

Abstract
Papillomavirus infection normally involves virion binding to cell surface heparan sulfate proteoglycans (HSPGs). However, we found that human papillomavirus type 16 pseudovirions efficiently bound and infected cells lacking HSPGs if their L2 capsid protein was precleaved by furin, a cellular protease required for infection. The inability of pseudovirions to efficiently bind and infect cultured primary keratinocytes was also overcome by furin precleavage, suggesting that the defect involves altered HSPG modification. We conclude that the primary function of HSPG binding is to enable cell surface furin cleavage of L2 and that binding to a distinct cell surface receptor(s) is a subsequent step of papillomavirus infection.
Virion attachment to cell surface receptor(s) and penetration into the cell are two obligatory early steps in viral infection. Although the identity of the receptors and the mechanism of internalization are known for many viruses, the interplay between these two steps is less well understood. We have been interested in examining the cell surface events that lead to productive entry for papillomaviruses (PV), a family of nonenveloped DNA viruses that includes the oncogenic human PV (HPV) types, such as HPV16, that are the causative agents of human cervical cancer (3). The PV capsid is composed of the following two structural proteins: the major capsid protein, L1, which can self-assemble into icosahedral viruslike particles, and the minor capsid protein, L2, whose functions are necessary for establishment of infection (4, 8, 16).
Cell surface heparan sulfate proteoglycans (HSPGs) have been shown to be the key attachment factor for most HPV types in vitro. For example, exogenous heparin can prevent infection, and the HSPG-null cell line, pgsa-745, is inefficiently infected (13, 15). Due to a deficiency in xylosyl transferase, these cells lack all proteoglycans (18).
L2 proteins of all sequenced PVs contain at their N termini a consensus cleavage motif for furin, a proprotein convertase, and furin cleavage is necessary for infection (23). However, mature virions in solution are resistant to furin cleavage. Cleavage is facilitated by cell surface binding and results in a conformational change in the viral capsid on the cell surface that can be monitored by the exposure of an L2 neutralization epitope (9). Therefore, we wondered if the initial interaction of the capsid with HSPG functions primarily to facilitate furin cleavage on the cell surface and, following this, a secondary receptor is engaged, or whether HSPG binding has additional roles in the infectious process. To examine this question, here we have compared infection efficiency of the following three types of HPV16 L1/L2 green fluorescent protein-expressing pseudovirus preparations: standard pseudovirus, standard pseudovirus in the presence of exogenous furin, and furin-precleaved (FPC) pseudovirus.
To produce FPC pseudovirus, we took advantage of the slow maturation of in vitro-produced pseudovirus and added furin to the pseudovirus extract prior to initiating the maturation process. After maturation, the FPC virus was isolated according to the standard methodology used to generate the other preparation (5). Western blot analysis (Fig. 1A) indicated that only some of the L2 in the capsids was cleaved by this procedure, although in the absence of L1 almost complete cleavage of the L2 protein can be achieved (23). It is unclear if L2 was partially cleaved in all of the pseudovirions or if a subset of pseudovirions contained completely cleaved L2. Partial cleavage of human immunodeficiency virus type 1 Env by furin has also been reported. This latter result was attributed to the conformation of the recognition site on the full-length Env protein (1). Interestingly, in vitro cleavage was greatly enhanced following a heparin-induced structural reorganization (21).
FIG. 1.

