

Abstract
Human cytomegalovirus (HCMV) is the most frequent cause of congenital viral infections in humans and frequently leads to long-term central nervous system (CNS) abnormalities that include learning disabilities, microcephaly, and hearing loss. The pathogenesis of the CNS infection has not been fully elucidated and may arise as a result of direct damage of CMV-infected neurons or indirectly secondary to inflammatory response to infection. We used a recently established model of mouse CMV (MCMV) infection in newborn mice to analyze the contribution of humoral immunity to virus clearance from the brain. In brains of MCMV-infected newborn mice treated with immune serum, the titer of infectious virus was reduced below detection limit, whereas in the brains of mice receiving control (nonimmune) serum significant amounts of virus were recovered. Moreover, histopathological and immunohistological analyses revealed significantly less CNS inflammation in mice treated with immune serum. Treatment with MCMV-specific monoclonal antibodies also resulted in the reduction of virus titer in the brain. Recipients of control serum or irrelevant antibodies had more viral foci, marked mononuclear cell infiltrates, and prominent glial nodules in their brains than mice treated with immune serum or MCMV-specific antibodies. In conclusion, our data indicate that virus-specific antibodies have a protective role in the development of CNS pathology in MCMV-infected newborn mice, suggesting that antiviral antibodies may be an important component of protective immunological responses during CMV infection of the developing CNS.
Human cytomegalovirus (HCMV) represents the most common cause of congenital viral infection in humans (41). Symptoms associated with congenital HCMV brain disease vary from mild perceptual deficits such as vision impairment and hearing loss to severe sequelae including microcephaly, cerebral dysplasia, and psychomotor retardation (6, 49). The mechanism of CMV-induced neuropathogenesis of CMV infection has not been elucidated, and it may arise secondary to direct viral damage to neurons or indirectly because of the host response to infection. Proposed mechanisms of the disease include interruption of blood supply to developing brain, migration deficits of developing neurons, and loss of neural progenitor cells in the subventicular zone due to inflammation coupled with delays in myelinization (2, 19, 29, 30, 52). Experimental evidence suggests that neurons in the developing brain are potentially more permissive for CMV infection than mature ones (22, 34, 59). In addition, susceptibility to murine CMV (MCMV) infection of central nervous system (CNS) diminishes with the age of mice, and MCMV infection in immunocompetent adult mice, in contrast to newborn mice, does not result in virus dissemination within the CNS (40, 54). In primary infection, CMV dissemination is considered mostly cell associated (43, 51), but little is known about the means of CMV dissemination into the developing CNS.
Both innate and adaptive immune responses play a role in the control of CMV infection in various organs and tissues (27). While NK cells and other components of innate immune response are responsible for containment of primary MCMV infection during the early period after infection, the components of adaptive cellular immune response are essential for termination of productive virus infection (26). In contrast, antiviral antibodies are dispensable for the resolution of primary CMV infection and establishment of latency. However, antibodies play a key role in prevention of virus dissemination after reactivation from latency or reinfection (21, 35). It is generally accepted that antiviral antibodies mediate their protective activity by direct neutralization or indirectly via complement activation and by promoting antibody-dependent cellular cytotoxicity (18). Therefore, it is conceivable that antibodies may suppress CMV dissemination by direct virus neutralization or by cytotoxicity of cells carrying the virus. Antiviral antibodies have been considered to be important components of the maternal immune response in the protection against congenital CMV infection (9, 15) although maternal seropositivity to CMV prior to pregnancy does not provide absolute protection against prenatal infection and disease (5). Early studies have shown that passively acquired maternal anti-CMV antibodies protected against transfusion-associated CMV infection in the immediate postnatal period (61). Passive transfer of CMV-specific antibodies in multiply transfused preterm infants has also been shown to be protective in terms of the reduction of CMV disease (48). Although maternal seropositivity prior to pregnancy does not provide full protection against prenatal infection and disease (5), more recent studies have claimed that treatment of pregnant women with CMV-specific hyperimmune globulin was effective in prevention of congenital CMV infection (32). In adult patients, the presence of antiviral antibodies has been correlated with slower progression of CMV disease in AIDS patients and transplant patients (7, 45). Finally, some studies reported that transplant recipients benefited from transfer of CMV-specific antibodies in the posttransplant period, whereas other studies have shown only minimal or no benefit from the transfer of anti-CMV antibodies in some transplant populations (36). Future studies should define the optimal conditions for the use of anti-CMV antibodies in the prevention and treatment of CMV infection.
Due to the HCMV species specificity, several animal models of HCMV infection, immunobiology, and disease have been established (4, 53). Among these, infection of mice with MCMV has been the most frequently used animal model of HCMV infection (20, 38). We have previously described a model of MCMV infection of newborn mice and showed that CNS susceptibility to MCMV diminishes with age since MCMV infection of newborn, but not adult immunocompetent mice, results in virus dissemination to the CNS (40, 54). More recently, we described a model of MCMV-induced encephalitis in the developing brain (24). After intraperitoneal (i.p.) inoculation of the virus into newborn mice, virus disseminated from peripheral tissues into the CNS, causing the development of focal, yet widespread, encephalitis defined by mononuclear cell infiltration. Furthermore, infected animals consistently exhibited developmental and morphological abnormalities in cerebellum.
Recent studies by Klenovsek et al. demonstrated the protective capacity of memory B cells. In contrast to previous findings, the investigators showed that memory B cells can proliferate, secrete antiviral antibodies in a T-cell independent manner, and efficiently control MCMV infection after transfer into immunodeficient hosts (23). The question arises whether passive immunization with specific antiviral antibodies has a protective role during the CMV infection in brain parenchyma. In the present study, we have described a role of antiviral antibodies in the control of MCMV infection in brain of newborn mice. Passive immunization of MCMV-infected newborn mice with immune serum or MCMV-specific monoclonal antibodies reduced both virus replication and virus-induced pathology in brain tissue, including developmental abnormalities and specific histopathological lesions.
MATERIALS AND METHODS
Animals, infection conditions, and virus titration.
BALB/c (H-2d) mice were bred under specific-pathogen-free conditions at the Central Animal Facility of the Faculty of Medicine, University of Rijeka. Handling of animals, experimental procedures, and administration of anesthesia were performed in accordance with the guidelines contained in the International Guiding Principles for Biomedical Research Involving Animals (31a). The Ethics Committee at the University of Rijeka approved all animal experiments described within this report. Newborn mice, 6 to 12 h postpartum, were inoculated i.p. with 500 PFU of wild-type (WT) MCMV (Smith strain, ATCC VR-194 [reaccessioned as VR-1399]). Mice were anesthetized by i.p. injection of ketamine (2.5 mg/mice) and sacrificed at various postnatal days (2 to 90). Blood from all groups of animals was collected by intracardial puncture, heparinized, and divided into cell fraction and plasma by centrifugation for 4 min at 4,000 rpm. Virus titers in the brain, liver, blood cells, and plasma were determined using standard plaque assay on mouse embryonic fibroblasts (8, 39). Individual mice from the same group were also analyzed for brain histology and cerebellar morphometry.
