

Abstract
A mutant poliovirus (PV) encoding a change in its polymerase (3Dpol) at a site remote from the catalytic center (G64S) confers reduced sensitivity to ribavirin and forms a restricted quasispecies, because G64S 3Dpol is a high-fidelity enzyme. A foot-and-mouth disease virus (FMDV) mutant that encodes a change in the polymerase catalytic site (M296I) exhibits reduced sensitivity to ribavirin without restricting the viral quasispecies. In order to resolve this apparent paradox, we have established a minimal kinetic mechanism for nucleotide addition by wild-type (WT) FMDV 3Dpol that permits a direct comparison to PV 3Dpol as well as to FMDV 3Dpol derivatives. Rate constants for correct nucleotide addition were on par with those of PV 3Dpol, but apparent binding constants for correct nucleotides were higher than those observed for PV 3Dpol. The A-to-G transition frequency was calculated to be 1/20,000, which is quite similar to that calculated for PV 3Dpol. The analysis of FMDV M296I 3Dpol revealed a decrease in the calculated ribavirin incorporation frequency (1/8,000) relative to that (1/4,000) observed for the WT enzyme. Unexpectedly, the A-to-G transition frequency was higher (1/8,000) than that observed for the WT enzyme. Therefore, FMDV selected a polymerase that increases the frequency of the misincorporation of natural nucleotides while specifically decreasing the frequency of the incorporation of ribavirin nucleotide. These studies provide a mechanistic framework for understanding FMDV 3Dpol structure-function relationships, provide the first direct analysis of the fidelity of FMDV 3Dpol in vitro, identify the β9-α11 loop as a (in)fidelity determinant, and demonstrate that not all ribavirin-resistant mutants will encode high-fidelity polymerases.
Viral polymerases are important targets for the therapeutic intervention of viral infections (3, 10, 15, 30, 33, 43). The best example is the reverse transcriptase from human immunodeficiency virus (5, 22, 23, 28, 47, 49). In most cases, (deoxy)nucleoside analogs that enter the clinic exploit the relaxed fidelity of viral polymerases relative to those of related cellular polymerases (3, 10, 15, 23, 28, 30, 33, 43, 47). The efficacy of an antiviral nucleoside is compromised by the development of resistance. In most cases, virus mutants that exhibit decreased sensitivity to antiviral nucleosides encode polymerases that exhibit the increased fidelity of substrate selection and/or incorporation (11, 24, 49). An understanding of the structural and kinetic bases for polymerase fidelity should reveal mechanisms that viruses will employ to escape the antiviral activity of (deoxy)nucleoside polymerase inhibitors. Importantly, this information may facilitate the development of antiviral nucleosides to which the emergence of viral resistance is less facile.
The purine analogue ribavirin (1-β-d-ribofuranosyl-1,2,4-triazole-carboxamide) is a synthetic antiviral ribonucleoside with activity against a broad spectrum of RNA viruses (26). Unlike most antiviral nucleosides, ribavirin does not function as a chain terminator. Ribavirin monophosphate (RMP) is a known inhibitor of IMP dehydrogenase, causing a twofold reduction in guanine nucleotide pools in cell culture (26). The inhibition of this cellular target may account for ribavirin's activity against some viruses (26). Ribavirin triphosphate (RTP) is the most abundant ribavirin metabolite in cells. RTP has been shown to act as an inhibitor and substrate of the viral RNA-dependent RNA polymerase (RdRp) (26). The utilization of RTP as a GTP analogue during the initiation of RNA synthesis can lead to uncapped transcripts that cannot be translated efficiently (26). The ambiguous utilization of RTP by the RdRp during genome replication leads to the lethal mutagenesis of the virus population (1, 13, 14, 48). Given the unique, and somewhat controversial, mechanisms of action proposed for this clinically important nucleoside, viral mutants capable of evading the antiviral activity of ribavirin should shed additional light on its mechanism of action and might suggest strategies for the design of more efficacious analogues of ribavirin (25, 26). Indeed, mutations in genes coding for functions other than that of the RdRp have been associated with ribavirin resistance (37).
The serial passage of poliovirus (PV) in cell culture in the presence of sublethal concentrations of ribavirin selected virus populations with decreased sensitivity to ribavirin, and the resistance was mapped to a Gly-to-Ser substitution at position 64 (G64S) of PV RdRp (3Dpol) (9, 38). PV G64S 3Dpol exhibits a threefold increase in nucleotide incorporation fidelity relative to that of wild-type (WT) 3Dpol, causing a corresponding reduction in viral quasispecies diversity (9, 36, 38, 45). Interestingly, the restricted quasispecies renders the resulting PV populations less adaptable to a complex biological environment and documents the behavior of the entire quasispecies as a unit of selection in vivo (36, 45, 46). The fact that the same, unstable PV mutant conferring reduced sensitivity to ribavirin was isolated by two laboratories (9, 38) suggests that few mechanisms exist to escape the antiviral activity of this compound. Whether or not this observation relates to the structure of the compound or its mechanism of action remains unclear.
Recently, we identified M296I as a change in 3Dpol of foot-and-mouth disease virus (FMDV) that confers decreased sensitivity to ribavirin (39). FMDV M296I 3Dpol became dominant in the population of two parallel lineages of FMDV passaged in the presence of ribavirin (but not in control populations passaged in the absence of ribavirin). Importantly, FMDV M296I 3Dpol incorporates RMP less efficiently than WT 3Dpol (39).
In contrast to Gly-64, which is remote from the catalytic site, Met-296 lies within the β9-α11 loop (residues 294 to 302) of the FMDV 3Dpol catalytic site (19) (Fig. 1). This loop could contribute to polymerase fidelity, given its interactions with the base of the incoming nucleotide as well as the template molecule (18). In order to elucidate the mechanistic basis for the decreased sensitivity of FMDV M296I 3Dpol, we have performed a detailed kinetic analysis of FMDV 3Dpol, yielding the first direct comparison of the minimal kinetic mechanism and the fidelity of nucleotide addition for two picornaviral polymerases from evolutionarily distant genera. Importantly, this comparative analysis revealed numerous differences in the biochemical properties of these enzymes that may have significance for the biology of these viruses. Consistently with our expectations, we find that FMDV M296I 3Dpol incorporated RMP less efficiently than WT FMDV 3Dpol. Surprisingly, FMDV M296I 3Dpol misincorporated GMP more efficiently than WT FMDV 3Dpol. Therefore, in contrast to the high-fidelity variant created by PV when faced with the antiviral activity of ribavirin, FMDV created a low-fidelity variant with a specific defect for ribavirin utilization. This observation suggests that counterintuitive solutions exist to the problem of the ribavirin antagonism of virus production. In addition, this study highlights the ability of the β9-α11 loop and/or elements with which it interacts to contribute to the (in)fidelity of nucleotide selection depending upon the nature of the nascent base pair.
FIG. 1.

Structure of FMDV 3Dpol-RNA-RTP catalytic complex. (A) FMDV 3Dpol is shown in gray, the template-primer molecule is shown in light green, and the RTP molecule is represented by dark green sticks. Loop β9-α11 of FMDV 3Dpol is shown in blue, and the residue Met-296 is represented by red sticks. (B) A closer view of the β9-α11 loop (blue), with the 3Dpol residue Met-296 (red) and the RTP molecule (green). Blue sticks are residues Ser-298 and Gly-299, which establish interactions with the incoming nucleotide base (18). Asp-245 and Asn-307, which interact with the ribose of the incoming nucleotide, are represented by gray sticks. (C) Superimposition of structures of different FMDV 3Dpol complexes. Shown are the binary complex with template-primer RNA (Protein Database accession no. [pdb] 1WNE; gray); ternary complex with UTP/ATP (pdb 2EC0; magenta); and ternary complex with ribavirin (pdb 2E9R; cyan). The loop of residues 294 to 301 is shown in yellow. The distance between Cα atoms of Gly299 in the different complexes shown (taken as a measure of the movement of the loop of residues 297 to 301) is about 0.9 and 1.4 Å in the complexes with UTP/ATP and ribavirin, respectively.
MATERIALS AND METHODS
Molecular cloning, expression, and purification of FMDV 3Dpol.
