

Abstract
A window of opportunity for immune responses to extinguish human immunodeficiency virus type 1 (HIV-1) exists from the moment of transmission through establishment of the latent pool of HIV-1-infected cells. A critical time to study the initial immune responses to the transmitted/founder virus is the eclipse phase of HIV-1 infection (time from transmission to the first appearance of plasma virus), but, to date, this period has been logistically difficult to analyze. To probe B-cell responses immediately following HIV-1 transmission, we have determined envelope-specific antibody responses to autologous and consensus Envs in plasma donors from the United States for whom frequent plasma samples were available at time points immediately before, during, and after HIV-1 plasma viral load (VL) ramp-up in acute infection, and we have modeled the antibody effect on the kinetics of plasma viremia. The first detectable B-cell response was in the form of immune complexes 8 days after plasma virus detection, whereas the first free plasma anti-HIV-1 antibody was to gp41 and appeared 13 days after the appearance of plasma virus. In contrast, envelope gp120-specific antibodies were delayed an additional 14 days. Mathematical modeling of the earliest viral dynamics was performed to determine the impact of antibody on HIV replication in vivo as assessed by plasma VL. Including the initial anti-gp41 immunoglobulin G (IgG), IgM, or both responses in the model did not significantly impact the early dynamics of plasma VL. These results demonstrate that the first IgM and IgG antibodies induced by transmitted HIV-1 are capable of binding virions but have little impact on acute-phase viremia at the timing and magnitude that they occur in natural infection.
The development of a preventive human immunodeficiency virus type 1 (HIV-1) vaccine is a global priority (12). A major roadblock in development of a preventive HIV-1 vaccine is the inability to induce protective antibodies by vaccines or natural infection. Studies in nonhuman primates have demonstrated that passive infusion of broadly neutralizing anti-HIV-1 monoclonal antibodies (MAbs) prevents infection by simian-human immunodeficiency viruses (29, 41, 64). Thus, if sufficiently high levels of broadly neutralizing antibodies were present at the time of transmission, protection from HIV-1 infection might be possible. However, to date there is no immunogen formulation that consistently induces broadly neutralizing anti-Env antibodies. Moreover, autologous neutralizing antibody responses do not occur until months after transmission (1, 24, 50, 60). The window of opportunity during which a protective antibody might extinguish HIV-1 after the initial transmission event is uncertain but is likely to be limited to the period of time prior to establishment of the latent pool of HIV-1-infected CD4+ T cells (34, 61). Although viral latency is certainly established at the time of seroconversion (6), it may be as early as a few days after transmission (18).
An important obstacle to the development of an effective HIV vaccine is the inability to induce antibodies that neutralize primary HIV-1 strains across all genetic subtypes (17, 42). While multiple forms of HIV-1 envelope-based vaccines express epitopes to which rare, broadly neutralizing human MAbs bind (i.e., Envs are antigenic), these vaccines have not been immunogenic and have failed to induce broadly neutralizing antibodies against the gp120 CD4 binding site shown to involved in neutralization breadth (38), the membrane proximal external region (MPER) of gp41 (44, 48), or against gp120 carbohydrate Env antigens (51) in animals or humans.
HIV-1 seroconversion has been reported to occur over a wide range of times when estimated from the onset of clinical acute HIV-1 infection (AHI) (5, 30, 45); however, the timing of seroconversion of HIV antibodies of particular specificities and isotypes has not been precisely quantified relative to the first time of detectable plasma viremia. Anti-HIV-1 immunoglobulin M (IgM) reactive with virus-infected cells has been detected during the course of AHI (10, 11), but the timing of these antibodies and the presence of IgM-virion immune complexes relative to the first detection of viral RNA in AHI have yet to be defined. It is known that autologous neutralizing antibodies arise only months after the first appearance of HIV-specific antibodies (1, 24, 50, 60). Critical questions for understanding the role of early HIV-1 antibodies in the control of HIV-1 are, first, what are the nature and timing of the earliest anti-HIV-1 antibodies and, second, what are the contributions of these antibodies in the control of viral replication after transmission?
In this study, we have investigated the timing of specific anti-envelope (Env) antibody responses from the eclipse phase (time between transmission and detectable viremia) (19) through 6 to 12 months of established infection and modeled the effect of B-cell responses on control of initial plasma viremia. We show that the earliest detectable antibodies to HIV-1 are in the form of virion-antibody immune complexes followed 5 days later by free anti-gp41 IgM plasma antibodies. Mathematical modeling of viral dynamics suggested that the initial Env gp41 IgM and IgG antibody responses had little effect on control of initial viral replication.
MATERIALS AND METHODS
Subjects studied.
A subset of subjects from four different acute-infection cohorts were examined: 21 plasma donors, 12 AHI subjects from the Trinidad cohort (clade B), 14 AHI subjects from the Centre for the AIDS Programme of Research in South Africa ([CAPRISA] clade C) cohort, and 10 AHI subjects from the CHAVI 001 acute infection cohort.
Viral load (VL) testing.
Plasma viral RNA was measured by Quest Diagnostics (HIV-1 RNA PCR Ultra; Lyndhurst, NJ).
Antigens used in antibody binding assays.
The antigens used for direct antibody binding assays were as follows: group M consensus Env CON-S gp140, consensus B gp140, and clade B wild-type IIIB, JRFL, and 89.6 Env gp120s (produced by either recombinant vaccinia [39] or 293T transfection). In addition, the following peptides (Primm Biotech Inc, Cambridge, MA) were used: SP400 (gp41 immunodominant region, RVLAVERYLRDQQLLGIWGCSGKLICTTAVPWNASWSNKSLNKI), SP62 (gp41 MPER, QQEKNEQELLELDKWASLWN), 4E10 P (SLWNWFNITNWLWYIK), consensus B V3 gp120 region (TRPNNNTRKSIHIGPGRAFYTTGEIIGDIRQAH), and consensus M V3 CON-S gp120 region (TRPNNNTRKSIRIGPGQAFYATGDIIGDIRQAH). Acute HIV-1 envelope gene sequences were derived from subtype B acute HIV-1-infected individuals (subjects 6246, 6240, and 9021) by single-genome amplification (35). To produce recombinant soluble gp140 proteins, a gp140 env gene construct named gp140C was designed, in which the transmembrane and cytoplasmic domains of HIV-1 Env were deleted and two critical Arg residues in the gp120-gp41 cleavage site were replaced with two Glu residues. All four gp140C env genes were codon optimized by employing the codon usage of highly expressed human housekeeping genes, synthesized de novo (Blue Heron Biotechnology, Bothell, WA) and cloned into pcDNA3.1/Hygro expression plasmid (Invitrogen, Carlsbad, CA) using standard molecular technology. Recombinant HIV-1 gp140C Env proteins were produced in 293T cells by transient transfection with the resulting plasmids and purified by Galanthus nivalis lectin-agarose (Vector Labs, Burlingame, CA) column chromatography (39). Autologous V3 peptides were made from these same Envs.
Antibody assay criteria.
The positivity criterion per antigen per antibody isotype was determined by screening ≥30 seronegative subjects. A standardized HIV-positive (HIV+) control was titrated on each assay (tracked with a Levy-Jennings plot with acceptance of titer only within 3 standard deviations of the mean), and the average optical density (OD) was plotted as a function of serum dilution to determine antibody titer using a four-parameter logistic equation (SoftMaxPro, Molecular Devices). The coefficient of variation per sample is ≤15%. Two negative sera and two HIV+ control sera were included in each assay to ensure specificity and for maintaining consistency and reproducibility between assays. To ensure the integrity of raw data acquisition, data analyses are electronically tracked (21 CFR part 11).
Direct ELISAs.
Direct enzyme-linked immunosorbent assays (ELISAs) were performed using consensus clade B or envelope glycoproteins, gp41 proteins, consensus V3 peptides, gp41 immunodominant proteins, and MPER epitopes, as well as autologous V3 and gp140 Env oligomers. ELISAs were conducted in 96-well ELISA plates (Costar 3369) coated with 0.2 μg/well antigen in 0.1 M sodium bicarbonate and blocked with assay diluent (phosphate-buffered saline [PBS] containing 4% [wt/vol] whey protein-15% normal goat serum-0.5% Tween 20-0.05% sodium azide). Serum was incubated for 1 h in twofold serial dilutions beginning at 1:25, followed by washing with PBS-0.1% Tween 20. A total of 100 μl of alkaline phosphatase-conjugated goat anti-human secondary antibody (Sigma A9544) was incubated at 1:4,000 for 1 h, washed, and detected with 100 μl of substrate (carbonate-bicarbonate buffer, 2 mM MgCl2, 1 mg/ml p-NPP [4-nitrophenyl phosphate di(2-amino-2-ethyl-1,3-propanediol) salt]). Plates were read at 405 nm at 45 min.
Competitive inhibition studies (antibody blocking assays).
Competitive inhibition studies (antibody blocking assays) were performed with 1b12 (CD4BS), 2G12 (anti-CHO), and the MPER MAbs 2F5 and 3H11. Ninety-six-well ELISA plates (Costar 3369) were coated with 0.2 μg/well JRFL in 0.1 M sodium bicarbonate and blocked with assay diluent (PBS containing 4% [wt/vol] whey protein-15% normal goat serum-0.5% Tween 20-0.05% sodium azide). All assay steps were conducted in assay diluent (except the substrate step) and incubated for 1 h at room temperature (13H11 assay at 37°), followed by washing with PBS-0.1% Tween 20. Serum was diluted 1:50 and incubated in triplicate wells. A total of 50 μl of biotinylated target MAb was added at the 50% effective concentration (determined by direct binding curve of biotinylated MAb to JRFL). The extent of biotin-MAb binding was detected with streptavidin-alkaline phosphatase at 1:1,000 (Promega V5591), followed by substrate (carbonate-bicarbonate buffer, 2 mM MgCl2, 1 mg/ml p-NPP)). Plates were read with a plate reader at 405 nm and 45 min. Triplicate wells were background subtracted and averaged. Percent inhibition was calculated as follows: 100 − (mean OD of triplicate serum wells/mean OD of no-inhibition control ×100.) CD4 binding site blocking assays were conducted as above, except that 100 μl of a saturating concentration of soluble CD4 (Progenics Pharm Inc.) was incubated between serum and biotin-MAb incubation steps.
