

Abstract
The septin family of GTPases, first identified for their roles in cell division, are also expressed in postmitotic tissues. SEPT3 (G-septin) and SEPT5 (CDCrel-1) are highly expressed in neurons, enriched in presynaptic terminals, and associated with synaptic vesicles. These characteristics suggest that SEPT3 or SEPT5 might be important for synapse formation, maturation, or synaptic vesicle traffic. Since Sept5−/− mice do not show any overt neurological phenotypes, we generated Sept3−/− and Sept3−/− Sept5−/− mice and found that SEPT3 and SEPT5 are not essential for development, fertility, or viability. Changes in the expression of septins were noted in the absence of SEPT3, SEPT5, and both septins. SEPT5 association with other septins in brain tissue was unaffected by the removal of SEPT3. No abnormalities were observed in the gross morphology and synapses of the hippocampus. Similarly, axon development and synapse formation were unaffected in vitro. In cultured hippocampal neurons, the size of the recycling synaptic vesicle pool was unaltered in the absence of SEPT3. Furthermore, synaptic transmission at two different central synapses was not significantly affected in Sept3−/− Sept5−/− mice. These results indicate that SEPT3 and SEPT5 are dispensable for neuronal development as well as for synaptic vesicle fusion and recycling.
The septins are a multigene family of guanine nucleotide-binding, filament-forming proteins. Several septins have well-established roles in cytokinesis in a variety of cells that span a broad variety of species from yeast to mammals. The septin gene family was originally identified in the budding yeast Saccharomyces cerevisiae in a genetic screen for critical determinants of cytokinesis (23). S. cerevisiae has seven septin genes (CDC3, CDC10, CDC11, CDC12, SHS1/SEP7, SPR3, and SPR28 genes), Caenorhabditis elegans has two (UNC-59 and UNC61 genes), Drosophila melanogaster has five (Pnut, Sep1, Sep2, Sep4 and Sep5 genes), and there are 14 genes in humans (SEPT1 to SEPT14) (21, 46).
The septins share a conserved GTPase domain that comprises the bulk of their sequence. The GTPase domain has characteristic amino acid motifs found in other P-loop GTPases (52): a P-loop for nucleotide binding [GXXXXGK(S/T)], a G3 domain for GTP hydrolysis (DXXG), and a G4 domain for stabilizing the guanine nucleotide binding site (XKXD). Septins bind and hydrolyze nucleotides in vitro (15, 17, 25, 42, 64) although the significance of this for their function is not known. At the C terminus of most mammalian septins is a predicted coiled-coil domain, with the exception of SEPT3, SEPT9, and SEPT12. Yeast two-hybrid and direct binding studies suggest that this region is important for septin-septin interactions (65). However, recent structural analysis of a mammalian septin complex by X-ray crystallography and electron microscopy revealed that the coiled-coil domains could be used for filament stabilization but are not essential for filament assembly (55). At the N-terminal side of the GTPase domain, most septins contain a stretch of polybasic amino acids, which mediates their association with phosphatidylinositol phospholipids (10, 50, 76).
Subsets of septins have a high affinity for each other and form large, stable heteromeric complexes in stoichiometric ratios (17, 19, 34). Mammalian septin complexes vary in composition, with identified complexes comprising SEPT4, SEPT5, and SEPT8 (SEPT4/5/8); SEPT7/9b/11; and SEPT2/6/7/9 (40, 43, 54, 60). Similar to other GTPases like tubulin and FtsZ, septin complexes assemble into filaments in vitro and these have a 4- to 10-nm diameter (17, 19, 24, 29). Although they do not have inherent curvature, septin filaments are flexible and tend to roll up into rings of 0.5- to 0.7-μm diameter (34). Septin rings are found in a variety of cellular contexts in vivo: at the mother-bud neck junction in yeast (9), at the yeast cell cortex (49), at the annulus of sperm tails (26, 36), as a circumferential band in platelets (39), and in cells where actin filament organization is disrupted with cytochalasin D (34, 69). Straight septin filaments are found along central actin stress fibers (35, 60).
Septins have a range of functions unrelated to cytokinesis that are only just beginning to emerge, including apoptosis, vesicular transport, cytoskeletal dynamics, cell polarity, and sperm motility (30, 33, 41, 57). Interestingly, septins are abundant in the central nervous system, which is comprised largely of postmitotic neurons. They are associated with many neurological diseases such as Parkinson's, Alzheimer's, schizophrenia, and hereditary neuralgic amyotrophy (3, 27, 31, 38, 56), but their neurobiological function is unknown.