The FMDV polymerase 3D coding region (cloned into pET-28a [pET28a3D]) was expressed in Escherichia coli BL21 and purified as previously described (4, 19). The polymerase was fused at its C terminus to a His tag sequence that allowed purification by Ni-nitrilotriacetic acid affinity chromatography (ProBond Resin; Invitrogen). FMDV polymerase encoding M296I (M296I 3Dpol) was obtained by the site-directed mutagenesis of pET-28a3D using the QuikChange site-directed mutagenesis kit (Stratagene) as previously described (17). Briefly, pET-28a3D was amplified by using mutagenic primers that include the substitution G7497A (sense primer) or C7497U (antisense primer). PCR amplification was carried out using Pfu Ultra (Roche); the amplified DNA was treated with DpnI at 37°C for 1 h and transformed into E. coli XL1-Blue-competent cells, which were provided by the manufacturer. Plasmids from kanamycin-resistant colonies were sequenced to ascertain that the whole 3D coding region was properly cloned and that the desired mutation was present. The M296I mutant was expressed and purified as described for WT 3Dpol. Proteins were dialyzed three times against 50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 10% glycerol. Both proteins obtained were >95% pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis.
Poly(U) synthesis activity assay.
The specific activity of purified WT FMDV 3Dpol was 160 pmol UMP incorporated/min/μg of protein in the standard incorporation assay. Reaction mixtures contained 30 mM morpholinepropanesulfonic acid, pH 7.0, 33 mM NaCl, 5 mM Mg(CH3COO)2, 40 ng/μl polyriboadenylate (average length of 300 residues) (Amersham), 2.4 μM oligo(dT)15 (Invitrogen), 500 μM [α-32P]UTP (200 mCi/mmol) (Amersham). Purity and specific activity were indistinguishable from those of previous preparations of the enzyme (4, 19, 39). M296I 3Dpol showed a specific activity similar to that of WT 3Dpol (>75% specific activity relative to that of WT 3Dpol).
Sym/sub incorporation assays.
Reaction mixtures contained 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 5 mM MgCl2, 500 μM nucleoside triphosphate (NTP), 5′-32P end-labeled sym/sub (s/s; a symmetrical substrate primer-template), and FMDV 3Dpol, as previously described for PV 3Dpol (6, 9). 5′-32P labeling and the annealing of s/s were performed as previously described (6, 9). Reactions were quenched by the addition of EDTA to a final concentration of 50 mM. Product was resolved from the substrate by electrophoresis through a denaturing 23% polyacrylamide gel, visualized by phosphorimaging, and quantified by using ImageQuant software (Molecular Dynamics) as previously described (6, 9). Specific concentrations of s/s, 3Dpol, and/or nucleotide are indicated in the figure legends. FMDV 3Dpol was diluted immediately before use in 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, and 20% glycerol. The volume of enzyme added to the reaction mixture was, at most, 1/10 of the total reaction volume. Reactions were performed at 30°C.
Rapid chemical quenched-flow experiments.
Rapid mixing/quenching experiments were performed by using a model RQF-3 chemical quench-flow apparatus (KinTek Corp., Austin, TX). Experiments were performed at 30°C by using a circulating water bath. FMDV 3Dpol (2 μM) was incubated with either s/s-AU or s/s-AC (2 μM; 1 μM duplex) and UTP (10 μM) for 900 to 1,200 s to allow the formation of 3Dpol-s/s product complexes. The 3Dpol-s/s product complex then was rapidly mixed with various concentrations of nucleotide substrate (apparent dissociation constant [Kd,app] of 0.2 to 5). After being mixed, reactant concentrations were reduced by 50%. At various times after mixing, reactions were quenched by the addition of EDTA to a final concentration of 0.3 M.
Determination of the kinetic parameters (kpol and Kd,app) for FMDV 3Dpol-catalyzed nucleotide incorporation.
Data were fit by nonlinear regression using the program KaleidaGraph (Synergy Software, Reading, PA). Time courses at fixed nucleotide concentrations were fit to the following equation:
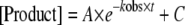 |
(1) |
where A is the amplitude of the burst, kobs is the observed first-order rate constant describing the burst, t is the time, and C is a constant. The kobs values were plotted as a function of nucleotide concentration, and the data were fitted to the following equation:
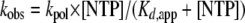 |
(2) |
where kpol is the maximal rate for nucleotide incorporation.
RESULTS
PV 3Dpol is the only picornavirus 3Dpol for which detailed kinetic and thermodynamic data exist (12, 40). This information has been invaluable to our understanding of the molecular mechanisms that govern the fidelity of RNA synthesis by RdRps (7-9, 27), the establishment of structure-function relationships for RdRps (8, 9, 20, 21, 27, 41, 42), the mechanism of action of antiviral compounds (13, 14), and the mechanisms of resistance to antiviral compounds (9). However, the extent to which the paradigms established for PV 3Dpol extend to other RdRps, especially other picornaviral 3Dpols, has yet to be evaluated empirically. The interpretation of recent structures for FMDV 3Dpol and its complexes with primed template in the absence and presence of natural and antiviral nucleotides has relied heavily on the mechanistic data available for PV 3Dpol (18, 19). Therefore, the initial motivation for this study was to create a mechanistic framework for the nucleotide addition cycle of FMDV 3Dpol that would illuminate both the unique and universal features of the picornaviral 3Dpol nucleotide addition cycle and permit the elucidation of FMDV structure-function relationships.
A minimal kinetic mechanism for single-nucleotide addition by PV 3Dpol is shown in Fig. 2. The binding of 3Dpol (oE) to primed template RNA (Rn) to form a 3Dpol-RNA complex (oERn) is in a rapid equilibrium. The partitioning of the 3Dpol-RNA complex into an activated complex (ERn) is slow and governed by the rate constant kassembly. The quantity of ERn formed at any given concentration of 3Dpol is attenuated by the inactivation of 3Dpol, which is governed by the temperature-dependent rate constant kinact. The activated 3Dpol-RNA complex binds nucleotides with an apparent binding affinity given by Kd,app to form the 3Dpol-RNA-nucleotide complex (ERnNTP). This complex undergoes catalysis at a rate governed by the rate constant kpol, yielding the product 3Dpol-extended RNA complex (ERn + 1) and pyrophosphate. 3Dpol is present in activated complexes; ERn and ERn + 1 dissociate from RNA at a rate governed by the rate constants koff,ERn and koff,ERn + 1, respectively. This mechanism is sufficient to explain the kinetics and mechanism of PV 3Dpol RNA synthesis in vitro and explains biological phenotypes observed for PV 3Dpol and its derivatives (6, 21). This framework guided our studies of FMDV 3Dpol.
FIG. 2.
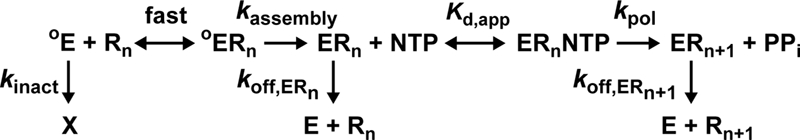
Mechanism of 3Dpol-RNA primer-template complex assembly and nucleotide incorporation. A minimal kinetic mechanism for single-nucleotide addition by PV 3Dpol is shown. X, inactivated 3Dpol.
Assembly of active FMDV 3Dpol-RNA complexes is slow and attenuated by thermal inactivation of 3Dpol.
We have employed a symmetrical primer-template (s/s) that permits nucleotide addition to be monitored by evaluating the extension of end-labeled primer (Fig. 3A) (6). We will maintain a convention in which the templating nucleotide(s) employed for an s/s in each experiment is indicated explicitly. For example, s/s-U refers to a substrate with uridine as the templating nucleotide.
FIG. 3.