AtheNA assay.
Antibody binding to proteins gp160, gp120, p66, p55, gp41, p31, p24, and p17 was measured on the Luminex platform (Luminex Corporation) using an AtheNA Multilyte HIV-1 Bead Blot kit (Zeus Scientific catalog number A71001G) following the kit manufacturer's protocol.
Cardiolipin and rheumatoid factor assays.
Anti-cardiolipin antibody assays were performed as described previously (2). Assays to measure IgM rheumatoid factor using IgG antigen were standardized using rheumatoid factor controls (kindly provided by Judy Fleming, Clinical Immunology Laboratory, Duke University Medical Center).
Isotype binding antibodies.
HIV antigens or purified IgM, IgG, and IgA proteins (used as controls) were precoated overnight onto the wells of microtiter plates (NUNC) and washed with an automated and calibrated plate washer (Bio-Tek). The serum/plasma test samples were diluted and incubated with the antigens bound to the plate. The plates were then washed, and the antigen-antibody complexes were incubated with isotype-specific anti-human IgG, IgA, and IgM conjugated to alkaline phosphatase. OD readings were measured using a VersaMax plate reader (Molecular Devices), and an average OD reading for each pair of replicates, with the background subtracted, was calculated. For each test sample, duplicate antigen-containing and non-antigen-containing wells of a microtiter plate were scored (i.e., OD of antigen-containing wells − OD of non-antigen-containing wells). A positive score is defined as an OD value of ≥0.1, with background subtracted, and also as at least threefold above the baseline, with a 15% coefficient of variation between replicates. As another level of validation, in the plasma donor samples we compared the HIV gp41-specific IgM binding antibody test with that of the third generation Abbott Diagnostics EIA (enzyme immunoassay; Abbott Park, IL) and found equal sensitivity to the commercially available kit for the first detection of any antibody response.
Specimen prep for MultiTrap IgG removal.
For detection of IgA and IgM antibodies, IgG was removed using protein G columns. Briefly, plasma was centrifuged (10,000 × g) for 10 min, diluted twofold in dilution buffer, and filtered in a 1.2-um-pore-size filter plate (Pall AcroPrep). The filtered and diluted samples were depleted of IgG using protein G high-performance MultiTrap plates (GE, Inc.) according to the manufacturer's instructions with minor modifications. IgG removal in the specimens was greater than 90% as assayed by HIV-specific binding assays.
Customized Luminex assay.
A total of 5 × 106 carboxylated fluorescent beads (Luminex Corp, Austin, TX) were covalently coupled to 25 μg of one of the purified HIV antigens used in ELISAs and incubated with patient samples at a 1:10 dilution. HIV-specific antibody isotypes were detected with goat anti-human IgA (Jackson Immunoresearch, West Grove, PA), mouse anti-human IgG (Southern Biotech, Birmingham, AL), or goat anti-human IgM (Southern Biotech, Birmingham, AL), each conjugated to phycoerythrin, at 4 μg/ml. Beads were then washed and acquired on a Bio-Plex instrument (Bio-Rad, Hercules, CA). Purified IgM, IgG, and IgA proteins (Sigma) and a constant HIV+ serum titration were utilized as positive controls in every assay. Background values (beads in the absence of detection antibody) and normal human plasma were utilized as the negative controls. A control for rheumatoid factor for IgM detection was an internal IgG protein standard.
HIV-1 immune complex capture assays.
ELISA plates (NUNC) were coated overnight at 4°C with anti-human IgM or IgG at a concentration of 1 μg/ml diluted in PBS. All subsequent steps were performed at room temperature. After incubation and washing, coated plates were blocked for 2 h with PBS supplemented with 5% fetal bovine serum, 5% milk, and 0.05% Tween. After blocking and washing, 90 μl of undiluted plasma was added to each well and incubated for 90 min, followed by four washes with PBS supplemented with 0.05% Tween. A total of 200 μl of AVL lysis buffer with carrier RNA (Qiagen) was added and shaken for 15 min, and viral RNA in the lysis was extracted by a Qiagen viral mini kit. HIV-1 RNA from the virion-antibody complexes was measured by Gag real-time reverse transcription-PCR. The detection of immune complexes by the ELISA capture assay was confirmed using protein G column absorption (Protein G HP column; Pierce, Inc.) to deplete IgG-virion immune complexes. IgG absorption was performed according the manufacturer's instructions. Plasma (90 μl) was added to the protein G column. After a mixing step and an incubation of 10 min, the column was centrifuged for 1 min at 5,000 × g. The presence of immune complexes was calculated by the percentage of viral RNA input divided by viral RNA flowthrough, similar to the method by Baron et al. (16). HIV immune globulin (reagent from the Division of AIDS, NIH) plus HIV-1 NL4-3 pseudotyped virus was the positive control for immune complex capture (81% ± 4%), and normal human serum ([NHS]Sigma) or RPMI 1640 medium plus HIV-1 NL4-3 was the negative control. The cutoff of non-HIV-1-specific capture (NHS plus NL-43) was 16.2% ± 0.8%; the background of virus-only control was 6.5% ± 4.6%.
Complement binding assays.
Virus and diluted plasma samples (1:40) were incubated at 37°C in the presence of 10% NHS (Sigma, St. Louis, MO) as a source of complement or with 10% heat-inactivated NHS. MT-2 cells, which express high levels of CR2, were then added, and the virus/cell suspensions were incubated for 2 h. Unbound virions were removed by successive washes. Bound virions were disrupted by treatment with 0.5% Triton X and the released p24 was measured by ELISA. To determine percent binding, the amount of p24 obtained was divided by the amount of p24 of the original virus after correcting for complement nonspecific binding (hi NHS).
Neutralizing antibody assays.
Antibody-mediated neutralization in the plasma donor cohort was measured as a function of reductions in luciferase reporter gene expression after a single round of infection in TZM-bl cells, as described previously (37). For assay of plasma for 2F- and 4E10-specific neutralizing antibodies, HIV-2 pseudoviruses expressing HIV-1 2F5 or 4E10 epitopes were used as described previously (24).
Statistical analyses and methods to classify simultaneous and sequential kinetics.
Statistical analyses were conducted using methods that included mixed-effects models (55, 58, 59), nonparametric regression (25), a binomial test, Kaplan-Meier curve, and an accelerated failure time model (13). For all four cohorts, smoothing spline-based nonparametric regression (25) was performed to obtain estimates of the VL and antibody curves. For the plasma donor cohort, where the acute burst of viremia was recorded, we defined an accurate time of origin (T0) to align different study panels for the joint analyses. For each patient, the T0 was estimated as a model parameter in the nonlinear mixed-effects model of the upswing VLs, accounting for censoring at the assay limits of detection (55). Right-censoring was used in the survival analysis such that data were suspended for subjects for whom no event occurred during the follow-up period (while the event could happen at a later time, i.e., to the right in the time scale). Two subjects were censored because of their short follow-up period (12 days after T0). For each analyte (e.g., anti-IgA/IgG/IgM gp41 antibody response), data recorded prior to T0 were fit to a linear mixed-effects model (58) to determine the background level for that analyte, where the upper 95% prediction limit of a future response (59) was used as a positivity threshold to define the last negative observation and the first positive observation. The statistical method of classifying simultaneous and sequential kinetics verified the results obtained from ELISA calculations based on the positivity criterion.
A Kaplan-Meier estimate was used to describe the distribution of the initial elevation timing. A two-sided binomial test of relative ranking in elevation timing between pairs of analytes was performed with a positive difference in timing as a success and the number of nonzero differences as the number of trials. Adjusted P values (q values) were computed to control for the false discovery rate of multiple testing (54). To assess association between the VLs and antibody markers in both the plasma donors and CHAVI 001 cohorts, accelerated failure time models (13) were used to correlate the expansion or decay of the VLs and the time to initial elevation (subject to censoring), and linear mixed-effects models (58) were used to correlate the downswing VLs and antibody response magnitudes over time. Additionally, statistical correlation and linear regression analyses were performed to identify the plausible association between different inhibition assays in the Trinidad and CAPRISA cohorts.
Modeling.
The following is the target cell-limited model used to mathematically model the plasma donor VL:
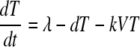 |
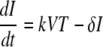 |
(1) |
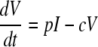 |
Cells that are susceptible to HIV infection are termed target cells (T). The model assumes that target cells are produced at a constant rate, λ, and die at rate d per cell. Upon interaction with HIV, these cells become productively infected cells (I)with infection rate constant k. Infected cells die at per cell rate δ and produce viral particles (virions), V, at rate (p) per infected cell. Virions are assumed to be cleared at a constant rate, c, per virion.
To incorporate enhanced virion clearance due to opsonization, we replaced the equation for V(t) in the model above with the following equation:
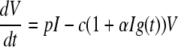 |
(2) |
Here, virion clearance is enhanced by a factor (1 + αIg(t)), where Ig(t) is either the measured concentration of anti-gp41 IgM or anti-gp41 IgG or the total of both Ig concentrations in plasma. If α = 0, then there is no opsonization effect.