Histopathology and immunohistochemistry.
Serial sagittal sections (4 μm thick) from formalin-fixed, paraffin-embedded brains (following intracardial perfusion with phosphate-buffered saline [PBS]) were stained with cresyl violet (CV). Approximately 80 to 120 slides per animal were stained, depending on the brain size. Histological evaluation of every 10th slide was performed by two observers who were unaware of experimental group design. Histopathological lesions such as inflammatory mononuclear cellular infiltrates, perivascular cuffing, areas of micronodular gliosis, and necrosis were monitored and counted per slide. Scores were averaged per group of animals and are presented as means ± standard deviations (9, 14).
MCMV-infected cells were visualized by immunohistochemistry by detection of viral IE1 protein using MCMV-specific monoclonal antibody CROMA 101 (55). Monoclonal antibody (MAb) 97.3 was visualized using biotinylated goat anti-mouse immunoglobulin G (IgG) antibody (BD Pharmingen, San Diego, CA). Purkinje cells were visualized using monoclonal anti-calbindin D-28K antibody (Sigma-Aldrich, Munich, Germany). The staining was performed by the avidin-biotin-peroxidase complex method, using biotinylated goat anti-mouse IgG as a secondary antibody (BD Pharmingen, San Diego, CA), followed by detection with an avidin-biotin-peroxidase complex (Roche Applied Science, Manheim, Germany). Aminoethylcarbazole (Dako Cytomation, Copenhagen Denmark) or diaminobenzidine (Sigma-Aldrich, Munich, Germany) was used as the substrate yielding red or brown precipitate. Counterstaining was performed with Shandon hematoxylin. Slides were analyzed on an Olympus BX40 microscope, and images were acquired by Olympus digital camera (C-3030).
Quantitative PCR.
DNA was extracted from mouse newborn brain (10 mg) by use of a DNA extraction kit (Wizard Promega) and dissolved in 100 μl of rehydration solution. Primers ie3/4fwd (5′-TGACTTAAACTCCCCAGGCAA-3′) and ie3/4rev (5′-TAGGTGAGGCCATAGTGGCAG-3′) were chosen to amplify a segment of exon 4 of the ie1 gene. A cellular gene was detected with primers glra1fwd (5′-TGCCTGTTCTTTGCAGTCTGT-3′) and glra1rev (5′-AGTCGAGTGAAGGGTAACGAGC-3′). Reaction mixtures of 20 μl consisted of 4 μl of LightCycler FastStart DNA MasterPlus Sybr Green I (Roche Applied Science), a 0.5 μM concentration of each primer, and 5 μl of sample DNA. PCR amplification was done for 45 cycles. Serial dilutions of pGEM-Teasy (Promega) expressing MCMV ie3/4 and of DNA extracted from murine embryonic fibroblasts were used as standards to determine the MCMV genome copy numbers and the number of cells, respectively. Five standards spanning 5 orders of magnitude were used in duplicate. Brain DNA samples from uninfected mice and multiple samples without template served as negative controls. Spectra were acquired in real time and were evaluated with sequence detector software (Applied Biosystems) and spectral compensation.
Cerebellar morphometry.
Images of CV-stained cerebellar sections were acquired by a Pulmix camera (TMC 76S; Japan) mounted on an Olympus BX 40 microscope, and morphometrical measurements were performed with ISSA software (VAMS, Zagreb, Croatia) and IPLab (Scanalytics, Inc., Fairfax, VA) software. Thickness of the cerebellar external granular layer (EGL) was measured at six points along the primary cerebellar fissure. The number of Purkinje cells was counted along 500 μm at both sides of the primary fissure.
MCMV immune serum and MAb 97.3 preparation.
Immune serum was collected from adult BALB/c mice infected with 2 × 105 PFU of WT MCMV for 3 to 4 months. Control serum was collected from uninfected age-matched BALB/c mice. MCMV-specific MAb 97.3 (IgG2a) was generated from MCMV-infected C57BL/6 mice challenged with 2 μg of MCMV particles intravenously 4 days before fusion essentially as described previously (11). Supernatant from growing hybridoma was used for an in vitro neutralization assay using the MCMV157luc virus (23). Supernatant showing strong neutralizing activity was further characterized for antigen specificity. The M55 gene encoding gB of WT MCMV (38) was cloned into the plasmid pcDNA3 and used in indirect immunofluorescence analysis after transient expression of the protein in Cos7 cells. Clone 97.3 showed strong reactivity, in contrast to control cells expressing unrelated MCMV glycoproteins that were negative in this assay. Ascites was produced by i.p. injection of 106 97.3 hybridoma cells into pristane-primed mice and purified with 30% ammonium sulfate. Protein concentration was determined by enzyme-linked immunosorbent assay.
Determination of neutralizing titer of anti-MCMV serum and MAbs.
Neutralization assays were performed as described previously (46). In brief, the titer of neutralizing antibody in serum of latently infected mice was defined as the maximal plaque reduction in a conventional plaque reduction assay. A total of 102 PFU of MCMV in 0.1 ml of plaque assay medium (3% Dulbecco's modified Eagle's medium) was incubated on ice for 30 min with 0.1 ml of serial dilutions of immune serum or MAbs, followed by plaque assay.
Neutralization units of MAb 97.3 were defined from in vitro neutralization assay as micrograms of MAb used to neutralize 100% of 102 virus PFU in a conventional plaque reduction assay (12).
In vivo inoculation of immune serum and MAb 97.3.
Newborn mice infected with MCMV were randomized and inoculated i.p. with immune serum, control serum (100 μl of either), or anti-MCMV MAb 97.3, isotype-matched control antibodies (IgG2a) (100 μg of either), or PBS (100 μl). The antibodies were administrated on day 5 or day 9 postinfection (p.i.).
Statistical evaluation.
Results from different groups of mice were compared by a Mann-Whitney exact rank sum test using Statistica software (version 7.0). For all analyses the significance level was set at a P value of <0.05.
RESULTS
MCMV replication kinetics and spread within the newborn brain parenchyma.
We have recently shown that MCMV infection of newborn mice results in brain infection, inflammation, and developmental abnormalities of the cerebellum (24). Here, we used MCMV infection of newborn mice to assess the protective capacity of antiviral antibodies with respect to virus replication and related neuropathology. In agreement with previously described results, we observed that i.p. injection of MCMV in newborn mice results in peak virus replication within the brain around day 11 p.i., and clearance of replicating virus occurs around day 17 p.i. (Fig. 1A). Notably, virus detection in the liver preceded the virus detection in the brain although the kinetics of virus replication were similar in both organs (Fig. 1B).
FIG. 1.