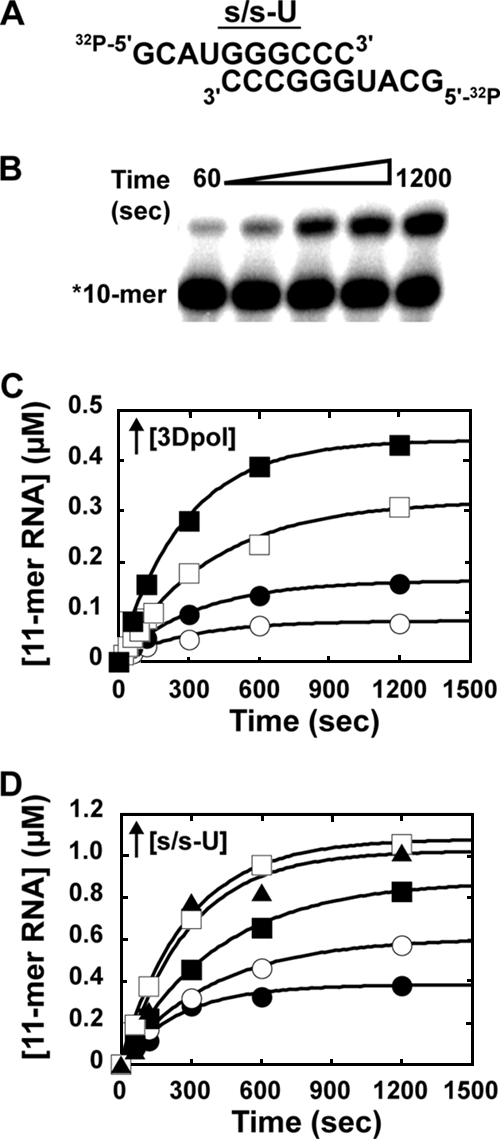
Kinetics of assembly of WT FMDV 3Dpol-s/s-U complexes. (A) Sequence of s/s-U. (B) AMP incorporation into s/s-U by WT FMDV 3Dpol. Products were resolved by electrophoresis on a denaturing 23% polyacrylamide gel. (C) Concentration dependence of WT FMDV 3Dpol. Reaction mixtures contained 1 μM s/s-U (0.5 μM duplex), 500 μM ATP, 5 mM MgCl2, and either 0.25 (○), 0.5 (•), 1 (□), or 5 (▪) μM 3Dpol. Reactions were initiated by the addition of 3Dpol, incubated at 30°C, and quenched at the indicated times by the addition of EDTA (50 mM). The solid lines represent the fit of the data to a single exponential. The observed rate of assembly was 0.003 s−1 for all concentrations of 3Dpol. (D) Concentration dependence of s/s-U. Reactions were performed as described above, but the mixtures contained 1 μM 3Dpol and either 1 (•), 2 (○), 4 (▪), 10 (□), or 20 (▴) μM s/s-U. The solid lines represent the fit of the data to a single exponential. The observed rates of assembly were 0.004, 0.004, 0.002, 0.002, and 0.004 s−1 for 20, 10, 4, 2, and 1 μM s/s-U, respectively (average, 0.003 s−1).
In order to determine if the FMDV 3Dpol utilization of s/s was limited by a slow assembly step, reaction mixtures containing s/s-U (0.5 μM duplex) and ATP (500 μM) were initiated by the addition of FMDV 3Dpol (0.25 to 5 μM). Reactions were quenched at various times by the addition of EDTA (50 mM). Products were resolved by denaturing polyacrylamide gel electrophoresis and visualized by phosphorimaging (Fig. 3B). The kinetics of AMP incorporation at the various concentrations of enzyme are shown in Fig. 3C. In no case did the end point reach the expected value of 0.5 μM. At 5 μM enzyme, 0.44 μM s/s-U was extended. Moreover, at all enzyme concentrations employed, the observed rate constant was the same (0.003/s). A concentration-independent rate constant suggests that a first-order process occurs after the formation of the bimolecular collision complex that limits the rate of 3Dpol activation.
The inability to reach the expected end point could have been caused by inactive enzyme present at the start of the experiment or inactivated during the course of the experiment. To distinguish between these two possibilities, the reaction was repeated under conditions in which the concentration of 3Dpol (1 μM) was held constant while varying the concentration of s/s-U (1 to 20 μM). In this experiment also, a concentration-independent rate constant of 0.003/s was observed (Fig. 3D). However, the expected end point of 1 μM was achieved by using high concentrations of s/s-U (Fig. 3D). The finding that both 10 and 20 μM s/s-U yielded essentially overlapping curves (Fig. 3D) can be interpreted only to mean that the observed rate constant reflects a step after s/s-U binding to 3Dpol. The ability to observe the stoichiometric utilization of enzyme (Fig. 3D) confirms that all of the enzyme in the preparation is active. Therefore, when lower concentrations of s/s-U are employed, the enzyme must be inactivating during the reaction to produce the lower-than-expected end points.
In order to examine enzyme inactivation directly, we performed the experiment shown in Fig. 4A. FMDV 3Dpol was incubated in buffer at different temperatures for various amounts of time, followed by the addition of s/s-U (0.5 μM) and ATP (500 μM). Primer extension was permitted for 10 min. In this experiment, FMDV 3Dpol was compared directly to PV 3Dpol as a control. The preincubation of both enzymes at 30°C prior to initiating the reaction caused a reduction in product formation (Fig. 4B). FMDV 3Dpol was clearly more stable than PV 3Dpol (Fig. 4B). Product formed was plotted as a function of preincubation time. Data were fit to a single exponential decay, yielding observed rate constants for inactivation (kinact) of 0.002 and 0.02/s for FMDV 3Dpol and PV 3Dpol, respectively (Fig. 4C). At temperatures of 22 and 4°C, FMDV 3Dpol remained one to two orders of magnitude more stable than PV 3Dpol (Table 1).
FIG. 4.
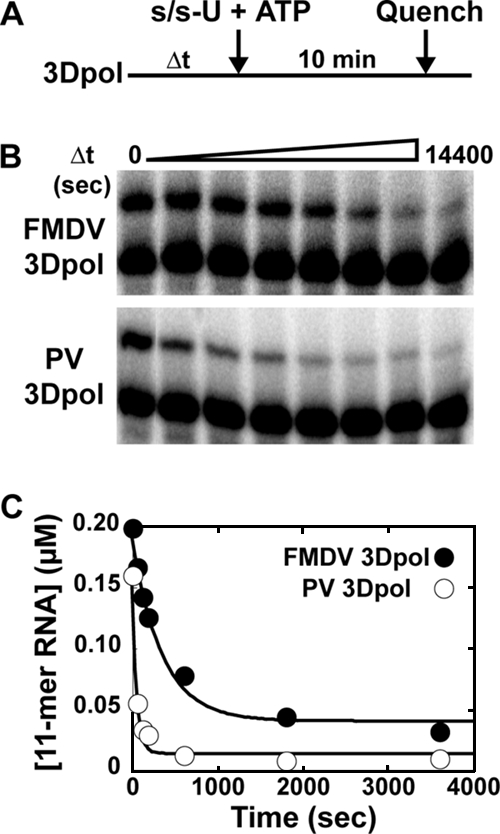
Inactivation of WT FMDV 3Dpol and PV 3Dpol. (A) Experimental design. WT FMDV or PV 3Dpol (2 μM) was incubated at 30°C in 1× reaction buffer for various amounts of time. The reaction was initiated by the addition of s/s-U (1 μM) and ATP (500 μM) and was allowed to proceed for an additional 10 min at 30°C, at which time the reaction was quenched by the addition of EDTA (50 mM). (B) Products from reactions described in panel A resolved by electrophoresis on a denaturing 23% polyacrylamide gel. (C) Kinetics of inactivation of WT FMDV (•) and PV (○) 3Dpol from the reactions described in panel A. The amount of 11-mer product was plotted as a function of the time of 3Dpol incubation. The solid lines represent the fit of the data to a single exponential and gives a kinact of 3 × 10−3 s−1 for FMDV 3Dpol and 20 × 10−3 s−1 for PV 3Dpol.
TABLE 1.
Kinetic parameters for the mechanism of FMDV and PV 3Dpol stability and 3Dpol primer-template complex assembly/stabilitya
| Virus 3Dpol | Rate of assembly (s−1) | Rate of dissociation for:
|
Inactivation at:
|
|||
|---|---|---|---|---|---|---|
| ERn (10−5 s−1) | ERn + 1 (10−5 s−1) | 4°C (10−5 s−1) | 22°C (10−5 s−1) | 30°C (10−3 s−1) | ||
| FMDV | 0.003 | 9 | 0.7 | 0.6c | 1c | 3 |
| PVb | 0.08 | 60 | 4 | 20 | 500 | 20 |
| Change (n-fold) | 30 | 7 | 6 | 30 | 500 | 7 |
All values are rounded to one significant figure. The error in each measurement is no greater than 20%, unless indicated otherwise. Experiments were performed as described in Materials and Methods.
Values for PV are taken from reference 6.
The error in the measurement was 50%.
FMDV 3D polymerase forms a very stable complex with RNA.
An important determinant of polymerase processivity is the rate constant for the dissociation of the enzyme from nascent RNA and template. In the system employed here, two FMDV 3Dpol-nucleic acid complexes form: 3Dpol-s/s-U substrate complex (ERn) and 3Dpol-s/s-U product complex (ERn + 1). The dissociation of 3Dpol from the latter complex will set the upper limit for primer extension under steady-state conditions (i.e., excess s/s-U).