To model the effects of antibody in neutralizing virus, we reduced the infectivity constant k in equation 1 by the factor (1 + βIg(t)). Here, if β = 0 there is no antibody-mediated viral neutralization. Lastly, to incorporate the possibility of antibody enhancing the rate of infected cell loss through antibody-dependent cellular cytotoxicity or complemented mediated lysis, we increased δ by the factor (1 + γ Ig(t)). If γ = 0, there is no enhanced death.
At T0, which we chose as time t = 0, the plasma VL by definition is 100 copies/ml. While some CD4+ depletion could have occurred by T0, for simplicity we assume the uninfected cell level is still 106 cells/ml. We estimated the number of infected cells at T0 as either 1 or 10 cells/ml based on preliminary fits. This low number of infected cells supports our assumption of little T-cell depletion by T0.
The target cell-limited model as well as the three variants of it that included antibody effects were fit to VL data of each plasma donor using a spline fit to the measured anti-gp41 concentrations for Ig(t). Fitting to the VL data was done using a nonlinear least squares method where loge V from the model was fit to the loge of the measured VL. An F-test was used to determine whether the target cell-limited model or one of the three variants fit the data best. For donor 9032 the best-fit target cell-limited model gave an extremely poor fit to the data unless we added to the sum of squared residuals a penalty function for not attaining a maximum at the time the VL was maximum. That is, we add an additional term to the function to be minimized equal to the square of the difference between the time the VL was maximum in the data and in the model.
RESULTS
Plasma donor cohort.
HIV-1 seroconversion plasma donors from the United States (clade B) were studied for the earliest antibody events in HIV-1 infection. These subjects donated plasma every 3 days, and the plasma units were held for weeks until tests for HIV-1, hepatitis B, or hepatitis C were completed (19). Once a donor was found to be positive for an infectious disease, the plasma sample donations were stopped; therefore, neither cells nor long-term follow-up of these plasma donors was available. Analysis of the U.S. plasma donor cohort provided for calculation of the earliest HIV-1 antibody response in relation to a defined point where viral RNA was first detected at transmission. A time zero (T0) that represented the initial time at which the VL trajectory crossed the assay lower limit of detection (100 HIV-1 copies/ml) was established to align each donor panel to a single reference time (Fig. 1A). The start of detectable plasma viremia, T0, is approximately 10 days (range, 7 to 21 days) (7, 11, 14, 21, 40, 52) after virus transmission and represents the end of the eclipse phase of HIV infection. The plasma samples studied from this cohort had the most frequent sampling for testing within the first 20 days before and the first 20 days after the start of detectable viremia (Fig. 1B).
FIG. 1.

(A) VL kinetics of 21 HIV+ seroconversion plasma donor panels (eclipse phase clade B infection) were determined. The alignment of the subjects was by T0, the first day that VL reached 100 copies/ml. (B) Histogram displaying the total number of samples studied for each day, relative to the first detectable day of viremia (T0). Bins represent intervals of 10 days.
Initial IgG anti-Env responses following transmission.
To ensure that the earliest antibodies were detected, both standardized ELISAs and more sensitive Luminex quantitative antibody assays were utilized. The lower level of sensitivity for the ELISAs for measurement of anti-HIV-1 Env IgG was 2.2 ng/ml, and the lower level of sensitivity of Luminex assays for measurement of anti-HIV-1 IgG was 0.2 ng/ml. For identification of initial antibody responses, we tested autologous, consensus B, and clade B wild-type Envs as antigens. For non-Env antigens, all antigens were wild-type clade B. To validate that consensus B Env antigens detected the earliest antibodies and to determine if earlier antibody responses could be detected using autologous Env antigens, gp140 and Env gp120 V3 peptides from four plasma donor subjects were produced and compared with consensus B Envs or V3 peptides for the ability to detect plasma Env antibodies.
First, using consensus or wild-type clade B Envs, we found that the earliest detectable anti-Env IgG plasma antibody responses following HIV-1 transmission were to envelope gp41 and occurred at a median of 13.0 days after T0. Figure 2A illustrates the earlier timing of the anti-gp41 antibody responses and the later and more variable timing of the antibody responses against gp120 (P < 0.01). Antibodies to gp41 developed in 90% of subjects by 18 days (Km estimate is 100%; two subjects were censored because they were lost to follow-up at 12 days after T0) in contrast to gp120 antibody responses, which came up in 33% of subjects during the follow-up period of between 12 and 67 days after T0 studied here (Table 1). There was no significant difference in the timing of the anti-gp120 antibody responses when two additional wild-type clade B gp120 envelope proteins, JRFL and 89.6 gp120 Envs, were examined (data not shown). Figure 2B shows the median time of appearance of gp41 and gp120 antibodies compared to time of appearance of antibodies to HIV-1 p24, p55, p66, p17, and p31 proteins. Pairwise comparison of the timing of each specificity of antibody demonstrated that HIV-1 structural component antibody timing (anti-Gag) was significantly later than that of HIV-1 gp41 antibodies.
FIG. 2.

(A) Kaplan Meier plot of anti-gp41 and anti-gp120 antibody responses in the eclipse phase clade B plasma donor cohort. The solid line shows the increasing percentage of the population that develops HIV-specific antibody responses at each time interval following the calculated T0. The dashed lines indicate the upper and lower point-wise confidence intervals, respectively. (B) Pairwise comparison of the timing of anti-Env antibody responses compared anti-Gag (p24, p17, and p55) and anti-Pol (p31) responses in the eclipse phase clade B plasma donor cohort. The solid line (from left to right) indicates the median day of antibody elevation from T0, and the gaps in the line indicate the HIV-specific antibody responses that group together relative to their time of elevation from T0. The values above the lines are q values for pairwise tests of differences between adjacent groups of antibody specificities. ns, no statistically significant pairwise difference within the group of antibody specificity. The median time for appearance of IgG anti-gp41 antibody was 13.5 days (A), while the median time for appearance of IgG gp120 antibody was 28 days (B).
TABLE 1.
Ontogeny: Env-specific IgG in eclipse phase clade B cohort
| Antigen | No. of subjectsa | Median time to antibody response (range [days])b | 0.95% LCL (days)c | 0.95% UCL (days)d |
|---|---|---|---|---|
| gp41 | 19 | 13 (9-18) | 12 | 14 |
| gp140 | 19 | 13 (6-17) | 12 | 15 |
| ID gp41 peptidee | 13 | 18 (13-34) | 16 | — |
| gp120 | 7 | 28 (13-41) | 26 | — |
| V3 | 7 | 34 (13-36) | 19 | — |
| MPER, CD4BS, CD4i | 0 | NA |
Number of plasma donor subjects that showed an elevation during the follow up period.
Median time from T0 (the first day viral load reaches 100 copies/ml).
LCL, lower confidence limit.
UCL, upper confidence limit. —, upper confidence limit is infinite (elevations can occur at >67 days from T0).
ID, immunodominant.
A summary of the timing of various anti-Env antibody responses detected in U.S. plasma donors is shown in Table 1. For the first antibody elicited against gp41, 13/19 (68%) of gp41 responses included the immunodominant region of gp41, and 7/7 of the plasma donor initial gp120 included responses that could be mapped to the V3 loop (i.e., 100% of the plasma donors that had gp120 antibodies within the first 40 days of transmission also had V3 antibodies). Antibodies that did not appear at all in U.S. plasma donor subjects (within 40 days after T0) were anti-MPER (neutralizing and nonneutralizing) antibodies, CD4 binding site antibodies (CD4bs), and CD4-induced antibodies (CD4i). Neutralizing antibodies to the easily neutralized tier 1 (37) HIV-1 Env pseudoviruses such as B.SF162 and antibody-dependent cell-mediated virus inhibition (ADCVI) activity (29) also did not appear within the first 40 days after T0 as well (data not shown). ADCVI has previously been reported to be present during the later stages of acute infection, so the time points examined here during acute viremia in the plasma donor subjects are likely just before the development of ADCVI (20). As expected, broadly neutralizing antibodies with specificities similar to the broadly neutralizing antibodies 2F5, 4E10, 1b12, and 2G12, as measured by a competitive ELISA with biotinylated MAbs, also did not appear during the first 40 days after T0 (not shown).
Analysis of isotype-specific gp41 antibody responses.
IgG antibodies are produced after Ig class switching and are classically produced after IgM antibodies. We assayed for HIV Env-specific IgM using Luminex assays with recombinant gp41, gp120, and consensus B and group M consensus gp140 protein antigens. As with IgG responses, the first IgM antibody against Env also targeted only gp41. The median time of rise in HIV-1-specific IgM antibodies was 13 days post-T0 (range, 5 to 18 days). The custom anti-IgM ELISA utilized in this study was equally sensitive to the third generation commercial ELISA (Abbott Anti-HIV-1/2 EIA; Abbott Park, IL) for detection of the first free HIV-specific antibody. The difference in timing of first antibody detection between the two assays was not significant (median difference in days to detection, 0; P value of 0.66, Wilcoxon signed rank test).
We expressed four autologous gp140 envelopes from four different plasma donor subjects (subjects 6246, 6240, 9021, and 63521) representing the transmitted or founder virus (35) as gp140C protein oligomers and also studied four subjects with autologous Env V3 loop peptides as targets for plasma antibody binding assays. Three of the gp140 Envs were chosen from subjects in whom an antibody response was detected with the consensus Env, while gp140 was expressed from one subject who did not have a detectable anti-gp41 response. A representative example from a single donor against the autologous and consensus clade B Env for IgM, IgG, and IgA is shown in Fig. 3A to C (see also Fig. S2 in the supplemental material). We found that, using both autologous Envs and autologous V3 peptides (not shown), we could not detect any IgM, IgG, and IgA antibody responses earlier than those detected using group M consensus Envs or consensus B V3 peptides (Fig. 3A to C). Interestingly, the gp41 antibody responses were greater in magnitude when tested with consensus Envs versus autologous envelopes.