We originally set out to test the function of septins in the mammalian central nervous system by genetically disrupting the Sept5 gene in mice (47). There were several good reasons to believe that SEPT5 had a role in neurotransmitter release. First, SEPT5 was isolated as a constituent of a synaptic vesicle protein complex associated with synaptophysin (4). Second, SEPT5 was localized to the presynaptic side of the synapse in close apposition with synaptic vesicles (32). Third, it was shown that SEPT5 directly binds to the synaptic vesicle fusion protein syntaxin 1 (4). Fourth, overexpression of SEPT5 inhibited the secretion of storage granules from an insulin-secreting cell line (4), and its absence enhanced the secretion of storage granules from platelets (11). Despite the overwhelming evidence supporting a role for SEPT5 in synaptic vesicle fusion, no alterations in synaptic transmission were observed in the hippocampus of Sept5−/− mice (47). Since septins form heteromeric protein complexes with each other, another septin could have overlapping functions with SEPT5.
SEPT3 was discovered as a substrate of cyclic GMP-dependent protein kinase (72, 74) and is the only septin known to date that is exclusively expressed in postmitotic neurons (73), implying that it has no role in cell division and is likely to function in a neuron-specific process. It is intriguing to think that SEPT3 has a critical role in the synaptic vesicle cycle because it is highly enriched in presynaptic terminals of hippocampal neurons, associates with synaptic vesicles, and colocalizes with synaptophysin and dynamin I (73). To determine whether SEPT3 is the critical septin essential for synaptic vesicle trafficking or whether SEPT3 and SEPT5 function together, we now report on the targeted disruption of the Sept3 gene as well as the generation of mice lacking both Sept3 and Sept5.
MATERIALS AND METHODS
Animal handling.
All procedures involving animals were in compliance with guidelines of the Animal Care Committee at The Hospital for Sick Children (Toronto, ON, Canada).
Antibodies.
Antibodies against the mammalian septins were made in-house as previously described (2, 60, 69). The SEPT5 monoclonal antibody (clone SP20) was a gift from W. G. Honer (University of British Columbia, Vancouver, BC, Canada). Antibodies to synaptophysin and MAP2 were from Chemicon (Temecula, CA). The antibody to neurofilament-H (clone SMI31) was from Covance Research Products (Berkeley, CA). The horseradish peroxidase-conjugated, Cy3-conjugated, and Cy5-conjugated anti-mouse and anti-rabbit secondary antibodies were from Jackson ImmunoResearch Laboratories (Westgrove, PA). AlexaFluor 488-conjugated secondary antibodies were from Invitrogen (Burlington, ON, Canada).
Sept3 gene targeting vector and generation of Sept3−/− mice.
Rat Sept3 (G-septin-α) cDNA was used as a radiolabeled probe to screen the RPCI 129/SvEvTACBr mouse bacterial artificial chromosome (BAC) library by Southern blot analysis. Exons 2 through 8 of the mouse Sept3 gene were mapped on a fragment of the 44M13 BAC clone by restriction digest analysis and by DNA sequencing. The long arm of homology from intron 5 to exon 9 was isolated on a 5.5-kb DNA fragment cut with KpnI. The short arm of homology was isolated by PCR amplification of a 2.6-kb DNA fragment from the 44M13 BAC clone using a forward primer to intron 1 (CGG AAT TCG GCA GGT ACA TCC CCC TAT CTT CTG AG, with a 5′ EcoRI extension) and a reverse primer to exon 2 (GCT CTA GAC ATG GGC ACC GCC GGC TTA GGC CTG GG, with a 3′ XbaI extension).
The 7.2-kb DNA plasmid pNT vector (63) was used as the backbone for the Sept3 gene targeting vector. This vector has a Neor gene for positive selection and a thymidine kinase gene for negative selection. The Neor gene is flanked by two multicloning sites that were used to insert the arms of homology and to linearize the targeting vector. The short arm of homology was directionally subcloned into the pNT vector using the EcoRI and XbaI restriction sites. The long arm of homology was subcloned into the XhoI restriction site using KpnI-XhoI adaptors, and directionality was checked by restriction digest analysis. The subcloning was in the opposite transcriptional orientation to the Neor and thymidine kinase genes. The Sept3 targeting vector was linearized with NotI, and 10 μg of linearized DNA was introduced by electroporation into the mouse embryonic stem (ES) cell line R1. Transfected ES cells were grown on gelatinized plates for several days in selective medium containing G418 and ganciclovir.