We measured the rate constant for the dissociation of FMDV 3Dpol from s/s-U by mixing 3Dpol and 5′-32P-labeled s/s-U and incubating it for 1,200 s to permit complexes to form (Fig. 5A). We next added a 1,000-fold molar excess of unlabeled s/s-U as a trap for free and dissociating enzyme (Fig. 5A). At various times after the addition of trap, ATP was added for 30 s (Fig. 5A). The extended primer provided an indication of the amount of complex remaining (Fig. 5B). The percentage of primer extended was plotted as a function of time after the addition of trap, and the data were fit to a single exponential decay (Fig. 5C). This analysis revealed a dissociation rate constant of 0.00009/s (half-life of 2 h). Dissociation from the s/s-U-product complex was measured by using the same approach; however, during the initial incubation of 3Dpol with s/s-U, a concentration of ATP stoichiometric with enzyme was added (Fig. 5D). After the addition of trap, complex stability was monitored by evaluating the formation of the two-nucleotide-extended product after the addition of UTP (Fig. 5E). Quantitative analysis yielded a dissociation rate constant of 0.000007/s (half-life of 27.5 h) (Fig. 5F).
FIG. 5.

Kinetics of dissociation of FMDV 3Dpol-s/s-U substrate and 3Dpol-s/s-U product complexes. (A) Experimental design. WT FMDV 3Dpol (1 μM) was incubated with stoichiometrically 32P-labeled s/s-U (0.1 μM; 0.05 μM duplex) for 1,200 s at 30°C, at which point trap (100 μM unlabeled s/s-U) was added to the reaction mixture. At fixed times after the addition of trap, ATP (500 μM) was added, and the reaction was allowed to proceed for 30 s and then quenched by the addition of EDTA (50 mM). (B) Products from reactions described in panel A were resolved by electrophoresis on a denaturing 23% polyacrylamide gel. (C) Kinetics of dissociation of FMDV 3Dpol-s/s-U complexes from the reactions described in panel A. The solid lines represent the fit of the data to a single exponential and give a koff,ERn of 9 × 10−5 s−1. (D) Experimental design. Reactions were performed as described for panel A but contained ATP (1 μM) during the assembly reaction, and at fixed times after the addition of trap, UTP (500 μM) was added. (E) Products from reactions described in panel D resolved by electrophoresis on a denaturing 23% polyacrylamide gel. (F) Kinetics of dissociation of FMDV 3Dpol-s/s-U product complexes from the reactions described in panel D. The solid lines represent the fit of the data to a single exponential and give a koff,ERn + 1 of 0.7 × 10−5 s−1.
These data reveal a very stable association of FMDV 3Dpol with s/s-U. Similarly to observations made for PV 3Dpol (6), the FMDV 3Dpol-s/s-U product complex is more stable than the initial complex. In the case of FMDV 3Dpol, a 13-fold difference was observed, whereas only a 6-fold difference was observed for PV 3Dpol (6). Also, the FMDV 3Dpol-s/s-U complexes ERn and ERn + 1 are 7- and 14-fold more stable than the corresponding PV 3Dpol-s/s-U complexes.
Nucleotide addition by FMDV 3Dpol is fast and faithful.
Given the substantial differences in the stability of 3Dpol-s/s-U substrate and product complexes (Fig. 5), we decided to evaluate the kinetics and fidelity of nucleotide incorporation after the first cycle of nucleotide addition. Additional justification for the evaluation of the ERn + 1 complex is provided by structural data that reveal more contacts in the 3Dpol-ERn + 1 complex than in the 3Dpol-ERn complex (18). We developed two new substrates for these experiments, s/s-AU and s/s-AC (Fig. 6A). These substrates permit the assembly of product complexes by incubating 3Dpol, s/s-AU, and UTP prior to the addition of ATP (correct for s/s-AU) or GTP (correct for s/s-AC).
FIG. 6.
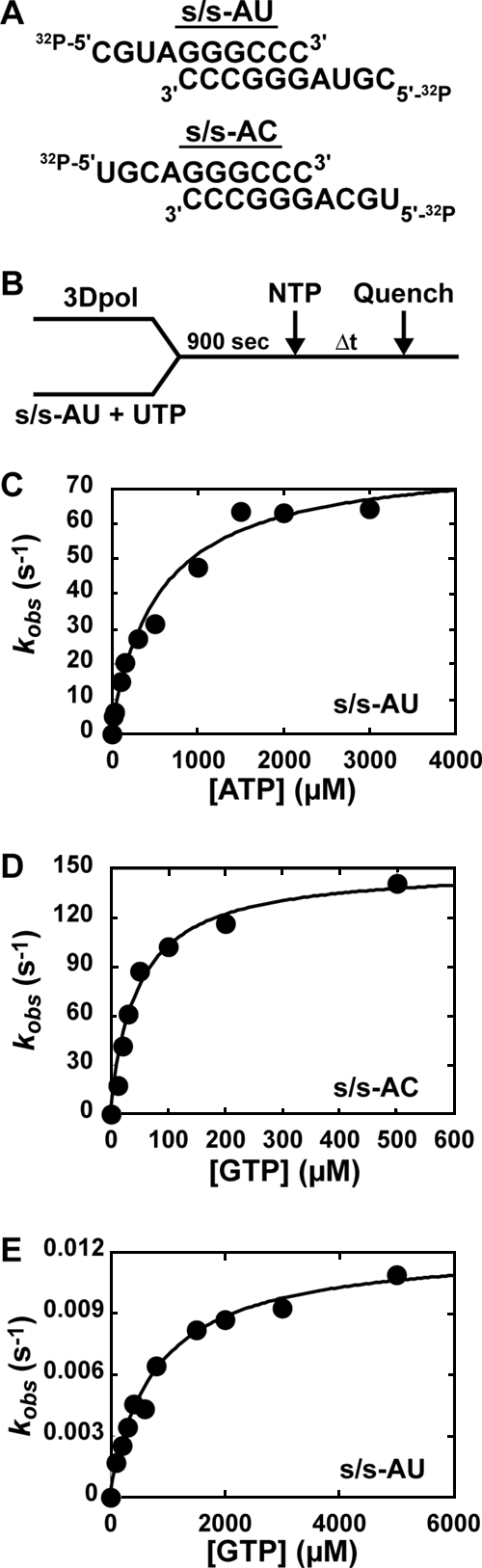
Pre-steady-state kinetic analysis of nucleotide incorporation into s/s-AU and s/s-AC by WT FMDV 3Dpol. (A) Sequences of s/s-AU and s/s-AC. (B) Experimental design. WT FMDV 3Dpol (2 μM) was incubated with either s/s-AU or s/s-AC (2 μM; 1 μM duplex) and UTP (10 μM) for 900 s to allow the formation of 3Dpol-s/s product complexes. The 3Dpol-s/s product complex then was rapidly mixed using a rapid chemical quench-flow apparatus with various concentrations of nucleotide substrate (either ATP or GTP, with Kd,apps of 0.2 to 5). At various times after being mixed, reactions were quenched by the addition of EDTA (0.3 M). Time courses at fixed nucleotide concentrations were fit to a single exponential to obtain the observed rate constant for nucleotide incorporation, kobs (see equation 1). The observed rate constants then were plotted as a function of nucleotide concentration, and the data were fit to a hyperbola (see equation 2) to obtain the maximal observed rate constant for nucleotide incorporation, kpol, and the apparent dissociation constant, Kd,app. (C) AMP incorporation into s/s-AU. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 80 s−1 and a Kd,app of 500 μM. (D) GMP incorporation into s/s-AC. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 200 s−1 and a Kd,app of 50 μM. (E) GMP incorporation into s/s-AU. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 0.01 s−1 and a Kd,app of 800 μM.
The general design of these experiments is presented in Fig. 6B. 3Dpol was assembled with the appropriate primed template in the presence of UTP, followed by the addition of the indicated nucleotide at different concentrations for various amounts of time. Reactions were quenched, and product analysis performed as described above. When correct nucleotide incorporation experiments were performed, the reactions occurred too fast for manual quenching, so a chemical quench-flow apparatus was employed as described in Materials and Methods. Product formed as a function of time was plotted, and data were fitted to a single exponential rise, yielding an observed rate constant (kobs). The observed rate constants were plotted as a function of nucleotide concentration and data were fitted to a hyperbola, yielding an apparent dissociation constant (Kd,app) and a maximal rate of nucleotide incorporation (kpol).