FIG. 3.

Anti-gp41 IgM antibodies are the first detectable HIV antibodies, and autologous gp140-transmitted Env or consensus Env gp140 proteins are equally sensitive for the detection of the first antibody isotypes in HIV infection. IgM antibodies (A), IgG antibodies (B), and IgA antibodies (C) were detected using either consensus gp140 (ConB) or autologous Env (6246 Env). The asterisk indicates the plasma sample from which the autologous gp140 Env was derived. The consensus gp160 oligomer detects anti-gp41 antibodies at the same time as autologous gp140 Env oligomers. MFI, mean fluorescence intensity.
We next examined Ig class switching patterns in plasma donors (15 subjects), four clade B CHAVI 001 subjects, and three clade B AHI subjects from the Trinidad/Tobago cohort, where the plasma sampling was sufficiently early to determine the first appearances of anti-gp41 IgM and IgG (see Materials and Methods and Fig. S1 in the supplemental material for VL and timing of samples from the CHAVI 001 and Trinidad/Tobago acute HIV-1 infection cohorts). Figure 4 shows representative subjects with either sequential class switching kinetics (Fig. 4A) or simultaneous class switching kinetics (Fig. 4B) in the plasma donor cohort. In both subjects, IgM responses were transient and decayed over a period of 20 to 40 days, while IgG responses rose over the same period. Anti-gp41 IgM responses appeared earlier than IgG responses in 9/22 (41%) of subjects; however, in 13/22 (59%) of subjects, IgM anti-gp41 was detected at the same time as IgG and IgA anti-gp41 antibodies (Fig. 4C). We also tested anti-IgM, anti-IgG, and anti-IgA responses to a gp41 immunodominant peptide in subject 6246 with simultaneous appearance of anti-gp41 IgM, IgG, and IgA to determine if the simultaneous appearance of the three isotypes could potentially be due to responses to different gp41 epitopes. We found that anti-immunodominant IgM, IgG, and IgA were simultaneously detected, demonstrating in this subject that the simultaneous appearance of antibodies could not be attributable solely to the development of antibody responses to multiple gp41 epitopes (data not shown). To determine if simultaneous IgM, IgA, and IgG antibody responses were unique to envelope or, rather, occurred in the response to other HIV-1 proteins, we also determined the pattern of isotype antibody responses to p55 Gag. We found that IgM, IgG, and IgA antibodies to p55 Gag were also detected simultaneously in subjects that had simultaneous appearance of IgM, IgG, and IgA antibodies to gp41 Env (Fig. 4D).
FIG. 4.

Kinetics of anti-gp41-specific antibody isotypes in acute HIV infection. Representative examples of sequential development (A) and simultaneous development (B) of early HIV-specific antibody responses are shown. (C) The percentage of patients in each of the three cohorts that displayed different kinetic patterns. (D) Simultaneous development of Gag-specific antibody responses. Anti-p55 antibodies of the IgM, IgG, and IgA isotypes were measured for all subjects in the eclipse phase clade B cohort. Subject 12007 could not be aligned to T0 due to the large interval between the first RNA-positive sample and the last RNA-negative sample. However, the short interval between antibody-positive and antibody-negative responses enabled measurement of antibody isotype kinetics, so the panel was aligned to T0 as the first RNA-positive sample. Pt, patient.
Detection of immune complexes in AHI.
We next assayed panels from U.S. plasma donors for earlier antibody responses in the form of antibody bound to virions to determine if earlier antibody was being made and was not in plasma but, rather, was present only in the form of virion-IgM or -IgG antibody complexes. In 6/6 of subjects tested with sufficiently high plasma viral RNA levels, immune complexes containing either IgG or IgM antibodies bound to virions were detected (Fig. 5). Immune complexes were detected at a median time of 8 days (range, 5 to 14 days) after T0. The detection of immune complexes preceded the detection of free antibodies by both commercial ELISA and custom ELISA by a median of 5 days (range, 5 to 7 days) (Table 2). Virion-antibody complexes were detected in subjects with either simultaneous or sequential Ig isotype kinetics, and this suggests that the presence of early immune complexes likely does not explain the simultaneous detection of IgM, IgG, and IgA anti-gp41 isotypes.
FIG. 5.

HIV immune complexes produced at a median time of 8.0 days after T0. The detection of immune complexes for patients (Pt) 9015, 12008, 9077, 9079, 9021, and 9076 are aligned to T0 and plotted in comparison to the detection of free antibody (Ab) responses.
TABLE 2.
Timing of immune complexes and free HIV antibodies relative to T0
| Subject | No. of days after T0 | Detection by immunoassay
|
Detection by PCR (RNA copies/ml)
|
|||
|---|---|---|---|---|---|---|
| Abbott anti-HIV-1/2a (sample net counts/cutoff) | Custom IgMb (OD values) | IgG ICc | IgM ICc | VLd | ||
| 12008 | 2 | 0.181 | 0.017 | — | — | 925 |
| 4 | 0.157 | 0.018 | — | — | 24,194 | |
| 9 | 0.173 | 0.014 | 32,833 | 25,389 | 5,631,550 | |
| 14 | 1.795 | 0.114 | 130,556 | 162,222 | 6,486,240 | |
| 16 | 9.528 | 0.795 | 132,778 | 89,056 | 2,296,060 | |
| 21 | 6.063 | 0.997 | 1,344 | 778 | 26,311 | |
| 23 | 4.646 | 0.872 | — | 2,350 | 17,425 | |
| 9079 | 4 | 0.108 | 0.045 | — | — | 56,508 |
| 6 | 0.135 | 0.037 | — | 989 | 731,600 | |
| 11 | 2.839 | 0.618 | 788 | 9,944 | 584,913 | |
| 13 | 10.763 | 1.186 | 190 | 652 | 2,170,000 | |
| 19 | 4.169 | 0.610 | — | — | 20,988 | |
| 26 | 6.331 | 0.353 | — | — | 3,594 | |
| 9076 | −3 | 0.164 | 0.001 | — | — | <50 |
| 3 | 0.155 | 0.000 | — | — | 5,393 | |
| 5 | NT | 0.039 | — | — | 352 | |
| 11 | 4.879 | 0.345 | — | — | 697,960 | |
| 14 | 17.241 | 0.958 | 3,256 | 1,489 | 507,740 | |
| 19 | 17.241 | 0.121 | — | — | 4,912 | |
| 9015 | 0 | 0.171 | 0.030 | — | — | 59 |
| 7 | 0.171 | 0.023 | — | 1,422 | 2,483,020 | |
| 9 | 0.171 | 0.036 | 1,933 | 2,067 | 2,912,250 | |
| 14 | 7.863 | 0.120 | — | 926 | 349,278 | |
| 16 | 11.538 | 0.255 | — | — | 276,354 | |
| 9021 | −3 | 0.483 | 0.055 | — | — | <50 |
| 1 | 0.793 | 0.069 | — | — | 387 | |
| 5 | 0.569 | 0.096 | — | 376 | 28,487 | |
| 8 | 0.138 | 0.085 | — | 4,922 | 404,218 | |
| 12 | 0.716 | 0.213 | — | 7,156 | 520,471 | |
| 15 | 2.345 | 1.251 | 1,314 | 29,833 | 5,417,090 | |
| 9077 | 2 | 0.153 | 0.071 | — | — | 1353 |
| 6 | 0.198 | 0.078 | — | — | 79,235 | |
| 11 | 0.135 | 0.091 | — | — | 798,590 | |
| 13 | 0.432 | 0.093 | 8,494 | 5,670 | 400,900 | |
| 18 | 8.793 | 1.682 | — | — | 1,114,040 | |
| 25 | 9.514 | 1.267 | — | — | 40,369 | |
| 28 | 10.225 | 0.861 | — | — | 17,838 | |
For the ABBOTT anti-HIV-1/2 EIA, the positivity cutoff is >1.0. Boldface indicates a positive value. NT, not tested.
Values were calculated as described in Materials and Methods; values that were ≥0.1 and at least threefold above baseline were considered positive, as indicated by boldface.
IC, immune complex. A dash indicates <200 copies/ml by quantitative real-time PCR of immune complexes; boldface indicates positive values.
HIV-1 RNA PCR Ultra, Quest Diagnostics, Lyndhurst, NJ.
The detection of IgG immune complexes was confirmed with a second assay of detection (data not shown) using a protein G column to capture antibodies bound to virions, followed by lysis of virions to measure viral RNA. Identical kinetics of the appearance and decline of immune complexes were observed using both methods for measurement of IgG immune complexes (data not shown). Taken together, these data suggest earlier production of anti-virion IgM and IgG on day 8 after T0 and before the appearance of free plasma anti-HIV IgM and IgG. The simultaneous appearance of both IgM and IgG virion immune complexes either suggests simultaneous induction of anti-virion IgM and IgG in these subjects or indicates yet earlier induction of IgM and IgG antibodies to HIV virion components with specificities that are not detectable with our current assays. The decline in detection of immune complexes may be due to clearance by the reticuloendothelial cell system. It is of interest that the detection of these antibody-virion complexes declines while virus (antigen) and antibody are still present. Further study of the specificities of the antibodies bound in immune complexes and whether they are able to alter infectivity by enabling binding to antigen-presenting cells is under investigation.
AHI anti-gp41 Env antibodies activate complement.
