysis of the Vibrio cholerae acfD gene reveals the presence of an overlapping reading frame, orfZ, which encodes a protein that shares sequence similarity to the FliA and FliC products of Salmonella. Gene 14679-82. [DOI] [PubMed] [Google Scholar]
- 19.Hulbert, R. R., and R. K. Taylor. 2002. Mechanism of ToxT-dependent transcriptional activation at the Vibrio cholerae tcpA promoter. J. Bacteriol. 1845533-5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hung, D. T., E. A. Shakhnovich, E. Pierson, and J. J. Mekalanos. 2005. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310670-674. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen, C. I., B. H. Kallipolitis, and P. Valentin-Hansen. 1998. DNA-binding characteristics of the Escherichia coli CytR regulator: a relaxed spacing requirement between operator half-sites is provided by a flexible, unstructured interdomain linker. Mol. Microbiol. 2741-50. [DOI] [PubMed] [Google Scholar]
- 22.Karaolis, D. K., J. A. Johnson, C. C. Bailey, E. C. Boedeker, J. B. Kaper, and P. R. Reeves. 1998. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc. Natl. Acad. Sci. USA 953134-3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krukonis, E. S., and V. J. DiRita. 2003. DNA binding and ToxR responsiveness by the wing domain of TcpP, an activator of virulence gene expression in Vibrio cholerae. Mol. Cell 12157-165. [DOI] [PubMed] [Google Scholar]
- 24.Krukonis, E. S., R. R. Yu, and V. J. DiRita. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol. Microbiol. 3867-84. [DOI] [PubMed] [Google Scholar]
- 25.Linn, T., and R. St. Pierre. 1990. Improved vector system for constructing transcriptional fusions that ensures independent translation of lacZ. J. Bacteriol. 1721077-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lonnroth, I., and J. Holmgren. 1973. Subunit structure of cholera toxin. J. Gen. Microbiol. 76417-427. [DOI] [PubMed] [Google Scholar]
- 27.Martin, R. G., and J. L. Rosner. 2001. The AraC transcriptional activators. Curr. Opin. Microbiol. 4132-137. [DOI] [PubMed] [Google Scholar]
- 28.Martin, R. G., W. K. Gillette, S. Rhee, and J. L. Rosner. 1999. Structural requirements for marbox function in transcriptional activation of mar/sox/rob regulon promoters in Escherichia coli: sequence, orientation and spatial relationship to the core promoter. Mol. Microbiol. 34431-441. [DOI] [PubMed] [Google Scholar]
- 29.Matson, J. S., J. H. Withey, and V. J. DiRita. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun. 755542-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matson, J. S., and V. J. DiRita. 2005. Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 10216403-16408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 32.Parsot, C., and J. J. Mekalanos. 1991. Expression of the Vibrio cholerae gene encoding aldehyde dehydrogenase is under control of ToxR, the cholera toxin transcriptional activator. J. Bacteriol. 1732842-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prouty, M. G., C. R. Osorio, and K. E. Klose. 2005. Characterization of functional domains of the Vibrio cholerae virulence regulator ToxT. Mol. Microbiol. 581143-1156. [DOI] [PubMed] [Google Scholar]
- 34.Reidl, J., and K. E. Klose. 2002. Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol. Rev. 26125-139. [DOI] [PubMed] [Google Scholar]
- 35.Sack, D. A., R. B. Sack, G. B. Nair, and A. K. Siddique. 2004. Cholera. Lancet 363223-233. [DOI] [PubMed] [Google Scholar]
- 36.Shah, I. M., and R. E. Wolf, Jr. 2004. Novel protein-protein interaction between Escherichia coli SoxS and the DNA binding determinant of the RNA polymerase alpha subunit: SoxS functions as a co-sigma factor and redeploys RNA polymerase from UP-element-containing promoters to SoxS-dependent promoters during oxidative stress. J. Mol. Biol. 343513-532. [DOI] [PubMed] [Google Scholar]
- 37.Shakhnovich, E. A., D. T. Hung, E. Pierson, K. Lee, and J. J. Mekalanos. 2007. Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc. Natl. Acad. Sci. USA 1042372-2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor, R. K., V. L. Miller, D. B. Furlong, and J. J. Mekalanos. 1986. Identification of a pilus colonization factor that is coordinately regulated with cholera toxin. Ann. Sclavo Collana Monogr. 351-61. [PubMed] [Google Scholar]
- 39.Thelin, K. H., and R. K. Taylor. 1996. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 642853-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waldor, M. K., and J. J. Mekalanos. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 2721910-1914. [DOI] [PubMed] [Google Scholar]
- 41.Withey, J. H., and V. J. DiRita. 2006. The toxbox: specific DNA sequence requirements for activation of Vibrio cholerae virulence genes by ToxT. Mol. Microbiol. 591779-1789. [DOI] [PubMed] [Google Scholar]
- 42.Withey, J. H., and V. J. DiRita. 2005. Vibrio cholerae ToxT independently activates the divergently transcribed aldA and tagA genes. J. Bacteriol. 1877890-7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Withey, J. H., and V. J. DiRita. 2005. Activation of both acfA and acfD transcription by Vibrio cholerae ToxT requires binding to two centrally located DNA sites in an inverted repeat conformation. Mol. Microbiol. 561062-1077. [DOI] [PubMed] [Google Scholar]
- 44.Wood, T. I., K. L. Griffith, W. P. Fawcett, K. W. Jair, T. D. Schneider, and R. E. Wolf, Jr. 1999. Interdependence of the position and orientation of SoxS binding sites in the transcriptional activation of the class I subset of Escherichia coli superoxide-inducible promoters. Mol. Microbiol. 34414-430. [DOI] [PubMed] [Google Scholar]
- 45.Yu, R. R., and V. J. DiRita. 2002. Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol. Microbiol. 43119-134. [DOI] [PubMed] [Google Scholar]