Kinetics of virus replication and spread in brain of MCMV-infected newborn mice. MCMV replication in the brains (A) and livers (B) of newborn mice. Mice were inoculated i.p. with 500 PFU of MCMV, 6 to 12 h after birth, and viral titers in their organs were assessed at various time points p.i. Productive infection in the brains was detected starting from day 7 p.i. and was visible through day 17 p.i. Titers of virus in individual mice (circles) and median values (horizontal bars) are shown. DL, detection limit. (C to G) MCMV antigen expression in the brains of newborn mice. Scattered infected cells located in the cerebral cortex, hippocampus, olfactory bulb, colliculi, and cerebellar cortex of infected mice are shown, as indicated on the figure, at day 11 p.i. Immunohistochemical staining with MCMV IE1-specific MAbs reactive with MCMV IE1 protein is shown. Magnification, ×20 (C, E, and F) and ×40 (D and G).
Immunohistological studies at the peak of virus replication (day 11 p.i.) revealed widespread distribution of infected cells with no particular preference to any region or cell type within the brain. According to nuclear staining for IE1 MCMV protein pp89, infected cells were detected in the cerebral cortex, hippocampus, olfactory bulb, thalamus, colliculi, and cerebellum (Fig. 1C to G). Virus-infected cells were also detected in brain stem, meninges, ependyma, and choroid plexuses of the ventricles (data not shown). Of note, although this type of infection led to readily observable abnormalities in the development of entire regions of the brain, such as cerebellum, foci of infected cells in the CNS were solitary and infrequent.
Anti-MCMV serum reduces MCMV titer in brains of infected newborn mice.
To determine the effect of antiviral antibodies on virus replication in the MCMV-infected brain, newborn BALB/c mice (day 0) were infected with 500 PFU of MCMV by i.p. inoculation and treated systemically with immune serum at days 5 or 9 p.i. Control animals received nonimmune mouse serum or PBS. Mice were sacrificed on days 11 and 14 p.i., and virus titers in brain and liver were determined. In agreement with previous results, all control mice given PBS showed significant virus replication on days 11 and 14 p.i., and injection of nonimmune serum did not affect the virus replication, findings that were similar to mice injected with PBS only. In contrast, the majority of mice inoculated with MCMV immune serum had no detectable replicating virus in the brain when the immune serum was given on day 5 p.i. (Fig. 2A). Although mice that received immune serum on day 9 p.i. showed only moderate protection at day 11 p.i., these animals efficiently controlled the infection in the CNS by day 14 p.i. when no infectious virus could be detected in the brain, confirming the antiviral activity of immune serum (Fig. 2B). The protective capacity of the immune serum was not restricted to the brain since serum administration also abrogated virus replication in the liver (data not shown). Finally, to rule out the unlikely possibility that residual antibody present in the parenchyma could have reduced virus titers in the infectivity assays, we also assayed brain tissue from treated animals for viral DNA by quantitative PCR. Results from assays of brain tissue from p.i. days 11 and 14 in mice treated with immune serum even on day 9 p.i. revealed reductions in viral genome copy numbers on day 14 p.i. of 1 log compared to tissue from mice given PBS only (Fig. 2C). These results demonstrated a strong protective capacity of immune serum in MCMV-infected newborn mouse brain that was associated with the reduction of both infectious virus and viral genome copy number. The observed differences in the protective effect of antibodies on the titers of infectious virus compared to viral DNA load in the brain was not unexpected since the quantitative PCR method allows detection of not only DNA of infectious virus but also the DNA in cells containing nonreplicating viral genome, including cells that populate the developing CNS such as infiltrating microglia. Immunohistochemical analysis of MCMV-infected newborn mice treated with immune serum confirmed the results obtained by virus titration and showed a reduced number of infected cells expressing viral IE1 protein (data not shown). Infected cells, although fewer in number, showed the same pattern of distribution in the brains of mice treated with immune serum as control mice.
FIG. 2.
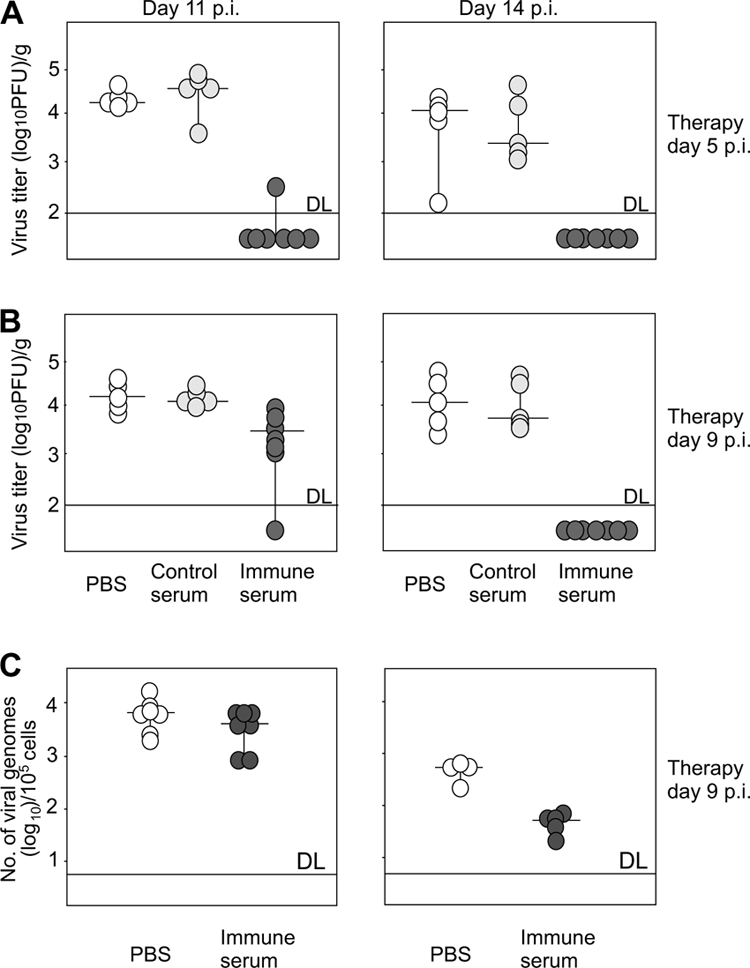
Immune serum limits the recovery of the infectious virus and reduces the load of viral genomes in the brains of MCMV-infected newborn mice. MCMV-infected newborn BALB/c mice received 100 μl of immune serum (black circles), control serum (gray circles), or equal amount of PBS (open circles) on day 5 (A) or day 9 p.i. (B). Virus titers were determined in the brain on days 11 and 14 p.i. (C) Viral DNA load was determined by quantitative PCR in the brains of mice receiving immune serum (black circles) or PBS (open circles) at day 9 p.i. on days 11 and 14 p.i. Horizontal bars represent the median value. DL, detection limit.
Anti-MCMV MAbs reduce virus replication in newborn mouse brains.