(A) Furin cleaved L2 within the FPC pseudovirus. Pseudovirions were generated according to standard procedures or with the inclusion of furin (20 U; Alexis Biochemicals) in the lysis/maturation buffer. Both preparations were purified through an OptiPrep gradient. Western blot analysis detection using an anti-L2 monoclonal antibody (11) revealed cleaved L2 in the FPC preparation. We determined that approximately 35% of the L2 is cleaved by furin in this pseudovirion preparation. Quantification of the bands was performed on a Fuji LAS4000 imaging system. (B) Infectivity of the pseudovirus preparations on FD11 and FD11+ cells. The CHO-derived cell lines FD11, which is furin-deficient, and FD11+, which was transfected with a cDNA-expressing furin, were infected with untreated pseudovirus (white bars), the FPC virus (light gray bars), or untreated virus with exogenous furin (dark gray bars). The preparations were equilibrated by L1 content and titrated, threefold dilutions starting at 50 ng/ml. Infection was quantified by flow cytometric quantification of green fluorescent protein expression at 48 h. Exogenous furin was delivered in supernatant form (to 30% of the total volume) from the CHO-derived cell line, Δfur, which secretes a functional, truncated furin (6). Quantification of these supernatants reveals approximately 8 U/ml of active furin. The efficiency of in vitro cleavage of HPV16 L2 (not assembled into viral particles) was compared to cleavage with commercially calibrated furin (data not shown). (C) Infectivity of the pseudovirus on pgsa-745 cells. The same procedure was followed to evaluate the infectivity of the pgsa-745 cells. A titration of pseudovirus infectivity is shown with untreated pseudovirus (white bars), the FPC virus (light gray bars), or untreated virus with exogenous furin (dark gray bars). (D) Infectivity of the pseudovirus on keratinocytes. The first group of the three shows the infection of primary HFKs under the various conditions. The second and third groups show the infection of Ect1 E6/E7 and End1 E6/E7, respectively (the white bars represent infection with untreated pseudovirus, the FPC virus is shown in light gray, and the untreated virus with exogenous furin is shown in dark gray). All cells were cultivated in keratinocyte serum-free medium (Invitrogen) with added epidermal growth factor and bovine pituitary extract. The infection with the untreated virus was performed in the presence of the supernatant from FD11 cells to compare it to the addition of exogenous furin from the Δfur cells. This indicated that the addition of conditioned CHO medium did not affect the infectivity of the keratinocyte cells without the presence of furin.
To evaluate the degree to which infectivity of the three pseudovirus preparations depended on cellular furin, furin-deficient FD11 cells and a furin-expressing derivative line, FD11+, were infected (Fig. 1B). The untreated pseudovirus did not efficiently infect the FD11 cells except in the presence of exogenous furin (conditioned medium from a CHO-derived cell line overexpressing secreted furin). The FPC virus also efficiently infected the cell lines, irrespective of cellular furin. This result establishes that the incomplete furin cleavage of the FPC pseudovirus population prior to exposure to the cell was sufficient for cellular furin-independent infection. It also demonstrates that furin cleavage coincident with binding is sufficient for infection. We consistently found that infection with FPC pseudovirus or infection in the presence of exogenous furin resulted in a more robust infection, even in cells that contain endogenous furin (FD11+ cells, shown in Fig. 1B), suggesting that furin is a limiting factor for PV infection.
To determine if exposure to excess furin might have rendered pseudovirus infection independent of cellular HSPG, we evaluated the infectivity of the three virus preparations on the HSPG-null pgsa-745 cell line. As expected, these cells were poorly infected with the untreated pseudovirus (Fig. 1C). However, they were efficiently infected by the standard pseudovirus in the presence of exogenous furin or by the FPC virus. The high infectivity of the furin-treated pseudovirions in the absence of cell surface HSPG rules out an obligatory role for HSPG in capsid internalization and suggests the existence of a second receptor molecule.
We thought it possible that the absence of the correct HSPG modifications could limit the ability to infect other cultured cells in vitro. In vivo, PVs replicate exclusively in keratinocytes. Paradoxically, primary keratinocytes are difficult to infect in vitro, generally requiring extremely sensitive reverse transcription-PCR methods for detection and a high multiplicity of infection (20). Therefore, we determined if these cells could be infected with the FPC pseudovirus, as we had found for the pgsa-745 cells. We examined the infection of primary human foreskin keratinocytes (HFKs) and two E6/E7-transformed keratinocyte cell lines, Ect1 E6/E7 and End1 E6/E7, which are also very poorly infected by untreated HPV16 pseudovirus. We found that, similar to the HSPG-deficient pgsa-745 cells, all three cell cultures could be efficiently infected with the FPC pseudovirus or the standard virus in the presence of exogenous furin (Fig. 1D), consistent with the hypothesis that HSPG modification is responsible for the poor infectivity of in vitro-cultured keratinocytes. It is well documented that variations in heparan sulfate structure can occur in response to altered culture conditions (25, 27).