A potentially important functional component of antibodies in AHI is their ability to fix complement. We determined if early anti-gp41 antibodies were capable of binding serum complement. Plasmas from six U.S. plasma donors were examined for complement activation/binding to CR2 using human peripheral blood mononuclear cells cocultured with HIV-1 virions. Complement-activating binding antibodies were present in all panels at every time point that plasma antibodies were detected as shown in Fig. 3. Moreover, the kinetics of appearance of complement-activating antibodies followed the same kinetics as gp41 binding antibodies. Both a laboratory-adapted HIV-1 strain (B.SF162) and an early transmitted virus strain (B.QH0692) were examined as targets of antibody and complement with similar results obtained with each virus (Fig. 6).
FIG. 6.

Ontogeny of complement binding antibodies during acute HIV-1 infection in times post-T0. Two representative patients (Pt) from the eclipse phase cohort (6240 and 6246) that had detectable HIV-specific antibodies were assessed for complement activation with an early virus isolate, HIV QH0692, and a laboratory-adapted isolate, HIV SF162.
Polyclonal B-cell activation following HIV-1 transmission in U.S. plasma donors.
HIV-1 Env gp120 has been suggested to be a polyclonal B-cell activator (3), to bind to VH3 Ig as a superantigen (23), and to induce polyclonal Ig class switching (28). Patients with chronic HIV-1 infection have polyclonal hypergammaglobulinemia (36), and a number of studies have reported hypergammaglobulinemia in early HIV-1 infection (47, 56). To examine whether polyclonal B-cell activation occurs during the initial antibody response to HIV-1 transmitted/founder Env, we measured quantitative IgM, IgG, and IgA levels on the initial and last plasma samples of the U.S. plasma donors. There was no significant elevation of IgM, IgG, or IgA during AHI in plasma donor panels (Fig. 7A). Similarly, the last plasma sample in each donor panel (range of 25 to 41 days after T0) was analyzed for the following autoantibodies: cardiolipin, SSA/Ro, SSB/La, Sm, RNP, Scl-70, Jo-1, double-stranded DNA, centromere B, and histones, and all were negative for all these specificities (data not shown). However, in a screen for rheumatoid factor autoantibodies (IgM antibodies that react with IgG) of 19 plasma donor samples and 10 CHAVI 001 acute infection cohort subjects studied, 8/29 (28%) tested positive for rheumatoid factor following HIV-1 transmission (Fig. 7B), with approximately half of these showing a decline in the level of rheumatoid factor antibody after initial detection during acute viremia. Thus, in some subjects, B-cell activation during acute HIV-1 infection can result in production of the autoantibody, rheumatoid factor (26).
FIG. 7.

(A) No hypergammaglobulinemia observed within the first 40 days of acute infection. Total antibody levels were measured at the first HIV-negative (HIV−)sample and the last sample in the panel (HIV+). The median concentration across panels is indicated. (B) Detection of rheumatoid factor (RF) during HIV acute phase viremia. IgM rheumatoid factor was measured using standard ELISA detection with positive rheumatoid factor controls. VL is measured in RNA copies/ml. Pt, patient.
Modeling of the initial gp41 antibody response with acute VL kinetics and assessment of antibody pressure on viral sequence evolution.
To determine the effects of initial anti-gp41 antibody responses on control of HIV-1 VL, we used mathematical modeling of the early viral dynamics. We first used the target cell-limited model (53), which does not include any effect of antibody, to analyze the plasma donor VL data obtained over the first 40 days after T0 for the six donors (6240, 6246, 9032, 9077, 9079, and 12008) for which both complete VL and antibody data were available over this time period. We found that for all donors except 9032, the target cell-limited model gave good agreement with the experimentally determined VL data (Fig. 8). We then fit to the same data three variants of this model that included enhanced virion clearance due to antibody opsonization, antibody-mediated viral neutralization, or antibody-dependent loss of HIV-1-infected cells. In these models (see Materials and Methods), we included the measured levels of anti-gp41 IgM, IgG, or the sum of IgM and IgG in mediating these effects. Anti-gp120-directed antibodies were not fit in the model since they did not appear in all subjects during the time period examined, and when they did, they appeared at a median time of 15 days later than the anti-gp41 antibodies. We found in all cases, except for donor 9032, that including anti-gp41 antibody-mediated effects did not improve the model fit (Table 3). Thus, it appears that for the majority of patients the basic target cell-limited model is the most compatible with the data; i.e., including the humoral immune response involving either anti-gp41 IgG, IgM, or both does not improve the model fit to the viral kinetics observed for the first 40 days after T0. This suggests that early in AHI either target cell limitation or cell-mediated responses play the predominant role in controlling VL. In support of this notion, no statistically significant association was identified between the magnitude of anti-gp41 IgG and the VL decay rate. There was also no statistically significant association identified between the VL decay rate and the time to the first elevation of antibody.
FIG. 8.

Modeling the effect of antibody on plasma viremia in AHI with the target cell-limited model. The target cell-limited model is the best-fitting model for the plasma donors studied except 9032. For 9032, a model with virion clearance enhanced by the sum of anti-gp41 IgM and IgG provides the best fit.
TABLE 3.
Comparison of the ability of the target cell-limited model and variants including anti-gp41 antibodies to fit early plasma VL kinetic data
| Subject no. | Fit of the indicated model (P value)a
|
Best-fitting model | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CE model:
|
ID model
|
CDE model
|
||||||||
| IgG | IgM | IgG+IgM | IgG | IgM | IgG+IgM | CDE IgG | CDE IgM | CDE IgG+ IgM | ||
| 6240 | 0.320 | 0.326 | 0.266 | 0.975 | 0.977 | 0.975 | 0.975 | 0.975 | 1.000 | Target cell limited |
| 6246 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | Target cell limited |
| 9032 | 0.004 | 0.006 | 0.001 | 0.005 | 0.006 | 0.006 | 0.89 | 0.011 | 0.923 | CE or ID |
| 9077 | 0.567 | 0.998 | 0.687 | 0.604 | 0.998 | 0.623 | 0.722 | 0.998 | 0.991 | Target cell limited |
| 9079 | 0.992 | 0.994 | 0.960 | 0.998 | 0.649 | 0.992 | 0.964 | 0.070 | 0.598 | Target cell limited |
| 12008 | 0.566 | 0.607 | 0.590 | 0.648 | 0.546 | 0.657 | 1.000 | 1.000 | 1.000 | Target cell limited |
The VL data from each donor was fit using the target cell-limited model and models that incorporated the effects of anti-gp41 IgG or IgM and IgG plus IgM. CE, clearance enhanced; ID, infectivity diminished; CDE, cell death enhanced. The models with antibody effects include one additional parameter, which when set to zero reduces the model to the target cell-limited model. An F-test was used to determine if any of the models including antibody fit the VL data significantly better than the target cell-model. The table gives the P values computed from the F-test.
Ontogeny of CD4i antibodies, CD4bs antibodies, and nonneutralizing cluster II (MPER) gp41 antibodies in plasma donors and in three additional AHI cohorts followed 6 to 12 months after transmission.
For analysis of antibody responses during the downslope of VL after transmission, we studied 12 clade B subjects from Trinidad/Tobago with heterosexual transmission, 14 acute clade C subjects from South Africa (the CAPRISA cohort) (24), and 10 clade B AHI subjects from the United States (3 untreated subjects and 7 subjects on antiretroviral treatment; CHAVI 001 cohort). The acute-phase viral kinetics and distribution of samples used in this study from a subset of subjects in the Trinidad/Tobago and the CHAVI 001 cohort are shown in Fig. S1 in the supplemental material. Subsequent to the initial B-cell response to HIV-1 infection that produces anti-gp41 antibodies, the anti-HIV-1 Env B-cell response eventually broadened to include other Env specificities. Antibodies that bind to the MPER gp41 (cluster II antibodies) (63) can be either neutralizing (e.g., MAbs 2F5, Z13, and 4E10) or nonneutralizing (e.g., MAbs 267D, 126-6, and 13H11) (reviewed in reference 2). Whereas nonneutralizing anti-gp41 MPER antibodies are commonly made in ∼80% of infected subjects (2), broadly neutralizing MPER antibodies are rarely made (27). CD4i antibodies bind at or near the coreceptor binding site and potently neutralize HIV-1 generally only after soluble CD4 is added to the in vitro neutralizing assay (15) due to the inability of a bivalent antibody to fit into the coreceptor binding site. Broadly neutralizing CD4bs antibodies are also rarely made (38). Previously described assays for these three anti-Env specificities were used to probe plasma donor panels (followed up to 40 days after T0) as well as to probe serial plasma samples from selected subjects in the clade C CAPRISA (24) and clade B Trinidad/Tobago (8) acute HIV-1 infection cohorts (both followed 6 to 12 months after transmission). As mentioned, CD4i, CD4bs, and nonneutralizing cluster II gp41 antibodies were not made during the first 40 days after T0 (Table 1). In the CAPRISA and Trinidad/Tobago AHI cohorts, CD4i antibodies, CD4bs antibodies, and nonneutralizing cluster II MPER gp41 antibodies arose at approximately the same time, from 5 to 10 weeks postenrollment into the acute infection study (see Fig. S3 in the supplemental material) (2).
Evaluation of anti-HIV-1 heterologous tier 1 and autologous neutralizing antibody responses in plasma donors and CAPRISA and Trinidad AHI cohorts.
As previously mentioned, using the highly neutralization-sensitive tier 1 clade B virus, SF162.LS, no neutralizing antibody responses were detected in plasma donors for up to 40 days after T0. Heterologous tier 1 neutralizing antibodies against HIV-1 MN were present in the Trinidad/Tobago cohort as early as 8 weeks after infection (see Table S1 in the supplemental material) and were likely primarily V3-directed since autologous V3 peptides competed for heterologous HIV-1 MN neutralization (M. L. Greenberg, unpublished data). Autologous neutralizing antibodies arose after a median of 32 weeks from the time of transmission in the Trinidad/Tobago clade B cohort (G. D. Tomaras and M. L. Greenberg, unpublished data) and at a mean of 19 weeks following transmission in the CAPRISA clade C cohort (24).