The efficacy of serum transfer into MCMV-infected newborn mice suggested that antibodies can control virus replication in the brain. To exclude an involvement of other immune serum components in addition to virus-specific antibodies, we tested the capacity of virus-specific MAbs to limit virus replication in the brain. MCMV-infected newborn BALB/c mice were injected i.p. with virus-neutralizing MAb 97.3 specific for MCMV gB (data not shown). The neutralizing capacity of this MAb was determined in vitro (see Fig. 1 posted at http://www.medri.hr/∼jstipan/data), and each mouse received 12.5 neutralizing units of purified MAbs i.p. Control animals received an irrelevant isotype-matched MAb. Virus-specific MAbs were also protective in vivo (Fig. 3). Mice injected with MAb 97.3 showed decreased viral loads in the brain of approximately 1 log compared to control animals treated with isotype-matched MAbs. However, in comparison to the protective capacity of MCMV-specific immune serum, MAbs did not limit virus replication below the level of detection in our assays (Fig. 3A and B).
FIG. 3.
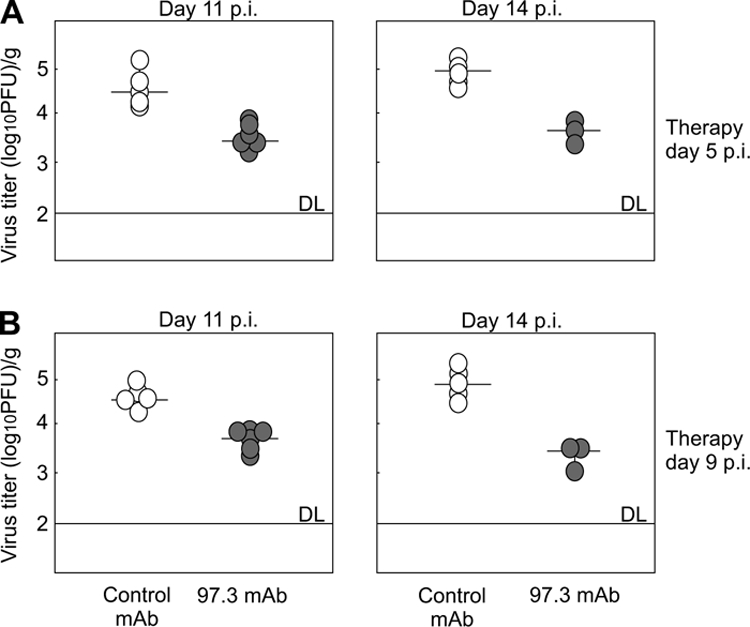
Anti-MCMV MAb 97.3 limits virus recovery from the brains of MCMV-infected newborn mice. MCMV-infected BALB/c mice received 100 μg of 97.3 (•) or control MAb of IgG2a isotype (○) on day 5 (A) or day 9 p.i. (B) Virus titers were determined in the brain on days 11 and 14 p.i. Titers of virus in individual mice (circles) and median values (horizontal bars) are shown. DL, detection limit.
The 97.3 MAbs were visualized in the parenchyma of the brains of infected animals treated on day 9 p.i. by immunohistochemistry. The passively transferred antibodies were detected surrounding the brain endothelium and, in some cases, around neurons, suggesting that passively acquired antibodies could enter the CNS of MCMV-infected young mice (see Fig. 2 posted at http://www.medri.hr/∼jstipan/data). Whether the effector function of passively acquired anti-MCMV antibodies is at the level of endothelium or within the infected parenchyma cannot be resolved in these experiments and will require additional study.
Protective capacity of MCMV-specific immune serum and MAbs correlates with reduction of virus-induced brain pathology.
Next, we analyzed the effects of passive transfer of antiviral antibodies on the inflammatory response in brains of MCMV-infected newborn mice. As described previously (24, 25), inflammatory lesions were observed in all parts of the brain of infected animals. These histopathological lesions varied in size, quantity, and distribution. The observed lesions included edema, infiltrates of mononuclear cells, micronodular gliosis, perivascular cuffing, and reactive gliosis. In some cases cortical involvement dominated whereas in other cases lesions were equally divided in gray and white matter.
The first pathological lesions observed on day 7 p.i. consisted of mononuclear cell infiltrations, localized to the meninges and choroid plexus (Fig. 4A and D). Inflammatory lesions were observed within the brain parenchyma by day 11 p.i. (Fig. 4B to F). The kinetics of brain inflammatory lesions resembled the kinetics of detection of infectious virus, but the peak number of lesions and the overall kinetics of their appearance were delayed in comparison to the kinetics of virus replication in the CNS. The productive infection terminated by day 17 p.i., but a focal encephalitic response persisted even by day 80 p.i. (Fig. 4G). To assess whether the protective capacity of MAbs influences the number and the severity of inflammatory lesions, we infected newborn BALB/c mice and subsequently treated the animals with immune serum or MAbs. Histological examination revealed a dramatic reduction in the number of histopathological lesions in brains of mice treated with immune serum or MAbs in contrast to control animals. Antibody transfer on day 5 p.i. resulted in a significant reduction of the number of inflammatory lesions by day 14 p.i., which was also observed through day 21 p.i. (Fig. 5A). Likewise, the transfer of MAbs resulted in pronounced reduction of inflammatory lesions in the infected brains (Fig. 5B), arguing that antiviral antibodies could limit not only virus replication in the CNS but also the MCMV-induced disease in the brain.
FIG. 4.

Histopathological lesions in brains of MCMV-infected newborn mice. CV staining of different brain regions show acute meningitis (A) and mononuclear cell infiltration in choroids plexus (D) on day 7 p.i., mononuclear cell infiltration in cerebellar cortex (B), perivascular cuffing in hippocampus (C) on day 11 p.i., and edema (E) and glial node (F) in hindbrain on day 14 p.i. Magnification, ×20 (A to E) and ×40 (F and insets). Histopathological lesions were observed in brain parenchyma even at day 80 p.i. (G).
FIG. 5.
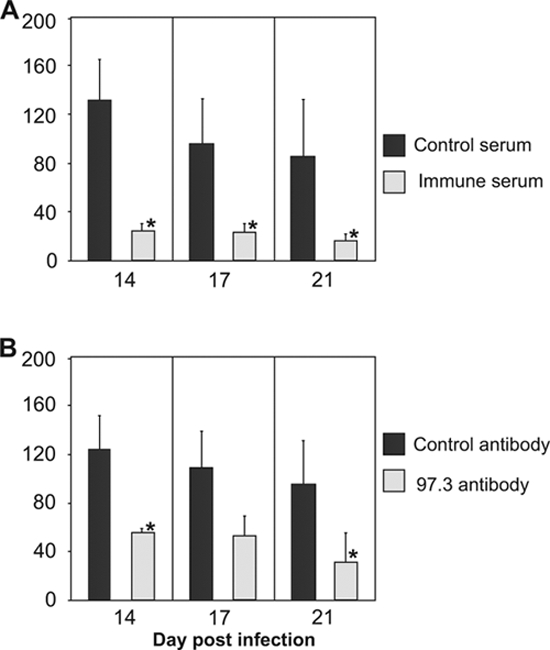
Protective effect of immune serum or anti-MCMV MAbs on the development of histopathology in infected newborn brain. MCMV-infected mice received 100 μl of immune serum and control serum (A) or 100 μg of MAb 97.3 and control antibody (B) on day 5 p.i. Histopathology was evaluated by counting the lesions in CV-stained slides of infected brains on days 14, 17, and 21 p.i. Each bar is representative of a minimum of three animals per group. *, P < 0.05.