We evaluated the incorporation of AMP (Fig. 6C) and GMP (Fig. 6D) as the correct nucleotide and the incorporation of GMP as the incorrect nucleotide (Fig. 6E). The values for Kd,app, kpol, and kpol/Kd,app are reported in Table 2. The Kd,app values for nucleotide binding ranged from 50 to 500 μM. As the correct nucleotide, GTP bound 10-fold more efficiently than ATP; a similar observation was made for PV 3Dpol (7). As the incorrect nucleotide, GTP bound to 3Dpol as efficiently as ATP. These data suggest that GTP binding as the correct nucleotide is a special case, and that the initial binding of both correct and incorrect nucleotides likely is driven by using the triphosphate moiety of incoming nucleotide instead of by base pairing. The kpol values for correct nucleotide incorporation ranged from 80 to 200/s, which is on par with values measured for PV 3Dpol (7). The kpol value for GMP misincorporation was 0.01/s. Thus, FMDV 3Dpol fidelity is determined by kinetic events reported by kpol. By using (kpol/Kd,app)incorr/(kpol/Kd,app)corr, where incorr is the incorrect nucleotide and corr is the correct nucleotide, to calculate the frequency of A→G transition mutations, a mutation frequency of 1/20,000 was obtained (Table 2). This observation predicts less than one A→G transition mutation per round of genome replication for FMDV. Clearly, this mutation frequency will contribute substantial sequence diversity at the population level. We conclude that FMDV 3Dpol is fast and quite faithful, even in the absence of a proofreading nuclease.
TABLE 2.
Kinetic constants for nucleotide incorporation by FMDV WT and M296I 3Dpola
| Template | Nucleotide | WT 3Dpol
|
M296I 3Dpol
|
||||
|---|---|---|---|---|---|---|---|
| Kd,app (μM) | kpol (s−1) | kpol/Kd,app (μM−1 s−1) | Kd,app (μM) | kpol (s−1) | kpol/Kd,app (μM−1 s−1) | ||
| s/s-AU | ATP | 500 | 80 | 0.2 | 400 | 30 | 0.08 |
| GTP | 800 | 0.01 | 1 × 10−5 | 500 | 0.006 | 1 × 10−5 | |
| RTP | 600 | 0.03 | 5 × 10−5 | 1000 | 0.01 | 1 × 10−5 | |
| s/s-AC | GTP | 50 | 200 | 4 | 70 | 100 | 1 |
| RTP | 500 | 0.02 | 4 × 10−5 | 800 | 0.009 | 1 × 10−5 | |
All values are rounded to one significant figure. The error in each measurement is no greater than 20%. Experiments were performed as described in Materials and Methods.
Nucleotide addition by FMDV M296I 3Dpol is slower and less faithful than that of WT 3Dpol.
To study the biochemical properties of FMDV M296I 3Dpol, the M296I-encoding mutation was engineered into the E. coli expression vector employed for WT FMDV 3Dpol as described in Materials and Methods. FMDV M296I 3Dpol was expressed and purified as previously reported for WT FMDV 3Dpol (4).
We evaluated the kinetics of nucleotide (mis)incorporation as described above for WT FMDV 3Dpol (Fig. 7 and Table 2). A two- to threefold reduction in the overall efficiency (kpol/Kd,app) of correct nucleotide incorporation was observed relative to that of WT enzyme (Table 2). This reduction was caused primarily by a decrease in the kpol value. Surprisingly, the overall efficiency of GMP misincorporation for FMDV M296I 3Dpol was unchanged relative to that for WT enzyme. However, the values for both Kd,app and kpol decreased somewhat (Table 2). We conclude that the fidelity of FMDV M296I is reduced relative to that of the WT enzyme, with a mutation frequency of 1/8,000, which is 2.5-fold higher than that for the WT (Table 3). These data suggest that FMDV encoding the M296I change in 3Dpol should exhibit a mutator phenotype. The reevaluation of the biological data (39) leads to a similar conclusion (Table 3).
FIG. 7.
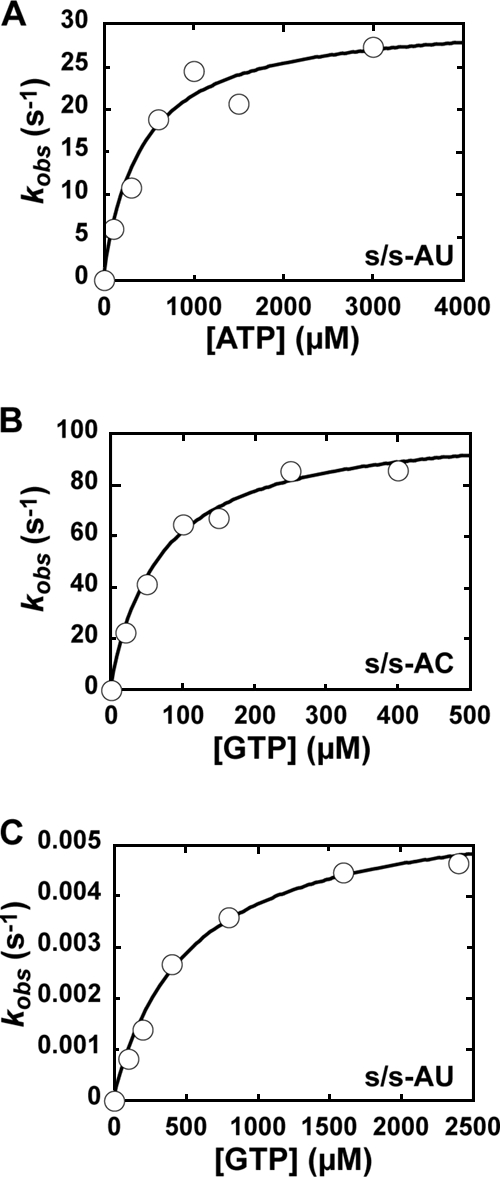
Pre-steady-state kinetic analysis of nucleotide incorporation into s/s-AU and s/s-AC by FMDV M296I 3Dpol. Reactions were performed as described in the legend to Fig. 6 but contained FMDV M296I 3Dpol. (A) AMP incorporation into s/s-AU. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 30 s−1 and a Kd,app of 400 μM. (B) GMP incorporation into s/s-AC. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 100 s−1 and a Kd,app of 70 μM. (C) GMP incorporation into s/s-AU. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 0.006 s−1 and a Kd,app of 500 μM.
TABLE 3.
Mutation frequency of FMDV WT and M296I 3Dpol
| FMDV type | Biochemical
|
Biological
|
|||
|---|---|---|---|---|---|
| Mutation frequencya
|
Transition mutation frequencyb
|
||||
| Transitionc | Ribavirin with s/s-AUd | Ribavirin with s/s-ACe | Without ribavirin | With ribivarin | |
| WT | 1/20,000 | 1/4,000 | 1/100,000 | 1/3,254 | 1/862 |
| M296I | 1/8,000 | 1/8,000 | 1/100,000 | 1/2,440 | 1/938 |
All values are rounded to one significant figure. The error in each measurement is no greater than 20%.
Values were calculated from data presented in Sierra et al. (39).
Transition mutation frequency is calculated as (kpol incorr/Kd,app incorr)/(kpol corr/Kd,app corr), using the kpol and Kd,app values for ATP and GTP as the correct (corr) or incorrect (incorr) nucleotide for s/s-AU.
Ribavirin mutation frequency is calculated as (kpol RTP/Kd,app RTP)/(kpolATP/Kd,app ATP).
Ribavirin mutation frequency is calculated as (kpol RTP/Kd,app RTP)/(kpol GTP/Kd,app GTP).
FMDV M296I 3Dpol incorporates ribavirin less efficiently than WT 3Dpol.
The observation that FMDV M296I 3Dpol is a low-fidelity enzyme was inconsistent with our expectations. FMDV encoding this change confers reduced sensitivity to ribavirin. The resistance of PV to ribavirin also was conferred by a mutation in its 3Dpol (G64S) (9, 38); however, PV G64S 3Dpol was a high-fidelity enzyme (9, 38).
In order to resolve this apparent paradox, we evaluated the kinetics and specificity of ribavirin incorporation by both FMDV WT and M296I 3Dpol enzymes (Fig. 8 and Table 2). For both enzymes, ribavirin utilization was templated by uridine and cytidine (Fig. 8 and Table 2). The overall efficiency of ribavirin incorporation by WT FMDV 3Dpol was four- to fivefold higher than that for GMP misincorporation (Table 2). In contrast, the overall efficiency of ribavirin incorporation by FMDV M296I 3Dpol was equal to that of GMP misincorporation (Table 2). Therefore, despite the low-fidelity behavior of the enzyme for GMP misincorporation, FMDV M296I 3Dpol is four- to fivefold less capable of incorporating ribavirin relative to the ability of WT FMDV 3Dpol. These data are consistent with the biological observation that FMDV encoding M296I 3Dpol is less sensitive to ribavirin (39), but they show that ribavirin resistance in this case comes with the penalty of higher frequencies of GMP misincorporation. We conclude that the molecular mechanism for FMDV resistance to ribavirin is unique. Therefore, at least two mechanisms exist for picornaviruses to evade the antiviral activity of ribavirin; the production of a high-fidelity polymerase is only one. Perhaps many other mechanisms remain to be discovered.
FIG. 8.