We also examined the binding of untreated pseudovirions to HFKs by confocal microscopy. As expected, relatively few capsids were bound to the cell surface. Instead, the majority of the capsids were associated with the extracellular matrix (ECM). The two focal planes in Fig. 2, which show ECM (Fig. 2A) and the cell surface (Fig. 2B), respectively, indicate the difference in the degree of binding to these locales.
FIG. 2.

Binding of pseudovirus preparations to HFK cells. Cells were seeded onto no. 01 thickness glass coverslips in a 24-well plate at a density of 1 × 105 cells/well and cultured overnight. To evaluate the capsid binding pattern, 50 ng of pseudovirus was added to the cells for 1 h. Unbound virus was removed by extensive washing, and the cells were fixed with 2% paraformaldehyde. Bound virus was detected with a rabbit polyclonal antiserum raised against HPV16 virus-like particles (24) and Alexa Fluor 488-conjugated donkey anti-rabbit immunoglobulin G (Molecular Probes). Alexa Fluor 594-conjugated phalloidin (Molecular Probes) was included in the secondary antibody stain at a dilution of 1/300. Untreated virus is shown in panels A and B. These two panels show the same cells in two different focal planes to emphasize the relative binding to the ECM (A) and the cell surface (B). FPC virus (C and D) was likewise imaged.
It has been demonstrated that HPV16 can bind to both the cell surface and the ECM in vitro (7). In addition, we have previously shown that certain neutralizing antibodies, or soluble heparin, prevent cell surface association, resulting in an accumulation of the virus on the ECM (10). Therefore, this result is consistent with the idea that the primary deficiency of the cells for PV infection is capsid attachment secondary to altered HSPG modification. The higher HFK infectivity with FPC pseudovirus than with standard pseudovirus is expected to parallel a concomitant increase in binding of the virus to the surfaces of the cells, and as expected, greater cell surface association of the FPC pseudovirus was clearly observed (Fig. 2B and D).
However, although furin expression has been reported previously in primary HFKs (2, 22), we wanted to confirm that the lack of HFK infectivity by standard pseudovirus could not be attributable to a deficiency in active furin at the cell surface. This possibility was ruled out by determining that HFKs under our culture conditions do possess functional cell surface furin by showing that the HFKs could cleave a known furin substrate, anthrax toxin protective antigen (PA), whose cleavage by furin occurs predominately on the cell surface (17). In this experiment, PA33, a genetically modified version of PA that is cleaved when exposed to furin but not to related proprotein convertases, was cleaved from its initial 83-kDa PA33 form to the 63-kDa form after incubation at 4°C or 37°C with all the cell lines except the FD11 cells, which lack furin (Fig. 3; data shown for 37°C).
FIG. 3.
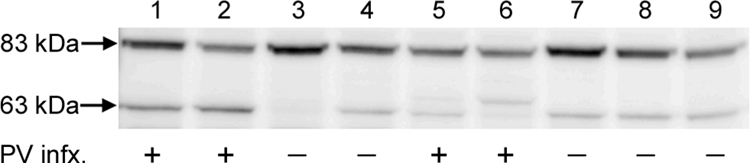
Cleavage of a furin-dependent ligand. Cells were seeded at 5 × 104 cells/well/ml of a 48-well plate and grown to confluence. Twenty-five micrograms of PA33 (17) was added for 1 h at 37°C, and furin cleavage was assessed by Western blot analysis using a polyclonal antiserum against PA (14). The lysates were loaded in the following order: lane 1, CHO-K1; lane 2, FD11+; lane 3, FD11; lane 4, pgsa-745; lane 5, HeLa; lane 6, HaCaT; lane 7, HFK; lane 8, Ect1 E6/E7; lane 9, End1 E6/E7. The ability of the cell line to be infected with untreated pseudovirus is indicated below the appropriate lane.