Altogether, the results confirmed that the transfer of antiviral antibodies reduced the virus load and, even more importantly, the development and the persistence of inflammatory pathology in the brain tissue.
Administration of anti-MCMV antibodies prevents developmental brain abnormalities in MCMV-infected newborn mice.
MCMV infection of the CNS has been shown to alter the normal postnatal development of the cerebellum, which is manifested by delayed migration of neurons from the cerebellar EGL into deeper parts of cerebellar cortex (24). To determine if the passive transfer of antiviral antibodies would also reduce cerebellar developmental abnormalities in CMV infection of the CNS, we compared the EGL thickness between groups of animals that received immune serum, control serum, or MCMV-specific MAbs on day 5 p.i. Mice treated with control serum manifested increased EGL thickness, similar to our previous observation in MCMV-infected mice (24). In mice treated with anti-MCMV immune serum or MAbs, the EGL thickness was reduced to levels comparable to those observed in uninfected control mice (Fig. 6B and C). Also, mice treated with immune serum or MAbs exhibited cerebellar area that was similar to that of the control, uninfected mice, whereas MCMV-infected mice receiving control serum exhibited a significantly reduced cerebellar area on days 11 and 14 p.i. (Fig. 6A). Immunohistological analysis using anti-calbindin MAbs specific for Purkinje cells demonstrated defective arborization and reduced numbers of these cells in the MCMV-infected cerebellum. Antibody-treated mice showed a significant reduction or even absence of these abnormalities (Fig. 6D).
FIG. 6.

Antiviral antibodies prevent MCMV-induced developmental abnormalities in the cerebella of newborn mice. (A) Cerebellar area in infected and uninfected newborn mice. Mice treated with immune serum on day 5 p.i. show significant increase (*, P < 0.05) in cerebellar area compared to untreated infected mice (days 11 and 14 p.i.). Mice treated with MAb 97.3 on day 5 p.i. show improvement in cerebellar area but without statistically significant difference to control infected mice. (B) EGL thickness in MCMV-infected and uninfected cerebellum. Mice treated with immune serum on day 5 p.i. show significantly decreased (*, P < 0.05) EGL thickness on day 11 p.i. compared to untreated infected mice. Mice treated with MAb 97.3 also have decreased EGL thickness but with no statistical difference relative to control infected animals. (C) EGL thickness in MCMV-infected cerebellum treated with control serum (left) or immune serum (right) on day 12 p.i. is shown in CV-stained sections. Magnification, ×20. (D) Purkinje cell alignment in MCMV-infected cerebellum is modified by treatment with antiviral antibodies. MCMV-infected cerebellum (left panel) and cerebellum of infected, immune serum-treated mice (right panel) are shown. Anti-calbindin MAb was used for immunohistochemical staining. Magnification, ×40.
These results demonstrated that MCMV-specific immune serum and MAbs reduced the viral load in the brain and as a consequence led to a decrease in the degree of inflammatory changes associated with this virus infection of the CNS. In addition, the decrease in levels of virus replication and inflammation was associated with lessening of the developmental abnormalities associated with this infection of the CNS in MCMV-infected newborn mice.
DISCUSSION
Congenital HCMV infection is an important public health issue, primarily due to long-term sensorimotor deficits that are observed after infection of the developing nervous system (28). The pathogenesis of CMV encephalitis is still insufficiently defined because of the limited number of autopsy cases with a comprehensive description of the pathological changes detected in infected infants. Since CMV can readily infect endothelial cells (13, 42), it has been postulated that a virus-induced vasculitis might be responsible for loss of vascular supply to regions of the developing brain, resulting in maldevelopment (30). Other investigators have postulated a direct cytopathic effect of CMV on developing neurons and glial cells (58). The immune response to CMV infection may also contribute to the pathogenesis of CNS disease. Specifically, elaboration of immune responses in this particular tissue may have immunopathological consequences as described for CNS infections with other viruses (17, 31, 60). Our recent studies indicate that infiltrating CD8+ T cells in MCMV-infected newborn brain mediate their antiviral activity mostly via a noncytolytic pathway (1). This finding could in part explain the histological characteristics of nonnecrotizing encephalitis following MCMV infection in newborn mouse brain (24). Importantly, the histopathology observed in the brains of mice infected with MCMV as newborns resembles findings reported in autopsied cases of human congenital CMV infections (33).
Antiviral antibodies are powerful effector functions for virus neutralization and prevention of virus spread. Although not required for resolution of primary infection, antibodies have proven to be essential for prevention of virus dissemination after recurrence or reinfection (21, 35). Using the mouse model of CMV-induced encephalitis, we tested the capacity of antiviral antibodies to reduce virus replication in brain and CMV-related brain pathology in newborn mice. We have demonstrated that anti-MCMV antibodies not only lowered the burden of infectious virus present in brain tissue but also dramatically reduced the associated neuropathology. Mice that received antiviral antibodies exhibited fewer and more limited histopathological lesions and had improved postnatal development of cerebellum compared to control infected mice.
Antibody-mediated neutralization of viruses has been extensively studied in vitro, but the precise mechanism that provides protection against viral infection in vivo still remains largely uncharacterized. Several studies have suggested that passive immunization reduces CMV infection. High levels of virus-neutralizing antibodies were associated with delayed progression of HCMV-induced retinitis in AIDS patients, and passive transfer of antiviral antibodies into solid organ transplant recipients has been correlated with improved outcome and reduced severity of HCMV infections (7, 47). Although the idea is controversial, it has recently been claimed that adoptive transfer of virus-neutralizing antibodies to women undergoing primary infection during pregnancy limited the rate of intrauterine transmission of HCMV to the developing fetus (32), whereas other studies have shown that antibodies play little, if any, role in the protection against virus-induced neurologic damage in the infected fetus following primary maternal infection (5). However, maternal preconceptual immunity, as measured by the presence of antiviral antibodies, does appear to reduce the rates of congenital CMV infection in subsequent pregnancies in both humans and the guinea pig model of human CMV infection (9, 15).