Ribavirin incorporation into s/s-AU and s/s-AC by FMDV WT and M296I 3Dpol. (A) RMP incorporation into s/s-AU by FMDV WT (•) and M296I (○) 3Dpol. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 0.03 s−1 and a Kd,app of 600 μM for WT FMDV 3Dpol and a kpol of 0.01 s−1 and a Kd,app of 1,000 μM for FMDV M296I 3Dpol. (B) RMP incorporation into s/s-AC by FMDV WT (•) and M296I (○) 3Dpol. Shown is a replot of the observed rate constants as a function of nucleotide concentration to yield a kpol of 0.02 s−1 and a Kd,app of 500 μM for WT FMDV 3Dpol and a kpol of 0.009 s−1 and a Kd,app of 800 μM for FMDV M296I 3Dpol.
DISCUSSION
The elaboration of a kinetic and thermodynamic description of the nucleotide addition cycle for a polymerase permits the establishment of structure-function relationships controlling catalysis and the fidelity of nucleotide selection and incorporation, as well as the elucidation of molecular mechanisms for antiviral compounds and corresponding resistance mechanisms (11, 12, 22, 24, 31, 40, 49). A wealth of biochemical and structural information exists for a large number of RdRps (32); however, PV 3Dpol is the only RdRp for which a complete kinetic and thermodynamic description of the nucleotide addition cycle exists (6-8, 12, 20, 40). The availability of mechanistic information for a second RdRp permits direct comparisons of RdRps in vitro that should illuminate similarities and differences. Significant differences observed in vitro may have implications for virus biology.
In this study, we have elucidated the minimal mechanism for nucleotide addition catalyzed by FMDV 3Dpol. We find that the kinetic pathway employed by FMDV 3Dpol is identical to that employed by PV 3Dpol (Fig. 2) (6). This observation is not surprising, given the conservation of kinetic pathways across diverse classes of nucleic acid polymerases (40). Importantly, numerous differences in the kinetics and/or thermodynamics of the steps comprising the pathway are revealed by comparing FMDV 3Dpol to PV 3Dpol.
During the first step of the reaction, 3Dpol must associate with primed template. We have employed the artificial primer-template s/s, which was developed for PV 3Dpol (6). This primer-template has been shown to recapitulate the biological elongation complex (6, 21). However, the in vitro assembly of 3Dpol onto duplex RNA does not have a known biological equivalent. During assembly, there is a race between 3Dpol binding to nucleic acid and temperature-dependent inactivation. The assembly of FMDV 3Dpol-s/s complexes was 30-fold slower than the rate observed for PV 3Dpol (Fig. 3; Table 1). Unexpectedly, FMDV 3Dpol produced more complexes than PV 3Dpol under identical conditions (Fig. 3; Table 1) (6). The reason for this apparent paradox was that the rate of the thermal inactivation of FMDV 3Dpol was at least 7-fold but as much as 500-fold slower than that of the thermal inactivation of PV 3Dpol depending upon the temperature employed (Table 1). These results suggest that a correlation exists between the rate of assembly and the rate of thermal inactivation. Note that we do not believe that the thermal inactivation of 3Dpol for s/s utilization equates to irreversible enzyme inactivation due to an unfolding event, because 3Dpol-catalyzed viral protein genome-linked uridylylation reactions can proceed linearly for hours (34, 35). The enhanced stability of FMDV 3Dpol on nucleic acid coupled with the enhanced thermal stability may provide explanations for the facile crystallization of this protein with so many ligands (17-19).
Peersen and colleagues originally suggested that PV 3Dpol assembly was limited by a cis-trans isomerization of Pro-119 (42). This possibility was ruled out by the finding that the structurally equivalent residue (Pro-120) in FMDV 3Dpol remains in the cis conformation when bound to s/s (19). Structures of FMDV 3Dpol and PV 3Dpol are essentially superimposable (19, 42). However, one exception has been noted recently. The β9-α11 (FMDV 3Dpol nomenclature [19]) loop of PV 3Dpol (residues 288 to 292) exists in two conformations: in and out (Olve Peersen, personal communication). The out conformation occludes the s/s binding site described for FMDV 3Dpol (19). Only the in conformation is observed for FMDV 3Dpol in the absence of nucleic acid and nucleotide (19). As discussed below, this loop exhibits changes in position in response to different template-incoming nucleotide pairs (18). Changes in the dynamic behavior and/or equilibrium position of this element in the absence of nucleic acid for 3Dpols from different picornaviruses may contribute to the rate of assembly and/or inactivation. Consistently with this hypothesis is the finding that some substitutions in this element, for example, Cys-290 to Val in PV 3Dpol, decrease the rate of assembly (J. J. Arnold and O. Peersen, unpublished observations).
The final complex formed after s/s binding to 3Dpol is very stable and becomes even more stable after the incorporation of the first nucleotide (Fig. 5; Table 1). The half-lives of the FMDV 3Dpol-s/s complexes before and after nucleotide incorporation are 2 and 27 h, respectively (Table 1). Complexes formed by FMDV 3Dpol are at least sixfold more stable than the corresponding PV complexes (Table 1). The half-lives for other polymerase-primed template complexes are on the order of seconds to minutes (2, 31). The structure of the FMDV 3Dpol-s/s complex does not reveal more interactions with s/s than those observed for other classes of nucleic acid polymerase (19). What is different, however, is the absence of large domain movements after binding (19). Therefore, the increased binding affinity may derive from the rigidity of the RNA binding site and the absence of large domain movements that could induce dissociation. We hypothesize that the restriction of interdomain movements is caused by the interaction of the 3Dpol fingertip subdomain with the thumb subdomain (41, 42).
After the formation of the 3Dpol-s/s complex, nucleotide binds to form the 3Dpol-s/s-NTP complex. The apparent dissociation constant, Kd,app, is derived from kinetic data. Our studies of PV 3Dpol have shown that nucleotide binding is a two-step process (7). The first step is driven by interactions of the triphosphate with motif F of 3Dpol (residues 155 to 176 for PV and 160 to 181 for FMDV). The data obtained for FMDV 3Dpol are consistent with this mechanism (Fig. 6 and 8 and Table 2). The Kd,app values measured for correct nucleotides, incorrect nucleotides, and nucleotide analogues are essentially the same. GTP appears to be a special case, as the Kd,app value of this nucleotide for FMDV 3Dpol was 10-fold lower than those of other nucleotides (Table 2); similar observations were made for PV 3Dpol (7). Interestingly, the Kd,app values measured for FMDV 3Dpol are 5- to 10-fold higher than those measured for PV 3Dpol (Table 2) (7). This observation suggests that FMDV multiplication should be more sensitive to changes in intracellular nucleotide concentration than PV. In addition, FMDV should be less susceptible to the antiviral action of nucleotide analogues than PV in direct comparisons. This difference may have contributed to a low-fidelity variant instead of a high-fidelity variant.
Finally, the formation of the 3Dpol-s/s-NTP complex leads to catalysis and the formation of one-nucleotide-extended RNA. The kinetics of nucleotide addition are governed by the macroscopic rate constant, kpol (7). This rate constant contains terms describing nucleotidyl transfer but also contains terms describing the second step of nucleotide binding (7). The second step of nucleotide binding is a conformational change of the complex that leads to the orientation of the triphosphate into a position suitable for catalysis (7, 8, 20). Importantly, this second step is used as a fidelity checkpoint (8, 9, 20). The kpol values measured for correct nucleotide incorporation by FMDV 3Dpol were fast (Fig. 6; Table 2) and on par with those measured for PV 3Dpol (7). The fidelity of nucleotide incorporation for FMDV 3Dpol is governed by steps reflected in kpol, as the kpol values for incorrect nucleotides and nucleotide analogues were decreased by a factor of ∼10,000 relative to those for correct nucleotides (Fig. 6, Table 2). Therefore, FMDV replication is as faithful as that of any DNA virus would be in the absence of proofreading and repair mechanisms. Nevertheless, over multiple rounds of replication, the calculated mutation frequency of 1/20,000 (Table 3) is still sufficient to create substantial genetic diversity in the virus population.