To provide further evidence that the FPC pseudovirus can bind in the absence of HSPG attachment, we examined capsid association to pgsa-745 cells (Fig. 4A and B). Similarly to that observed with the HFKs, significant capsid association was observed only with the FPC capsids. Additionally, we treated HaCaT cells with sodium chlorate for 2 days prior to pseudovirus binding. Sodium chlorate prevents the sulfation of HSPGs and has been shown to decrease HPV11 virus-like particle binding to the cell surface (15). The untreated pseudovirus was unable to bind to the cell surface under these conditions (Fig. 4C). In contrast, the FPC pseudovirus showed strong cell surface association (Fig. 4D). These data confirm the role of HSPG for the initial binding of untreated capsids and also provide direct support for the existence of an additional non-HSPG cell surface PV receptor.
FIG. 4.

Binding of pseudovirus in the absence of HSPG. The binding of untreated pseudovirus to pgsa-745 cells is shown in panel A. FPC virus is shown in panel B. Panels C and D show binding to sodium chlorate-treated HaCaT cells. Cells were grown in 50 mM sodium chlorate for 2 days prior to being plated on coverslips. Untreated virus is shown in panel C. FPC virus is shown in panel D.
Our results suggest the following model of the early events in PV infection. Initial attachment to HSPG moieties functions primarily to facilitate the critical step of L2 cleavage by furin. HSPGs have been well described to sequester ligands at the cell surface and ECM and also to facilitate modification of protein conformation (27). Interestingly, there are increasing data on the proprotein convertase cleavage of HSPG ligands, indicating a possible enrichment of convertases in the vicinity of the cellular glycocalyx (19, 26). It was recently found that endothelial lipase has decreased affinity for HSPG following furin cleavage (12). Similarly, in our model for PV, following cleavage and a conformational change in the capsid, the capsid would detach from HSPG and associate with a putative second receptor. Utilization of either the FPC pseudovirus or exposure to exogenous furin can bypass the requirement for HSPG engagement, permitting direct binding of the secondary receptor. Thus, it appears that HSPGs function as more than a simple attachment factor in PV infection in that this interaction promotes essential conformational changes, but they are clearly not the cell surface receptor that mediates virion internalization or later events in infection.
Acknowledgments
We thank Stephen Leppla and Shihui Lui for the anthrax toxin PA and suggestions to design the PA cleavage assay. We also thank David Fitzgerald for the furin-secreting CHO-based cell line.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
Published ahead of print on 1 October 2008.
REFERENCES
- 1.Binley, J. M., R. W. Sanders, A. Master, C. S. Cayanan, C. L. Wiley, L. Schiffner, B. Travis, S. Kuhmann, D. R. Burton, S. L. Hu, W. C. Olson, and J. P. Moore. 2002. Enhancing the proteolytic maturation of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 762606-2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bitoun, E., A. Micheloni, L. Lamant, C. Bonnart, A. Tartaglia-Polcini, C. Cobbold, T. Al Saati, F. Mariotti, J. Mazereeuw-Hautier, F. Boralevi, D. Hohl, J. Harper, C. Bodemer, M. D'Alessio, and A. Hovnanian. 2003. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum. Mol. Genet. 122417-2430. [DOI] [PubMed] [Google Scholar]
- 3.Bosch, F. X., A. Lorincz, N. Munoz, C. J. Meijer, and K. V. Shah. 2002. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 55244-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bossis, I., R. B. Roden, R. Gambhira, R. Yang, M. Tagaya, P. M. Howley, and P. I. Meneses. 2005. Interaction of tSNARE syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J. Virol. 796723-6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 119445-462. [DOI] [PubMed] [Google Scholar]
- 6.Chiron, M. F., C. M. Fryling, and D. FitzGerald. 1997. Furin-mediated cleavage of Pseudomonas exotoxin-derived chimeric toxins. J. Biol. Chem. 27231707-31711. [DOI] [PubMed] [Google Scholar]
- 7.Culp, T. D., L. R. Budgeon, and N. D. Christensen. 2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 347147-159. [DOI] [PubMed] [Google Scholar]