In animal models antibodies have been shown to be essential for virus clearance from infected brain (16). The mechanism of antibody-mediated clearance of the virus in this model remains incompletely defined. Both virus-neutralizing and -nonneutralizing antibodies could affect virus clearance, and immunoglobulin subclass appears important for virus clearance (56). A series of experiments has been consistent with these findings, showing that nonneutralizing antibodies (as determined in in vitro assays) can protect mice from lethal neurovirulent Sindbis virus encephalitis (50). Although we have not extended our studies to include MAbs of different subclasses and different neutralization capacities, in this report we show that the administration of antibodies with virus-neutralizing activity reduced the number of infected cells detected in the CNS. In the current study we also observed that the neutralizing capacity of a MAb in vitro does not necessarily correlate with its antiviral activity in vivo. In mice that received antibodies, the distribution of virus-infected cells was similar to findings in control mice, indicating that antiviral antibodies did not alter the pattern of virus infection of the CNS but more likely reduced the amount of the virus in the brain. We also observed a lower viral burden in brains of mice that received antibodies on day 5 p.i., supporting the possibility that antibody-mediated neutralization of cell-free virus in blood of newborn mice could effectively limit the access of MCMV to CNS. It is important to point out that both cell-associated and free virus could be detected in blood of newborn mice (see Fig. 3 posted at http://www.medri.hr/∼jstipan/data). This finding suggests that antibodies applied on day 5 p.i. may prevent virus spread into developing brain by both direct neutralization of the virus or by mechanisms which clear virus-infected cells such as antibody-dependent cellular cytotoxicity. However, antibodies given to infected mice on day 9 p.i., a time that coincided with the peak of virus replication in the brain, were also protective, suggesting that neutralization of cell-free virus in the blood of infected animals and limitation of seeding the brain were not the only mechanisms of antiviral activity in this model and that antiviral antibodies could be functioning within the CNS to limit virus spread and/or replication as shown for other viruses (57). Recent studies show that antibodies are capable of crossing the blood-brain barrier after i.p. inoculation (44). In animals that received MAbs, we detected antibodies around blood vessel endothelium as well as around neuronal cells, suggesting that antibodies infiltrate the CNS (see Fig. 4 posted at http://www.medri.hr/∼jstipan/data). Although the mechanism of transfer of antiviral IgG antibodies into the CNS in these young animals has not been fully determined, preliminary studies using Evans blue to document leakage of the blood-brain barrier did not indicate that diffusion of IgG into the brain secondary to disruption of the blood-brain barrier contributed to the antiviral activity in CNS (data not shown). Other mechanisms of IgG transport such as facilitated transport through the endothelium or perhaps binding to mononuclear cells through interactions with Fc remain potential explanations (37).
We also found that adoptive transfer of antibodies limited development of brain lesions. Histopathological findings in congenital HCMV infections include ventriculoencephalitis consisting of periventricular necrosis, intraventricular hemorrhage, and calcifications (3, 33). In a murine model of congenital CMV infection, widespread encephalitis is a common finding following direct intracranial inoculation of the virus (10, 25). In this model histopathological lesions were comprised of infected cells which were associated with infiltration of inflammatory cells, including mono- and polymorphonuclear leukocytes, or activation of resident microglia (25, 59). In our model we also observed histopathological lesions characteristic of meningoencephalitis. The first observed lesion was leptomeningitis, whose appearance coincided with detection of the virus in the brain, whereas lesions in the parenchyma appeared later. The most obvious response in the brain parenchyma was perivascular cuffing composed of mononuclear cells. In contrast, glial nodules represented a major local response. Lesions persisted even when replicating virus was no longer detected in the brain, suggesting that immune cells resided in the brain parenchyma despite the termination of productive virus infection. We have recently shown that CD8+ T lymphocytes are a predominant immune cell population in MCMV-infected newborn brain and are present in the CNS after the resolution of an acute infection (1). In infected newborn mice that received antibody treatment, histopathological findings in brain tissue showed distinctive differences among the extent and severity of parenchyma destruction compared to control mice. Administration of immune serum reduced both the number and severity of inflammatory lesions. Moreover, treatment with immune serum shortened the interval required for reestablishment of the normal brain architecture in infected mice.
Newborn mice infected with MCMV exhibit altered cerebellar morphogenesis in terms of foliation delay, impaired morphology of Purkinje cells, and persistence of EGL (24). Antibody transfer into MCMV-infected newborn mice resulted in improved development of infected cerebellum. These mice showed EGL thickness comparable to uninfected mice, normal Purkinje cell morphology, and increased cerebellar area compared to untreated infected mice. Therefore, antiviral antibodies also appeared to be effective in preventing virus-associated developmental abnormalities and altered neuronal migration, suggesting that limiting virus replication and presumably the host inflammatory response also decreased the developmental alterations associated with virus replication in the CNS.
In summary, this experimental model may provide insight into the pathogenesis of CMV injury and support the efforts toward devising optimal antibody-based therapeutic approaches.
Acknowledgments
We thank J. Trgovcich for critically reading the manuscript.
This work was supported by NIH grant R01 HD044721 and Croatian Ministry of Science, Education and Sport grants 062-0621261-1263 and 062-0621261-1269.
Footnotes
Published ahead of print on 8 October 2008.
REFERENCES
- 1.Bantug, G. R., D. Cekinovic, R. Bradford, T. Koontz, S. Jonjic, and W. J. Britt. 2008. CD8+ T lymphocytes control murine cytomegalovirus replication in the central nervous system of newborn animals. J. Immunol. 1812111-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barkovich, A. J., and C. E. Lindan. 1994. Congenital cytomegalovirus infection of the brain: imaging analysis and embryologic considerations. AJNR Am. J. Neuroradiol. 15703-715. [PMC free article] [PubMed] [Google Scholar]