DISCUSSION
In this study we show that the initial B-cell response to the transmitted/founder virus is to HIV-1 gp41 Env, with responses to gp120 delayed by an additional 14 days. Mathematical modeling of the effects of initial IgM and IgG gp41 antibodies on viral kinetics in the plasma donor cohort revealed little if any effect of the initial antibody response on control of acute phase plasma viremia.
The timing and specificity of the initial antibody response to HIV-1 Env were of interest for several reasons. First, the window of opportunity for a vaccine to extinguish the transmitted or founder strain of HIV-1 is likely quite short, and the timing of this window will vary from subject to subject depending on the time of establishment of the latent pool of CD4+ T cells. That postexposure prophylaxis does not protect beyond 24 h after simian immunodeficiency virus challenge in rhesus macaques (18) implies that the window of opportunity may be 10 days or less in humans (61). Moreover, early appearance of evidence of systemic inflammation and acute phase reactants in plasma at 5 to 7 days before T0 (B. Kessler, A. McMichael, and P. Borrow, personal communication), as well as the appearance of plasma analytes of apoptosis 7 days after T0 (22), adds support to this short estimated window of opportunity for vaccine efficacy. Given that a narrow window of time might exist for antibodies to be protective and given that immune complexes only arise ∼18 days after transmission (8 days after T0 with an estimate of time from transmission to T0 of a mean of 10 days; range, 7 to 21 days) (7, 11, 14, 21, 40, 52), then the first antibody response to HIV-1 is quite delayed relative to when it optimally should occur to either extinguish or control transmitted/founder HIV-1 strains.
Our study is the first demonstration of virion-antibody complexes during the initial phase of viremia in acute HIV-1 infection. Previous work looking at later times in acute infection did not find immune complexes early in HIV-1 infection but, rather, found immune complexes only in chronic infection (16). The presence of these early immune complexes during acute infection raises the question of whether antibody-coated virus remains infectious; work is ongoing to determine the infectivity of opsonized virus (D. C. Montefiori, G. D. Tomaras, and B. F. Haynes, unpublished data). However, it is well established that the likelihood of HIV-1 transmission is highest during acute infection. Taken together, these data suggest an HIV-1 evasion strategy wherein the transmitted/founder virus initially induces antibodies that bind virions yet are nonneutralizing. Further work is needed to decipher the specificities and avidities of the antibodies in the immune complexes present at 8 days after T0 to determine if these initial antibody responses could be protective if a vaccine were able to prime for an early boost of the timing and magnitude of these antibodies after HIV-1 transmission.
That the initial B-cell response to Env selectively recognized gp41 was also of interest. Li et al. recently demonstrated that when broadly neutralizing antibodies do appear, they appear late and include antibodies against the CD4bs on gp120 (38). While there are broadly neutralizing epitopes on gp41 in the MPER, like broadly neutralizing CD4bs antibodies, neutralizing antibodies to the MPER are rarely made, and when they are made, require >1 year after transmission to arise (X. Shen and G. D. Tomaras, unpublished observations). Thus, the host-pathogen interactions occurring during and immediately after transmission result in a delay in recognition of the HIV-1 envelope by host B cells until after the latent pool of infected cells is likely established.
Polyclonal activation of B cells occurs in chronic HIV-1 infection and as well has been reported in early HIV-1 infection (47). We found no polyclonal hypergammaglobulinemia in plasma donors but did find plasma rheumatoid factor in ∼30% of subjects. Thus, polyclonal B-cell activation does occur early on, as signaled by the detection of this autoantibody, likely indicating triggering of CD5+ B cells that are producers of rheumatoid factor autoantibodies (26).
Also of interest is that there was heterogeneity in the pattern of Ig class switching seen following HIV-1 transmission. We have shown that the simultaneous appearance of anti-HIV IgM, IgG, and IgA is unlikely to be due to the presence of immune complexes that mask detection of antibodies because immune complexes appeared at the same time as free antibody in half of the subjects. Other potential explanations for simultaneous appearance of IgM, IgG, and IgA anti-HIV-1 antibodies in plasma include prior exposure to HIV-1 and primary T-cell-independent B1 and marginal zone B-cell responses to HIV-1 following transmission (9).
If the simultaneous appearance of IgM, IgG, and IgA to Env and Gag represents prior exposure of ∼60% of subjects to HIV-1 antigens and represents a rapid secondary response to HIV-1 full infection, then an atypical aspect of the response is that the putative “secondary” response occurs at exactly the same time as the primary IgM response (day 13.0 after T0) occurs in those with sequential appearance of anti-Env and anti-Gag IgM, IgG, and IgA. If the simultaneous response is indeed secondary from prior HIV-1 exposure, then it should occur approximately 7 days earlier than observed. Thus, we believe it is unlikely that the simultaneous appearance of IgM and class-switched antibodies in plasma in more than two-thirds of AHI subjects studied indicates prior exposure to HIV-1.
Soon after transmission in both humans and nonhuman primates when infection is established, there is a severe depletion of CD4 T lymphocytes (4, 57) that could lead to a lack of sufficient CD4 help for stimulation of B-cell responses. The early depletion of CD4+ CCR5+ T lymphocytes with massive apoptosis could, in addition to altering T-cell homeostasis, lead as well to suppression of an initial protective B-cell response (22, 43). Thus, elicitation of initial T-cell-independent antibody responses, in the setting of T-cell depletion, could be responsible for simultaneous appearance of IgM, IgG, and IgA anti-HIV-1 antibodies. A similar T-cell-independent pattern with simultaneous appearance of IgM, IgG, and IgA anti-pneumococcal antibody occurs following pneumococcal vaccine (9).
It was important to model both the antibody timing and the VL dynamics to determine any salutary or detrimental effects of early antibody responses on control of plasma viremia. As a null model we used a simple target cell-limited model that includes no effect of humoral or cellular immune responses (53). We found that for five of the six plasma donors for which we had VL data extending past the VL peak and out to day 40 after T0, this model gave good agreement with the VL data. Nonetheless, we asked if the fit could be improved by using a model that incorporated any of a variety of known functional effects of antibody. Not surprisingly, it was only for the one plasma donor, 9032, for which the target cell-limited model did not give good agreement with the VL data that an improvement was seen when antibody was included in the model. Interestingly, this donor was unusual in that the peak VL was significantly lower than in the other donors (3.4 × 104 copies/ml). Taken together, these analyses suggested that for most donors, early antibody had little functional consequence for the control of viremia.
If early antibody induced by the transmitted virus had any antiviral effects, then antibody-induced viral escape should be detected after the appearance of complexed or free antibody in plasma. In this regard, Keele et al. (35) have recently sequenced the transmitted founder virus for the plasma donors studied in this report and found that virus sequences at 14 days after T0 conformed to a model of random viral evolution, thus showing no evidence of early antibody-induced selection.
For HIV-1, functional consequences of antibody binding could include virus neutralization on T lymphocytes or macrophages (31, 32), ADCVI/antibody-dependent cellular cytotoxicity, complement-mediated neutralization, antibody Fc-mediated effector functions, virolysis, and/or inhibition of transcytosis. A recent study (29) suggested that the concentrations of antibodies mediating the different antiviral functions may be an important consideration for complete virus elimination since Fcγ receptor-binding function requires higher antibody concentrations than are required for virus neutralization. In addition, antibody- and complement-mediated virion lysis can develop in acute infection and can correlate with plasma VL during the acute stage of infection (33). This antiviral activity did not correlate with neutralizing antibodies and is thought to be an antiviral component of nonneutralizing antibodies. Dendritic cells are positioned in mucosae, where they are thought to be one of the first cell types to help establish infection (reviewed in reference 62). It will be important to determine whether the very earliest antibodies elicited in acute HIV-1 infection can block HIV-1 infection in dendritic cells at mucosal surfaces.
From the present study, it is clear that the initial B-cell response to the transmitted/founder virus does not control initial virus levels during the first 40 days of infection. However, we cannot rule out that some antibody specificities elicited after virus transmission may affect a subset of virions but are not substantial enough to significantly affect plasma viremia at the time they appear. A critical question is whether these types of nonneutralizing antibodies could be protective if present before infection or, alternatively, if completely different types of inhibitory antibodies will need to be induced by future HIV-1 vaccines. Autologous neutralizing antibodies target envelope variable loops (46, 49, 50, 60) that can arise long after any window of opportunity to extinguish HIV-1 has passed. Thus, an effective HIV-1 vaccine will need to induce antibodies prior to infection that bind native virion envelope molecules and lead as well to maturation of a rapid secondary neutralizing antibody response within the first week after transmission.
Supplementary Material
Acknowledgments
We sincerely thank the patients and staff of the Trinidad, CAPRISA, CHAVI, and Plasma Donor Cohorts along with Farley Cleghorn, Noreen Jack, and Koleka Mlisana. We thank Bette Korber for the consensus envelope design and Feng Gao and the Duke Center for AIDS Research Molecular Virology Core for sequence analysis. We also thank Judy Fleming, Regina Eschette, Steve Plonk, R. Glenn Overman, Ashleigh M. Nagel, Lei Jin, and Charlene McDanal for expert technical assistance; Darcy McMullin, Xuesong Yu, Linda Harris, and Doug Groves for database and statistical support; and Robin Shattock, Michael M. Frank, Peter F. Wright, Stephanie A. Freel, and Xiaoying Shen for helpful discussions.