With this minimal description of the WT FMDV 3Dpol in hand, we were in a position to probe the molecular mechanism for the decreased ribavirin sensitivity of an FMDV variant encoding M296I 3Dpol (39). Our expectation was that FMDV M296I 3Dpol would be a higher-fidelity polymerase than the WT based on PV G64S 3Dpol, which also confers decreased sensitivity to ribavirin (9). Surprisingly, FMDV M296I 3Dpol exhibited a lower fidelity than that of WT FMDV 3Dpol. The overall efficiency (kpol/Kd,app) of correct nucleotide incorporation for FMDV M296I 3Dpol was decreased 2.5-fold relative to that of the WT enzyme without exhibiting any change in the overall efficiency of incorrect nucleotide incorporation (Fig. 7; Table 2). This change in fidelity decreases the calculated transition mutation frequency (Table 3). Importantly, evidence for low-fidelity replication by this variant is apparent in the transition mutation frequency observed in cell culture (Table 3) (39). The mutation frequency calculated from biochemical data is higher than that observed in cell culture for both FMDV WT and M296I 3Dpols (Table 3). The difference is due, in part, to the fact that all four possible mispaired combinations have not been considered. In addition, intracellular conditions, for example, nucleotide pools, may be conducive to more faithful RNA synthesis. Finally, selection occurs during virus multiplication in cell culture, which will reduce the observed mutation frequency. These data provide another correlation between polymerase error frequency in vitro and virus population diversity in cell culture. However, future experiments will be required to prove that the increased error frequency of M296I 3Dpol observed in vitro is the cause of the reduced fitness of M296I FMDV observed in cell culture.
This is the first FMDV polymerase variant with a mutator phenotype. A PV 3Dpol variant that changes Asn-297 in motif B to Glu also exhibits a mutator phenotype (27). The corresponding residue in FMDV 3Dpol is Asn-307. As shown in Fig. 1B, Asn-307 also interacts with the nucleotide. This illustration also conveys the ability of Asn-307 to influence the β9-α11 loop and vice versa. The kinetic data show that substitutions in both motif B and the β9-α11 loop cause the mutator phenotype by decreasing the efficiency of correct nucleotide incorporation, not by increasing the efficiency of incorrect nucleotide incorporation. This observation suggests that the RdRp active site employs two distinct conformations for correct versus incorrect nucleotide incorporation.
Why is M296I FMDV less sensitive to ribavirin than the WT virus? Although FMDV M296I 3Dpol is more efficient at GMP misincorporation than the WT enzyme, FMDV M296I 3Dpol is less efficient at ribavirin incorporation (Fig. 8). In fact, the overall efficiency of ribavirin incorporation by FMDV M296I 3Dpol is four- to fivefold lower than that observed for WT FMDV 3Dpol (Table 2). The ability to decrease the efficiency of ribavirin incorporation without changing the efficiency of GMP misincorporation suggests that the active-site conformations supporting these incorporation events are different. We now have to conclude that there are at least three distinct conformations assumed by the RdRp that are competent for nucleotide incorporation. Therefore, substantial flexibility exists in the active-site conformations employed for nucleotide incorporation, and these active-site conformations are induced by the nascent base pair. This interpretation is consistent with recent reports on 2′-deoxynucleotide selection by DNA polymerases (29, 44).
Residue 296 is located on the β9-α11 loop (Fig. 1). This loop moves as much as 1.5 Å in response to nucleotide binding and the nature of the nascent base pair (Fig. 1C) (18). It is likely that changes in the position of this loop contribute to the induced-fit mechanism revealed by the biochemical studies reported here. In the context of our other studies of RdRp structure-function relationships (9, 20, 21, 27), we conclude that cross-talk between residues in the ribose binding pocket, including motifs A and B, and the β9-α11 loop determines RdRp (in)fidelity.
The M296I substitution was identified as a common change encoded by a variety of FMDV 3Dpol genes present in populations serially passaged in the presence of ribavirin that exhibited reduced sensitivity to the drug (39). The distribution of mutations observed in the presence of ribavirin for the population was biased heavily toward G→A and C→U transition mutations, suggesting that ribavirin was incorporated preferentially opposite C in the template (13). In contrast, FMDV encoding the M296I substitution exhibited a bias for A→G and U→C transition mutations, suggesting that ribavirin was incorporated preferentially opposite U in the template (39). Our biochemical data explain that the bias for FMDV M296I 3Dpol as the frequency of ribavirin incorporation opposite C is 1/100,000, a value 12.5-fold lower than that observed for incorporation opposite U (Table 3). Therefore, even more insight into RdRp fidelity may be obtained from the further analysis of mutations in the 3Dpol coding sequence present in the original populations of FMDV selected in the presence of ribavirin.
The viral RdRp is responsible for generating genomic diversity during replication, leading to the phenotypic flexibility of RNA virus quasispecies. Viral quasispecies dynamics depends not only on the genomic diversity of the population but also on selection imposed by the environment and changes therein (recently reviewed in reference 16). Although not proven, it is likely that viral quasispecies dynamics are optimized for viral pathogenesis. Clearly, even in the absence of extinction or even detectable mutagenesis, nucleotide analogues like ribavirin could alter viral quasispecies dynamics, leading to an antiviral effect, especially in the complex environment of a mammalian host. Consistently with this possibility is the finding that the ribavirin-resistant PV variant encoding the G64S 3Dpol exhibits a restricted quasispecies (36, 45) and is attenuated in a mouse model for PV infection (36, 45). G64S PV grows as well as the WT virus in cell culture, and the population diversity is decreased by only threefold (9, 38, 45). G64S PV is capable of protecting mice against a lethal challenge with WT PV (46). Therefore, processes selecting for virus variants displaying a restricted quasispecies may represent processes for viral attenuation and vaccine development.
Is the selection of viral mutants conferring resistance to ribavirin a general approach for viral attenuation by quasispecies restriction? Clearly, multiple mechanisms for resistance to ribavirin exist. FMDV has found a counterintuitive solution: a low-fidelity polymerase with a specific defect in ribavirin utilization. In the case of hepatitis C virus, mutations in both the RdRp (NS5b) (50) and another replicase component (NS5a) (37) have been implicated in ribavirin resistance, but the molecular mechanism(s) is not known. The study presented here underscores the impact of mechanistic studies of the RdRp in vitro on defining the mechanisms of resistance in cell culture and the elucidation of structural elements controlling RdRp fidelity/genomic diversity, which are potential targets for viral attenuation and vaccine development.
Acknowledgments
We thank Ibrahim Moustafa for preparing Fig. 1C. We are indebted to C. Ferrer-Orta and N. Verdaguer for many valuable discussions of FMDV polymerase structure. We thank R. Agudo and C. Escarmís for their many helpful suggestions.
This study was supported by the following grants: AI45818 from NIAID/NIH (to C.E.C.), BFU 2005-00863 from MEC, Proyecto Intramural de Frontera 2005-20F-0221 from CSIC, and Fundación Ramón Areces. CIBERehd is funded by the Instituto de Salud Carlos III. A.A. is a recipient of an I3P contract from CSIC, and M.S. was supported by a predoctoral fellowship from the Ministerio de Educación y Ciencia.
Footnotes
Published ahead of print on 1 October 2008.
REFERENCES
- 1.Airaksinen, A., N. Pariente, L. Menendez-Arias, and E. Domingo. 2003. Curing of foot-and-mouth disease virus from persistently infected cells by ribavirin involves enhanced mutagenesis. Virology 311339-349. [DOI] [PubMed] [Google Scholar]
- 2.Anand, V. S., and S. S. Patel. 2006. Transient state kinetics of transcription elongation by T7 RNA polymerase. J. Biol. Chem. 28135677-35685. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, J. P., R. Daifuku, and L. A. Loeb. 2004. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58183-205. [DOI] [PubMed] [Google Scholar]
- 4.Arias, A., R. Agudo, C. Ferrer-Orta, R. Perez-Luque, A. Airaksinen, E. Brocchi, E. Domingo, N. Verdaguer, and C. Escarmis. 2005. Mutant viral polymerase in the transition of virus to error catastrophe identifies a critical site for RNA binding. J. Mol. Biol. 3531021-1032. [DOI] [PubMed] [Google Scholar]
- 5.Arnold, E., K. Das, J. Ding, P. N. Yadav, Y. Hsiou, P. L. Boyer, and S. H. Hughes. 1996. Targeting HIV reverse transcriptase for anti-AIDS drug design: structural and biological considerations for chemotherapeutic strategies. Drug Des. Discov. 1329-47. [PubMed] [Google Scholar]
- 6.Arnold, J. J., and C. E. Cameron. 2000. Poliovirus RNA-dependent RNA polymerase (3Dpol). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub). J. Biol. Chem. 2755329-5336. [DOI] [PubMed] [Google Scholar]
- 7.Arnold, J. J., and C. E. Cameron. 2004. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+. Biochemistry 435126-5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnold, J. J., D. W. Gohara, and C. E. Cameron. 2004. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mn2+. Biochemistry 435138-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold, J. J., M. Vignuzzi, J. K. Stone, R. Andino, and C. E. Cameron. 2005. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J. Biol. Chem. 28025706-25716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll, S. S., and D. B. Olsen. 2006. Nucleoside analog inhibitors of hepatitis C virus replication. Infect. Disord. Drug Targets 617-29. [DOI] [PubMed] [Google Scholar]
- 11.Carvalho, A. P., P. A. Fernandes, and M. J. Ramos. 2006. Molecular insights into the mechanisms of HIV-1 reverse transcriptase resistance to nucleoside analogs. Mini. Rev. Med. Chem. 6549-555. [DOI] [PubMed] [Google Scholar]
- 12.Castro, C., J. J. Arnold, and C. E. Cameron. 2005. Incorporation fidelity of the viral RNA-dependent RNA polymerase: a kinetic, thermodynamic and structural perspective. Virus Res. 107141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crotty, S., C. E. Cameron, and R. Andino. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 986895-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crotty, S., D. Maag, J. J. Arnold, W. Zhong, J. Y. N. Lau, Z. Hong, R. Andino, and C. E. Cameron. 2000. The broad-spectrum antiviral ribonucleotide, ribavirin, is an RNA virus mutagen. Nat. Med. 61375-1379. [DOI] [PubMed] [Google Scholar]
- 15.Domingo, E. (ed.). 2005. Virus entry into error catastrophe as a new antiviral strategy. Virus Res. 107115-228. [Google Scholar]
- 16.Domingo, E., C. Parrish, and J. J. Holland (ed.). 2008. Origin and evolution of viruses, 2nd ed. Elsevier, Oxford, United Kingdom.