- 3.Becroft, D. M. O. 1981. Prenatal cytomegalovirus infection: epidemiology, pathology and pathogenesis, p. 203-241. In H. S. Rosenberg and J. Bernstein (ed.), Perspective in pediatric pathology. Masson Press, New York, NY. [PubMed]
- 4.Booss, J., P. R. Dann, B. P. Griffith, and J. H. Kim. 1988. Glial nodule encephalitis in the guinea pig: serial observations following cytomegalovirus infection. Acta Neuropathol. 75465-473. [DOI] [PubMed] [Google Scholar]
- 5.Boppana, S. B., and W. J. Britt. 1995. Antiviral antibody responses and intrauterine transmission after primary maternal cytomegalovirus infection. J. Infect. Dis. 1711115-1121. [DOI] [PubMed] [Google Scholar]
- 6.Boppana, S. B., K. B. Fowler, R. F. Pass, L. B. Rivera, R. D. Bradford, F. D. Lakeman, and W. J. Britt. 2005. Congenital cytomegalovirus infection: association between virus burden in infancy and hearing loss. J. Pediatr. 146817-823. [DOI] [PubMed] [Google Scholar]
- 7.Boppana, S. B., M. A. Polis, A. A. Kramer, W. J. Britt, and S. Koenig. 1995. Virus-specific antibody responses to human cytomegalovirus (HCMV) in human immunodeficiency virus type 1-infected persons with HCMV retinitis. J. Infect. Dis. 171182-185. [DOI] [PubMed] [Google Scholar]
- 8.Brune, W., H. Hengel, and U. H. Koszinowski. 1999. A mouse model for cytomegalovirus infection. John Wiley & Sons, New York, NY. [DOI] [PubMed]
- 9.Chatterjee, A., C. J. Harrison, W. J. Britt, and C. Bewtra. 2001. Modification of maternal and congenital cytomegalovirus infection by anti-glycoprotein b antibody transfer in guinea pigs. J. Infect. Dis. 1831547-1553. [DOI] [PubMed] [Google Scholar]
- 10.Cheeran, M. C., G. Gekker, S. Hu, X. Min, D. Cox, and J. R. Lokensgard. 2004. Intracerebral infection with murine cytomegalovirus induces CXCL10 and is restricted by adoptive transfer of splenocytes. J. Neurovirol. 10152-162. [DOI] [PubMed] [Google Scholar]
- 11.Farrell, H. E., and G. R. Shellam. 1990. Characterization of neutralizing monoclonal antibodies to murine cytomegalovirus. J. Gen. Virol. 71655-664. [DOI] [PubMed] [Google Scholar]
- 12.Farrell, H. E., and G. R. Shellam. 1991. Protection against murine cytomegalovirus infection by passive transfer of neutralizing and non-neutralizing monoclonal antibodies. J. Gen. Virol. 72149-156. [DOI] [PubMed] [Google Scholar]
- 13.Fish, K. N., C. Soderberg-Naucler, L. K. Mills, S. Stenglein, and J. A. Nelson. 1998. Human cytomegalovirus persistently infects aortic endothelial cells. J. Virol. 725661-5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flanagan, E. B., T. R. Schoeb, and G. W. Wertz. 2003. Vesicular stomatitis viruses with rearranged genomes have altered invasiveness and neuropathogenesis in mice. J. Virol. 775740-5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fowler, K. B., S. Stagno, and R. F. Pass. 2003. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 2891008-1011. [DOI] [PubMed] [Google Scholar]
- 16.Griffin, D., B. Levine, W. Tyor, S. Ubol, and P. Despres. 1997. The role of antibody in recovery from alphavirus encephalitis. Immunol. Rev. 159155-161. [DOI] [PubMed] [Google Scholar]
- 17.Hallensleben, W., M. Schwemmle, J. Hausmann, L. Stitz, B. Volk, A. Pagenstecher, and P. Staeheli. 1998. Borna disease virus-induced neurological disorder in mice: infection of neonates results in immunopathology. J. Virol. 724379-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hangartner, L., R. M. Zinkernagel, and H. Hengartner. 2006. Antiviral antibody responses: the two extremes of a wide spectrum. Nat. Rev. Immunol. 6231-243. [DOI] [PubMed] [Google Scholar]
- 19.Hayward, J. C., D. S. Titelbaum, R. R. Clancy, and R. A. Zimmerman. 1991. Lissencephaly-pachygyria associated with congenital cytomegalovirus infection. J. Child Neurol. 6109-114. [DOI] [PubMed] [Google Scholar]
- 20.Hudson, J. B. 1979. The murine cytomegalovirus as a model for the study of viral pathogenesis and persistent infections. Arch. Virol. 621-29. [DOI] [PubMed] [Google Scholar]
- 21.Jonjic, S., I. Pavic, B. Polic, I. Crnkovic, P. Lucin, and U. H. Koszinowski. 1994. Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J. Exp. Med. 1791713-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawasaki, H., I. Kosugi, Y. Arai, and Y. Tsutsui. 2002. The amount of immature glial cells in organotypic brain slices determines the susceptibility to murine cytomegalovirus infection. Lab. Investig. 821347-1358. [DOI] [PubMed] [Google Scholar]
- 23.Klenovsek, K., F. Weisel, A. Schneider, U. Appelt, S. Jonjic, M. Messerle, B. Bradel-Tretheway, T. H. Winkler, and M. Mach. 2007. Protection from CMV infection in immunodeficient hosts by adoptive transfer of memory B cells. Blood 1103472-3479. [DOI] [PubMed] [Google Scholar]
- 24.Koontz, T., M. Bralic, J. Tomac, E. Pernjak-Pugel, G. Bantug, S. Jonjic, and W. J. Britt. 2008. Altered development of the brain after focal herpesvirus infection of the central nervous system. J. Exp. Med. 205423-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosugi, I., H. Kawasaki, Y. Arai, and Y. Tsutsui. 2002. Innate immune responses to cytomegalovirus infection in the developing mouse brain and their evasion by virus-infected neurons. Am. J. Pathol. 161919-928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koszinowski, U. H., M. J. Reddehase, and S. Jonjic. 1991. The role of CD4 and CD8 T cells in viral infections. Curr. Opin. Immunol. 3471-475. [DOI] [PubMed] [Google Scholar]
- 27.Krmpotic, A., I. Bubic, B. Polic, P. Lucin, and S. Jonjic. 2003. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 51263-1277. [DOI] [PubMed] [Google Scholar]
- 28.Malm, G., and M. L. Engman. 2007. Congenital cytomegalovirus infections. Semin. Fetal Neonatal Med. 12154-159. [DOI] [PubMed] [Google Scholar]
- 29.Malm, G., E. H. Grondahl, and I. Lewensohn-Fuchs. 2000. Congenital cytomegalovirus infection: a retrospective diagnosis in a child with pachygyria. Pediatr. Neurol. 22407-408. [DOI] [PubMed] [Google Scholar]
- 30.Marques Dias, M. J., G. Harmant-van Rijckevorsel, P. Landrieu, and G. Lyon. 1984. Prenatal cytomegalovirus disease and cerebral microgyria: evidence for perfusion failure, not disturbance of histogenesis, as the major cause of fetal cytomegalovirus encephalopathy. Neuropediatrics 1518-24. [DOI] [PubMed] [Google Scholar]
- 31.Moore, M. L., C. C. Brown, and K. R. Spindler. 2003. T cells cause acute immunopathology and are required for long-term survival in mouse adenovirus type 1-induced encephalomyelitis. J. Virol. 7710060-10070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31a.National Research Council. 2004. International guiding principles for biomedical research involving animals, p. 240-248. In The development of science-based guidelines for laboratory animal care. Proceedings of the November 2003 International Workshop. National Academies Press, Washington, DC. [PubMed]