This study was supported by NIH/NIAID grant UO1 AI-0678501, the Center for HIV/AIDS Vaccine Immunology grant AI64518, the Duke Center for AIDS Research, the Collaboration for AIDS Vaccine Discovery Vaccine Immune Monitoring Center, the Bill and Melinda Gates Foundation, and the R37 Merit Award DK 49381 (M.C.). The Trinidad Cohort was initially supported by NIH/National Institute of Allergy and Infectious Diseases grants PO1-AI40237 and 5-D43 TN 01041. N.L.Y. was supported by the Duke Interdisciplinary Research Training Program in AIDS 5T32 AI007392-17.
Footnotes
Published ahead of print on 8 October 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aasa-Chapman, M. M., A. Hayman, P. Newton, D. Cornforth, I. Williams, P. Borrow, P. Balfe, and A. McKnight. 2004. Development of the antibody response in acute HIV-1 infection. Aids 18371-381. [DOI] [PubMed] [Google Scholar]
- 2.Alam, S. M., R. M. Scearce, R. J. Parks, K. Plonk, S. G. Plonk, L. L. Sutherland, M. K. Gorny, S. Zolla-Pazner, S. Vanleeuwen, M. A. Moody, S. M. Xia, D. C. Montefiori, G. D. Tomaras, K. J. Weinhold, S. A. Karim, C. B. Hicks, H. X. Liao, J. Robinson, G. M. Shaw, and B. F. Haynes. 2008. Human immunodeficiency virus type 1 gp41 antibodies that mask membrane proximal region epitopes: antibody binding kinetics, induction, and potential for regulation in acute infection. J. Virol. 82115-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berberian, L., L. Goodglick, T. J. Kipps, and J. Braun. 1993. Immunoglobulin VH3 gene products: natural ligands for HIV gp120. Science 2611588-1591. [DOI] [PubMed] [Google Scholar]
- 4.Brenchley, J. M., T. W. Schacker, L. E. Ruff, D. A. Price, J. H. Taylor, G. J. Beilman, P. L. Nguyen, A. Khoruts, M. Larson, A. T. Haase, and D. C. Douek. 2004. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J. Exp. Med. 200749-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carne, C. A., R. S. Tedder, A. Smith, S. Sutherland, S. G. Elkington, H. M. Daly, F. E. Preston, and J. Craske. 1985. Acute encephalopathy coincident with seroconversion for anti-HTLV-III. Lancet 21206-1208. [DOI] [PubMed] [Google Scholar]
- 6.Chun, T. W., D. Engel, M. M. Berrey, T. Shea, L. Corey, and A. S. Fauci. 1998. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 958869-8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark, S. J., M. S. Saag, W. D. Decker, S. Campbell-Hill, J. L. Roberson, P. J. Veldkamp, J. C. Kappes, B. H. Hahn, and G. M. Shaw. 1991. High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N. Engl. J. Med. 324954-960. [DOI] [PubMed] [Google Scholar]
- 8.Cleghorn, F. R., N. Jack, J. K. Carr, J. Edwards, B. Mahabir, A. Sill, C. B. McDanal, S. M. Connolly, D. Goodman, R. Q. Bennetts, T. R. O'Brien, K. J. Weinhold, C. Bartholomew, W. A. Blattner, and M. L. Greenberg. 2000. A distinctive clade B HIV type 1 is heterosexually transmitted in Trinidad and Tobago. Proc. Natl. Acad. Sci. USA 9710532-10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clutterbuck, E. A., P. Salt, S. Oh, A. Marchant, P. Beverley, and A. J. Pollard. 2006. The kinetics and phenotype of the human B-cell response following immunization with a heptavalent pneumococcal-CRM conjugate vaccine. Immunology 119328-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper, D. A., J. Gold, P. Maclean, B. Donovan, R. Finlayson, T. G. Barnes, H. M. Michelmore, P. Brooke, and R. Penny. 1985. Acute AIDS retrovirus infection. Definition of a clinical illness associated with seroconversion. Lancet 1537-540. [DOI] [PubMed] [Google Scholar]
- 11.Cooper, D. A., A. A. Imrie, and R. Penny. 1987. Antibody response to human immunodeficiency virus after primary infection. J. Infect. Dis. 1551113-1118. [DOI] [PubMed] [Google Scholar]
- 12.Coordinating Committee of the Global HIV/AIDS Vaccine Enterprise. 2005. The Global HIV/AIDS Vaccine Enterprise: scientific strategic plan. PLoS Med. 2e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox, D. R., and D. Oakes. 1984. Analysis of survival data. Chapman and Hall, New York, NY.
- 14.Daar, E. S., T. Moudgil, R. D. Meyer, and D. D. Ho. 1991. Transient high levels of viremia in patients with primary human immunodeficiency virus type 1 infection. N. Engl. J. Med. 324961-964. [DOI] [PubMed] [Google Scholar]
- 15.Decker, J. M., F. Bibollet-Ruche, X. Wei, S. Wang, D. N. Levy, W. Wang, E. Delaporte, M. Peeters, C. A. Derdeyn, S. Allen, E. Hunter, M. S. Saag, J. A. Hoxie, B. H. Hahn, P. D. Kwong, J. E. Robinson, and G. M. Shaw. 2005. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J. Exp. Med. 2011407-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dianzani, F., G. Antonelli, E. Riva, O. Turriziani, L. Antonelli, S. Tyring, D. A. Carrasco, H. Lee, D. Nguyen, J. Pan, J. Poast, M. Cloyd, and S. Baron. 2002. Is human immunodeficiency virus RNA load composed of neutralized immune complexes? J. Infect. Dis. 1851051-1054. [DOI] [PubMed] [Google Scholar]
- 17.Douek, D. C., P. D. Kwong, and G. J. Nabel. 2006. The rational design of an AIDS vaccine. Cell 124677-681. [DOI] [PubMed] [Google Scholar]
- 18.Emau, P., Y. Jiang, M. B. Agy, B. Tian, G. Bekele, and C. C. Tsai. 2006. Post-exposure prophylaxis for SIV revisited: animal model for HIV prevention. AIDS Res. Ther. 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiebig, E. W., D. J. Wright, B. D. Rawal, P. E. Garrett, R. T. Schumacher, L. Peddada, C. Heldebrant, R. Smith, A. Conrad, S. H. Kleinman, and M. P. Busch. 2003. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS 171871-1879. [DOI] [PubMed] [Google Scholar]
- 20.Forthal, D. N., G. Landucci, and E. S. Daar. 2001. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural-killer effector cells. J. Virol. 756953-6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaines, H., M. von Sydow, A. Sonnerborg, J. Albert, J. Czajkowski, P. O. Pehrson, F. Chiodi, L. Moberg, E. M. Fenyo, B. Asjo, et al. 1987. Antibody response in primary human immunodeficiency virus infection. Lancet 11249-1253. [DOI] [PubMed] [Google Scholar]
- 22.Gasper-Smith, N., D. M. Crossman, J. F. Whitesides, N. Mensali, J. S. Ottinger, S. G. Plonk, M. A. Moody, G. Ferrari, K. J. Weinhold, S. E. Miller, C. F. Reich III, L. Qin, S. G. Self, G. M. Shaw, T. N. Denny, L. E. Jones, D. S. Pisetsky, and B. F. Haynes. 2008. Induction of plasma (TRAIL), TNFR-2, Fas ligand, and plasma microparticles after human immunodeficiency virus type 1 (HIV-1) transmission: implications for HIV-1 vaccine design. J. Virol. 827700-7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodglick, L., N. Zevit, M. S. Neshat, and J. Braun. 1995. Mapping the Ig superantigen-binding site of HIV-1 gp120. J. Immunol. 1555151-5159. [PubMed] [Google Scholar]
- 24.Gray, E. S., P. L. Moore, I. A. Choge, J. M. Decker, F. Bibollet-Ruche, H. Li, N. Leseka, F. Treurnicht, K. Mlisana, G. M. Shaw, S. S. Karim, C. Williamson, and L. Morris. 2007. Neutralizing antibody responses in acute human immunodeficiency virus type 1 subtype C infection. J. Virol. 816187-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green, P. J., and B. W. Silverman. 1994. Nonparametric regression and generalized linear models: a roughness penalty approach. Chapman and Hall, New York, NY.