- 17.Ferrer-Orta, C., A. Arias, R. Agudo, R. Perez-Luque, C. Escarmis, E. Domingo, and N. Verdaguer. 2006. The structure of a protein primer-polymerase complex in the initiation of genome replication. EMBO J. 25880-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrer-Orta, C., A. Arias, R. Perez-Luque, C. Escarmis, E. Domingo, and N. Verdaguer. 2007. Sequential structures provide insights into the fidelity of RNA replication. Proc. Natl. Acad. Sci. USA 1049463-9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrer-Orta, C., A. Arias, R. Perez-Luque, C. Escarmis, E. Domingo, and N. Verdaguer. 2004. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J. Biol. Chem. 27947212-47221. [DOI] [PubMed] [Google Scholar]
- 20.Gohara, D. W., J. J. Arnold, and C. E. Cameron. 2004. Poliovirus RNA-dependent RNA polymerase (3Dpol): kinetic, thermodynamic, and structural analysis of ribonucleotide selection. Biochemistry 435149-5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gohara, D. W., S. Crotty, J. J. Arnold, J. D. Yoder, R. Andino, and C. E. Cameron. 2000. Poliovirus RNA-dependent RNA polymerase (3Dpol): structural, biochemical, and biological analysis of conserved structural motifs A and B. J. Biol. Chem. 27525523-25532. [DOI] [PubMed] [Google Scholar]
- 22.Götte, M. 2006. Effects of nucleotides and nucleotide analogue inhibitors of HIV-1 reverse transcriptase in a ratchet model of polymerase translocation. Curr. Pharm. Des. 121867-1877. [DOI] [PubMed] [Google Scholar]
- 23.Gotte, M. 2004. Inhibition of HIV-1 reverse transcription: basic principles of drug action and resistance. Expert Rev. Anti Infect. Ther. 2707-716. [DOI] [PubMed] [Google Scholar]
- 24.Götte, M., and M. A. Wainberg. 2000. Biochemical mechanisms involved in overcoming HIV resistance to nucleoside inhibitors of reverse transcriptase. Drug Resist. Updat. 330-38. [DOI] [PubMed] [Google Scholar]
- 25.Graci, J. D., and C. E. Cameron. 2004. Challenges for the development of ribonucleoside analogues as inducers of error catastrophe. Antivir. Chem. Chemother. 151-13. [DOI] [PubMed] [Google Scholar]
- 26.Graci, J. D., and C. E. Cameron. 2006. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 1637-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korneeva, V. S., and C. E. Cameron. 2007. Structure-function relationships of the viral RNA-dependent RNA polymerase: fidelity, replication speed, and initiation mechanism determined by a residue in the ribose-binding pocket. J. Biol. Chem. 28216135-16145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kukhanova, M., A. Krayevsky, W. Prusoff, and Y. C. Cheng. 2000. Design of anti-HIV compounds: from nucleoside to nucleoside 5′-triphosphate analogs. Problems and perspectives. Curr. Pharm. Des. 6585-598. [DOI] [PubMed] [Google Scholar]
- 29.Luo, G., M. Wang, W. H. Konigsberg, and X. S. Xie. 2007. Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc. Natl. Acad. Sci. USA 10412610-12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magden, J., L. Kaariainen, and T. Ahola. 2005. Inhibitors of virus replication: recent developments and prospects. Appl. Microbiol. Biotechnol. 66612-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menéndez-Arias, L. 2002. Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptases. Prog. Nucleic Acid Res. Mol. Biol. 7191-147. [DOI] [PubMed] [Google Scholar]
- 32.Ng, K. K., J. J. Arnold, and C. E. Cameron. 2008. Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 320137-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oberg, B. 2006. Rational design of polymerase inhibitors as antiviral drugs. Antiviral Res. 7190-95. [DOI] [PubMed] [Google Scholar]
- 34.Pathak, H. B., J. J. Arnold, P. N. Wiegand, M. R. Hargittai, and C. E. Cameron. 2007. Picornavirus genome replication: assembly and organization of the VPg uridylylation ribonucleoprotein (initiation) complex. J. Biol. Chem. 28216202-16213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pathak, H. B., S. K. Ghosh, A. W. Roberts, S. D. Sharma, J. D. Yoder, J. J. Arnold, D. W. Gohara, D. J. Barton, A. V. Paul, and C. E. Cameron. 2002. Structure-function relationships of the RNA-dependent RNA polymerase from poliovirus (3Dpol). A surface of the primary oligomerization domain functions in capsid precursor processing and VPg uridylylation. J. Biol. Chem. 27731551-31562. [DOI] [PubMed] [Google Scholar]
- 36.Pfeiffer, J. K., and K. Kirkegaard. 2005. Increased fidelity reduces poliovirus fitness under selective pressure in mice. PLoS Pathogens 1102-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeiffer, J. K., and K. Kirkegaard. 2005. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J. Virol. 792346-2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfeiffer, J. K., and K. Kirkegaard. 2003. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 1007289-7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sierra, M., A. Airaksinen, C. González-López, R. Agudo, A. Arias, and E. Domingo. 2007. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: implications for error catastrophe. J. Virol. 812012-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smidansky, E. D., J. J. Arnold, and C. E. Cameron. 2008. Nucleic acid polymerase fidelity and viral population fitness. In E. Domingo, C. Parrish, and J. J. Holland (ed.), Evolution and origin of viruses. Elsevier, Oxford, United Kingdom.
- 41.Thompson, A. A., R. A. Albertini, and O. B. Peersen. 2007. Stabilization of poliovirus polymerase by NTP binding and fingers-thumb interactions. J. Mol. Biol. 3661459-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson, A. A., and O. B. Peersen. 2004. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J. 233462-3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsai, C. H., P. Y. Lee, V. Stollar, and M. L. Li. 2006. Antiviral therapy targeting viral polymerase. Curr. Pharm. Des. 121339-1355. [DOI] [PubMed] [Google Scholar]
- 44.Tsai, Y. C., and K. A. Johnson. 2006. A new paradigm for DNA polymerase specificity. Biochemistry 459675-9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vignuzzi, M., J. K. Stone, J. J. Arnold, C. E. Cameron, and R. Andino. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439344-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vignuzzi, M., E. Wendt, and R. Andino. 2008. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 14154-161. [DOI] [PubMed] [Google Scholar]
- 47.Vivet-Boudou, V., J. Didierjean, C. Isel, and R. Marquet. 2006. Nucleoside and nucleotide inhibitors of HIV-1 replication. Cell Mol. Life Sci. 63163-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vo, N. V., K. C. Young, and M. M. C. Lai. 2003. Mutagenic and inhibitory effects of ribavirin on hepatitis C virus RNA polymerase. Biochemistry 4210462-10471. [DOI] [PubMed] [Google Scholar]
- 49.Yin, P. D., D. Das, and H. Mitsuya. 2006. Overcoming HIV drug resistance through rational drug design based on molecular, biochemical, and structural profiles of HIV resistance. Cell Mol. Life Sci. 631706-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young, K. C., K. L. Lindsay, K. J. Lee, W. C. Liu, J. W. He, S. L. Milstein, and M. M. Lai. 2003. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 38869-878. [DOI] [PubMed] [Google Scholar]