- 32.Nigro, G., S. P. Adler, R. La Torre, and A. M. Best. 2005. Passive immunization during pregnancy for congenital cytomegalovirus infection. N. Engl. J. Med. 3531350-1362. [DOI] [PubMed] [Google Scholar]
- 33.Perlman, J. M., and C. Argyle. 1992. Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann. Neurol. 3164-68. [DOI] [PubMed] [Google Scholar]
- 34.Poland, S. D., L. L. Bambrick, G. A. Dekaban, and G. P. Rice. 1994. The extent of human cytomegalovirus replication in primary neurons is dependent on host cell differentiation. J. Infect. Dis. 1701267-1271. [DOI] [PubMed] [Google Scholar]
- 35.Polic, B., H. Hengel, A. Krmpotic, J. Trgovcich, I. Pavic, P. Luccaronin, S. Jonjic, and U. H. Koszinowski. 1998. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J. Exp. Med. 1881047-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puius, Y. A., and D. R. Snydman. 2007. Prophylaxis and treatment of cytomegalovirus disease in recipients of solid organ transplants: current approach and future challenges. Curr. Opin. Infect. Dis. 20419-424. [DOI] [PubMed] [Google Scholar]
- 37.Ravetch, J. V., and S. Bolland. 2001. IgG Fc receptors. Annu. Rev. Immunol. 19275-290. [DOI] [PubMed] [Google Scholar]
- 38.Rawlinson, W. D., H. E. Farrell, and B. G. Barrell. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 708833-8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reddehase, M. J., F. Weiland, K. Munch, S. Jonjic, A. Luske, and U. H. Koszinowski. 1985. Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J. Virol. 55264-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reuter, J. D., D. L. Gomez, J. H. Wilson, and A. N. Van Den Pol. 2004. Systemic immune deficiency necessary for cytomegalovirus invasion of the mature brain. J. Virol. 781473-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross, S. A., and S. B. Boppana. 2005. Congenital cytomegalovirus infection: outcome and diagnosis. Semin. Pediatr. Infect. Dis. 1644-49. [DOI] [PubMed] [Google Scholar]
- 42.Rue, C. A., M. A. Jarvis, A. J. Knoche, H. L. Meyers, V. R. DeFilippis, S. G. Hansen, M. Wagner, K. Fruh, D. G. Anders, S. W. Wong, P. A. Barry, and J. A. Nelson. 2004. A cyclooxygenase-2 homologue encoded by rhesus cytomegalovirus is a determinant for endothelial cell tropism. J. Virol. 7812529-12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saederup, N., and E. S. Mocarski, Jr. 2002. Fatal attraction: cytomegalovirus-encoded chemokine homologs. Curr. Top. Microbiol. Immunol. 269235-256. [DOI] [PubMed] [Google Scholar]
- 44.Sas, A., R. Jones, and W. Tyor. 7 May 2008. Intra-peritoneal injection of polyclonal anti-interferon alpha antibodies cross the blood brain barrier and neutralize interferon alpha. Neurochem. Res. 332281-2287. [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 45.Schoppel, K., C. Schmidt, H. Einsele, H. Hebart, and M. Mach. 1998. Kinetics of the antibody response against human cytomegalovirus-specific proteins in allogeneic bone marrow transplant recipients. J. Infect. Dis. 1781233-1243. [DOI] [PubMed] [Google Scholar]
- 46.Shimamura, M., M. Mach, and W. J. Britt. 2006. Human cytomegalovirus infection elicits a glycoprotein M (gM)/gN-specific virus-neutralizing antibody response. J. Virol. 804591-4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snydman, D. R. 2001. Historical overview of the use of cytomegalovirus hyperimmune globulin in organ transplantation. Transpl. Infect. Dis. 3(Suppl. 2)6-13. [DOI] [PubMed] [Google Scholar]
- 48.Snydman, D. R., B. G. Werner, H. C. Meissner, S. H. Cheeseman, J. Schwab, F. Bednarek, J. L. Kennedy, Jr., M. Herschel, A. Magno, M. J. Levin, et al. 1995. Use of cytomegalovirus immunoglobulin in multiply transfused premature neonates. Pediatr. Infect. Dis. J. 1434-40. [DOI] [PubMed] [Google Scholar]
- 49.Stagno, S., and W. J. Britt. 2006. Cytomegalovirus. In J. S. Remington and J. O. Klein (ed.), Diseases of the fetus and newborn infant, 6th ed. W. B. Saunders, Philadelphia, PA.
- 50.Stanley, J., S. J. Cooper, and D. E. Griffin. 1986. Monoclonal antibody cure and prophylaxis of lethal Sindbis virus encephalitis in mice. J. Virol. 58107-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stoddart, C. A., R. D. Cardin, J. M. Boname, W. C. Manning, G. B. Abenes, and E. S. Mocarski. 1994. Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J. Virol. 686243-6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stranska, R., R. Schuurman, M. Toet, M. Verboon-Maciolek, L. S. de Vries, and A. M. van Loon. 2006. Application of UL144 molecular typing to determine epidemiology of cytomegalovirus infections in preterm infants. J. Clin. Microbiol. 441108-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tarantal, A. F., M. S. Salamat, W. J. Britt, P. A. Luciw, A. G. Hendrickx, and P. A. Barry. 1998. Neuropathogenesis induced by rhesus cytomegalovirus in fetal rhesus monkeys (Macaca mulatta). J. Infect. Dis. 177446-450. [DOI] [PubMed] [Google Scholar]
- 54.Trgovcich, J., E. Pernjak-Pugel, J. Tomac, U. Koszinowski, and S. Jonjic. 1998. Pathogenesis of murine cytomegalovirus infection in neonatal mice, p. 42-53. In M. Scholz, H. F. Rabenau, H. W. Doerr, and J. Cinatl, Jr. (ed.), CMV-related immunopathology. Monographs in virology, vol. 21. Karger, Basel, Switzerland. [Google Scholar]
- 55.Trgovcich, J., D. Stimac, B. Polic, A. Krmpotic, E. Pernjak-Pugel, J. Tomac, M. Hasan, B. Wraber, and S. Jonjic. 2000. Immune responses and cytokine induction in the development of severe hepatitis during acute infections with murine cytomegalovirus. Arch. Virol. 1452601-2618. [DOI] [PubMed] [Google Scholar]
- 56.Tyor, W. R., and D. E. Griffin. 1993. Virus specificity and isotype expression of intraparenchymal antibody-secreting cells during Sindbis virus encephalitis in mice. J. Neuroimmunol. 4837-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyor, W. R., S. Wesselingh, B. Levine, and D. E. Griffin. 1992. Long term intraparenchymal Ig secretion after acute viral encephalitis in mice. J. Immunol. 1494016-4020. [PubMed] [Google Scholar]
- 58.van den Pol, A. N., E. Mocarski, N. Saederup, J. Vieira, and T. J. Meier. 1999. Cytomegalovirus cell tropism, replication, and gene transfer in brain. J. Neurosci. 1910948-10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van den Pol, A. N., J. D. Reuter, and J. G. Santarelli. 2002. Enhanced cytomegalovirus infection of developing brain independent of the adaptive immune system. J. Virol. 768842-8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang, Y., M. Lobigs, E. Lee, and A. Mullbacher. 2003. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 7713323-13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yeager, A. S., F. C. Grumet, E. B. Hafleigh, A. M. Arvin, J. S. Bradley, and C. G. Prober. 1981. Prevention of transfusion-acquired cytomegalovirus infections in newborn infants. J. Pediatr. 98281-287. [DOI] [PubMed] [Google Scholar]