- 26.Hardy, R. R., K. Hayakawa, M. Shimizu, K. Yamasaki, and T. Kishimoto. 1987. Rheumatoid factor secretion from human Leu-1+ B cells. Science 23681-83. [DOI] [PubMed] [Google Scholar]
- 27.Haynes, B. F., and D. C. Montefiori. 2006. Aiming to induce broadly reactive neutralizing antibody responses with HIV-1 vaccine candidates. Expert Rev. Vaccines 5347-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He, B., X. Qiao, P. J. Klasse, A. Chiu, A. Chadburn, D. M. Knowles, J. P. Moore, and A. Cerutti. 2006. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J. Immunol. 1763931-3941. [DOI] [PubMed] [Google Scholar]
- 29.Hessell, A. J., L. Hangartner, M. Hunter, C. E. Havenith, F. J. Beurskens, J. M. Bakker, C. M. Lanigan, G. Landucci, D. N. Forthal, P. W. Parren, P. A. Marx, and D. R. Burton. 2007. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449101-104. [DOI] [PubMed] [Google Scholar]
- 30.Ho, D. D., M. G. Sarngadharan, L. Resnick, F. Dimarzoveronese, T. R. Rota, and M. S. Hirsch. 1985. Primary human T-lymphotropic virus type III infection. Ann. Intern. Med. 103880-883. [DOI] [PubMed] [Google Scholar]
- 31.Holl, V., M. Peressin, T. Decoville, S. Schmidt, S. Zolla-Pazner, A. M. Aubertin, and C. Moog. 2006. Nonneutralizing antibodies are able to inhibit human immunodeficiency virus type 1 replication in macrophages and immature dendritic cells. J. Virol. 806177-6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holl, V., M. Peressin, S. Schmidt, T. Decoville, S. Zolla-Pazner, A. M. Aubertin, and C. Moog. 2006. Efficient inhibition of HIV-1 replication in human immature monocyte-derived dendritic cells by purified anti-HIV-1 IgG without induction of maturation. Blood 1074466-4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huber, M., M. Fischer, B. Misselwitz, A. Manrique, H. Kuster, B. Niederost, R. Weber, V. von Wyl, H. F. Gunthard, and A. Trkola. 2006. Complement lysis activity in autologous plasma is associated with lower viral loads during the acute phase of HIV-1 infection. PLoS Med. 3e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnston, M. I., and A. S. Fauci. 2007. An HIV vaccine—evolving concepts. N. Engl. J. Med. 3562073-2081. [DOI] [PubMed] [Google Scholar]
- 35.Keele, B. F., E. E. Giorgi, J. F. Salazar-Gonzalez, J. M. Decker, K. T. Pham, M. G. Salazar, C. Sun, T. Grayson, S. Wang, H. Li, X. Wei, C. Jiang, J. L. Kirchherr, F. Gao, J. A. Anderson, L. H. Ping, R. Swanstrom, G. D. Tomaras, W. A. Blattner, P. A. Goepfert, J. M. Kilby, M. S. Saag, E. L. Delwart, M. P. Busch, M. S. Cohen, D. C. Montefiori, B. F. Haynes, B. Gaschen, G. S. Athreya, H. Y. Lee, N. Wood, C. Seoighe, A. S. Perelson, T. Bhattacharya, B. T. Korber, B. H. Hahn, and G. M. Shaw. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1057552-7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lane, H. C., H. Masur, L. C. Edgar, G. Whalen, A. H. Rook, and A. S. Fauci. 1983. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N. Engl. J. Med. 309453-458. [DOI] [PubMed] [Google Scholar]
- 37.Li, M., F. Gao, J. R. Mascola, L. Stamatatos, V. R. Polonis, M. Koutsoukos, G. Voss, P. Goepfert, P. Gilbert, K. M. Greene, M. Bilska, D. L. Kothe, J. F. Salazar-Gonzalez, X. Wei, J. M. Decker, B. H. Hahn, and D. C. Montefiori. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 7910108-10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li, Y., S. A. Migueles, B. Welcher, K. Svehla, A. Phogat, M. K. Louder, X. Wu, G. M. Shaw, M. Connors, R. T. Wyatt, and J. R. Mascola. 2007. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat. Med. 131032-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao, H. X., L. L. Sutherland, S. M. Xia, M. E. Brock, R. M. Scearce, S. Vanleeuwen, S. M. Alam, M. McAdams, E. A. Weaver, Z. Camacho, B. J. Ma, Y. Li, J. M. Decker, G. J. Nabel, D. C. Montefiori, B. H. Hahn, B. T. Korber, F. Gao, and B. F. Haynes. 2006. A group M consensus envelope glycoprotein induces antibodies that neutralize subsets of subtype B and C HIV-1 primary viruses. Virology 353268-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Little, S. J., A. R. McLean, C. A. Spina, D. D. Richman, and D. V. Havlir. 1999. Viral dynamics of acute HIV-1 infection. J. Exp. Med. 190841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mascola, J. R., M. G. Lewis, G. Stiegler, D. Harris, T. C. VanCott, D. Hayes, M. K. Louder, C. R. Brown, C. V. Sapan, S. S. Frankel, Y. Lu, M. L. Robb, H. Katinger, and D. L. Birx. 1999. Protection of macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J. Virol. 734009-4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mascola, J. R., S. W. Snyder, O. S. Weislow, S. M. Belay, R. B. Belshe, D. H. Schwartz, M. L. Clements, R. Dolin, B. S. Graham, G. J. Gorse, M. C. Keefer, M. J. McElrath, M. C. Walker, K. F. Wagner, J. G. McNeil, F. E. McCutchan, D. S. Burke, et al. 1996. Immunization with envelope subunit vaccine products elicits neutralizing antibodies against laboratory-adapted but not primary isolates of human immunodeficiency virus type 1. J. Infect. Dis. 173340-348. [DOI] [PubMed] [Google Scholar]
- 43.Moir, S., and A. S. Fauci. 2008. Pathogenic mechanisms of B-lymphocyte dysfunction in HIV disease. J. Allergy Clin. Immunol. [DOI] [PMC free article] [PubMed]
- 44.Montero, M., N. E. van Houten, X. Wang, and J. K. Scott. 2008. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol. Mol. Biol. Rev. 7254-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore, J. P., Y. Cao, D. D. Ho, and R. A. Koup. 1994. Development of the anti-gp120 antibody response during seroconversion to human immunodeficiency virus type 1. J. Virol. 685142-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore, P. L., E. S. Gray, I. A. Choge, N. Ranchobe, K. Mlisana, S. S. Abdool Karim, C. Williamson, and L. Morris. 2008. The C3-V4 region is a major target of autologous neutralizing antibodies in human immunodeficiency virus type 1 subtype C infection. J. Virol. 821860-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris, L., J. M. Binley, B. A. Clas, S. Bonhoeffer, T. P. Astill, R. Kost, A. Hurley, Y. Cao, M. Markowitz, D. D. Ho, and J. P. Moore. 1998. HIV-1 antigen-specific and -nonspecific B cell responses are sensitive to combination antiretroviral therapy. J. Exp. Med. 188233-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phogat, S., K. Svehla, M. Tang, A. Spadaccini, J. Muller, J. Mascola, I. Berkower, and R. Wyatt. 2008. Analysis of the human immunodeficiency virus type 1 gp41 membrane proximal external region arrayed on hepatitis B surface antigen particles. Virology 37372-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinter, A. 2007. Roles of HIV-1 Env variable regions in viral neutralization and vaccine development. Curr. HIV Res. 5542-553. [DOI] [PubMed] [Google Scholar]
- 50.Richman, D. D., T. Wrin, S. J. Little, and C. J. Petropoulos. 2003. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 1004144-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scanlan, C. N., J. Offer, N. Zitzmann, and R. A. Dwek. 2007. Exploiting the defensive sugars of HIV-1 for drug and vaccine design. Nature 4461038-1045. [DOI] [PubMed] [Google Scholar]
- 52.Schacker, T., A. C. Collier, J. Hughes, T. Shea, and L. Corey. 1996. Clinical and epidemiologic features of primary HIV infection. Ann. Intern. Med. 125257-264. [DOI] [PubMed] [Google Scholar]
- 53.Stafford, M. A., L. Corey, Y. Cao, E. S. Daar, D. D. Ho, and A. S. Perelson. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203285-301. [DOI] [PubMed] [Google Scholar]
- 54.Storey, J. D. 2002. A direct approach to false discovery rates. J. Royal Stat. Soc. 64479-498. [Google Scholar]
- 55.Thiebaut, R., and H. Jacqmin-Gadda. 2004. Mixed models for longitudinal left-censored repeated measures. Comput. Methods Programs Biomed. 74255-260. [DOI] [PubMed] [Google Scholar]
- 56.Titanji, K., F. Chiodi, R. Bellocco, D. Schepis, L. Osorio, C. Tassandin, G. Tambussi, S. Grutzmeier, L. Lopalco, and A. De Milito. 2005. Primary HIV-1 infection sets the stage for important B lymphocyte dysfunctions. AIDS 191947-1955. [DOI] [PubMed] [Google Scholar]
- 57.Veazey, R. S., P. M. Acierno, K. J. McEvers, S. H. Baumeister, G. J. Foster, M. D. Rett, M. H. Newberg, M. J. Kuroda, K. Williams, E. Y. Kim, S. M. Wolinsky, E. P. Rieber, M. Piatak, Jr., J. D. Lifson, D. C. Montefiori, C. R. Brown, V. M. Hirsch, and J. E. Schmitz. 2008. Increased loss of CCR5+ CD45RA− CD4+ T cells in CD8+ lymphocyte-depleted simian immunodeficiency virus-infected rhesus monkeys. J. Virol. 825618-5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verbeke, G., and G. Molenberghs. 2000. Linear mixed models for longitudinal data. Springer, New York, NY.
- 59.Vidoni, P. 2006. Response prediction in mixed effects models. J. Stat. Plan. Inference 1363948-3966. [Google Scholar]
- 60.Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422307-312. [DOI] [PubMed] [Google Scholar]
- 61.Wong, S. B. J., and R. F. Siliciano. 2007. Biology of early infection and impact on vaccine design, p. 17-22. In P. K. Wayne C. Koff, and Ian D. Gust (ed.), AIDS vaccine development: challenges and opportunities. Caister Academic Press, Norfold, United Kingdom.
- 62.Wu, L., and V. N. KewalRamani. 2006. Dendritic-cell interactions with HIV: infection and viral dissemination. Nat. Rev. Immunol. 6859-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu, J. Y., M. K. Gorny, T. Palker, S. Karwowska, and S. Zolla-Pazner. 1991. Epitope mapping of two immunodominant domains of gp41, the transmembrane protein of human immunodeficiency virus type 1, using ten human monoclonal antibodies. J. Virol. 654832-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu, W., R. Hofmann-Lehmann, H. M. McClure, and R. M. Ruprecht. 2002. Passive immunization with human neutralizing monoclonal antibodies: correlates of protective immunity against HIV. Vaccine 201956-1960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.