- 8.Day, P. M., C. C. Baker, D. R. Lowy, and J. T. Schiller. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. USA 10114252-14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Day, P. M., R. Gambhira, R. B. Roden, D. R. Lowy, and J. T. Schiller. 2008. Mechanisms of human papillomavirus type 16 neutralization by L2 cross-neutralizing and L1 type-specific antibodies. J. Virol. 824638-4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day, P. M., C. D. Thompson, C. B. Buck, Y. Y. Pang, D. R. Lowy, and J. T. Schiller. 2007. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J. Virol. 818784-8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gambhira, R., B. Karanam, S. Jagu, J. N. Roberts, C. B. Buck, I. Bossis, H. Alphs, T. Culp, N. D. Christensen, and R. B. Roden. 2007. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J. Virol. 8113927-13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gauster, M., A. Hrzenjak, K. Schick, and S. Frank. 2005. Endothelial lipase is inactivated upon cleavage by the members of the proprotein convertase family. J. Lipid Res. 46977-987. [DOI] [PubMed] [Google Scholar]
- 13.Giroglou, T., L. Florin, F. Schafer, R. E. Streeck, and M. Sapp. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 751565-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon, V. M., K. R. Klimpel, N. Arora, M. A. Henderson, and S. H. Leppla. 1995. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 6382-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joyce, J. G., J. S. Tung, C. T. Przysiecki, J. C. Cook, E. D. Lehman, J. A. Sands, K. U. Jansen, and P. M. Keller. 1999. The L1 major capsid protein of human papillomavirus type L1 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 2745810-5822. [DOI] [PubMed] [Google Scholar]
- 16.Kämper, N., P. M. Day, T. Nowak, H. C. Selinka, L. Florin, J. Bolscher, L. Hilbig, J. T. Schiller, and M. Sapp. 2006. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 80759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klimpel, K. R., S. S. Molloy, G. Thomas, and S. H. Leppla. 1992. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 8910277-10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lugemwa, F. N., and J. D. Esko. 1991. Estradiol β-d-xyloside, an efficient primer for heparan sulfate biosynthesis. J. Biol. Chem. 2666674-6677. [PubMed] [Google Scholar]
- 19.Nour, N., G. Mayer, J. S. Mort, A. Salvas, M. Mbikay, C. J. Morrison, C. M. Overall, and N. G. Seidah. 2005. The cysteine-rich domain of the secreted proprotein convertases PC5A and PACE4 functions as a cell surface anchor and interacts with tissue inhibitors of metalloproteinases. Mol. Biol. Cell 165215-5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozbun, M. A. 2002. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 7611291-11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasquato, A., M. Dettin, A. Basak, R. Gambaretto, L. Tonin, N. G. Seidah, and C. Di Bello. 2007. Heparin enhances the furin cleavage of HIV-1 gp160 peptides. FEBS Lett. 5815807-5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearton, D. J., W. Nirunsuksiri, A. Rehemtulla, S. P. Lewis, R. B. Presland, and B. A. Dale. 2001. Proprotein convertase expression and localization in epidermis: evidence for multiple roles and substrates. Exp. Dermatol. 10193-203. [DOI] [PubMed] [Google Scholar]
- 23.Richards, R. M., D. R. Lowy, J. T. Schiller, and P. M. Day. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 1031522-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roden, R. B., H. L. Greenstone, R. Kirnbauer, F. P. Booy, J. Jessie, D. R. Lowy, and J. T. Schiller. 1996. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 705875-5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt, A., A. Skaletz-Rorowski, and E. Buddecke. 1995. Basic fibroblast growth factor controls the expression and molecular structure of heparan sulfate in corneal endothelial cells. Eur. J. Biochem. 234479-484. [DOI] [PubMed] [Google Scholar]
- 26.Seidah, N. G., G. Mayer, A. Zaid, E. Rousselet, N. Nassoury, S. Poirier, R. Essalmani, and A. Prat. 2008. The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell. Biol. 401111-1125. [DOI] [PubMed] [Google Scholar]
- 27.Turnbull, J., A. Powell, and S. Guimond. 2001. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 1175-82. [DOI] [PubMed] [Google Scholar]