Recombination in ES cell clones was confirmed by Southern blot analysis. Proper integration at the 5′ end of the homologous target sequence was determined by mobility shift of an SphI-digested DNA fragment. ES cells undergoing proper homologous recombination were prepared for aggregation. ES cells were grown sparsely on gelatinized plates to produce 8- to 16-cell clumps. These clumps were mixed with embryos at the morula stage (8 cells) isolated from 12 superovulated CD1 females, and both cell populations were allowed to aggregate for 1 day. Chimeric embryos (20 to 30) were reimplanted into pseudopregnant female mice. The chimeric male mice were bred with CD1 female mice to produce the F1 generation. F1 mice heterozygous for the mutation were then bred to get the F2 generation. Germ line transmission of the mutation was verified by Southern blot analysis.
Genotyping.
PCR analysis was used to routinely genotype mouse tails using primers to regions in the short arm of homology (Ex2 start, GGG CTC CCA GAG GCT AGG ACG GAC AC) and the neomycin gene (Neo, CGC ATG CCC GAC GGC GAG GAT CTC GTC) to yield a 0.74-kb DNA fragment from the recombinant locus. Primers to exon 5 (Ex5, ATT GCC AGG AAG AAA CGC ATC CCT GAC) and exon 6 (Ex6, GTT TCA TGA ACT CAA GAT CCA GTG GTC) were used to yield a 1.4-kb DNA fragment from the wild-type locus.
Generation of Sept3−/− Sept5−/− mice.
Compound disruption of both the Sept3 and Sept5 genes was created by interbreeding Sept3−/− and Sept5−/− mice (47).
Tissue preparation and Western blot analysis.
Brains from wild-type and Sept3−/− mice at various stages of development were homogenized in homogenization buffer (10 mM HEPES [pH 7.4], 150 mM NaCl, 320 mM sucrose, and 2 mM EDTA plus protease inhibitors [1 μg/ml leupeptin, 1 μg/ml pepstatin, 20 μg/ml phenylmethylsulfonyl fluoride, and 1 mM EDTA]). Aliquots were solubilized in 2× concentrated sodium dodecyl sulfate (SDS) loading buffer (100 mM Tris-HCl [pH 6.8], 4% SDS, 0.2% bromophenol blue, and 20% glycerol) with 5% β-mercaptoethanol when reducing conditions were required. As previously described (60), 20 μg of each sample was electrophoresed through a 10% or 12% SDS-polyacrylamide gel, transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA), and blocked in 5% milk powder in phosphate-buffered saline (PBS)-Tween. Following primary and secondary antibody incubations, immobilized antibodies were detected with an ECL Western blotting detection reagent (GE Healthcare Biosciences Corp., Piscataway, NJ) and exposure to Kodak film (Rochester, NY).
Immunoprecipitation.
Mouse brains were flash-frozen with liquid nitrogen, crushed using a mortar and pestle, resuspended in 10 ml HKA buffer (10 mM HEPES-KOH [pH 7.4], 140 mM potassium acetate, 1 mM magnesium chloride and protease inhibitors), and homogenized using a Dounce homogenizer. The homogenized brain tissue was centrifuged at 800 × g for 10 min at 4°C to remove cell debris and nuclei. The resulting supernatant was incubated with 2% Triton X-100 in HKA buffer for 1 h at 4°C and passed through a 27-gauge needle three times. Triton X-100 insoluble material was pelleted at 14,000 rpm at 4°C. Protein concentrations were determined by the bicinchoninic assay (Pierce, Rockford, IL), and 2 mg of protein lysate was used for immunoprecipitation. A 10-μl bed volume of protein G beads (Invitrogen Canada Inc, Burlington, ON, Canada) was washed with 1 ml of HKA buffer. Nonspecific binding sites were blocked with 0.1% gelatin and 0.1% bovine serum albumin in HKA buffer for 30 min at room temperature and then incubated with 2 μg of SEPT5 antibody (clone SP20) for 4 h at 4°C. Following immunoprecipitation, the beads were washed three times with 1 ml of HKA buffer containing 1% Triton X-100 and 0.1% gelatin, followed by a wash with the Triton X-100 concentration increased to 2%. Protein complexes were eluted from the beads with 2× SDS loading buffer containing 5% β-mercaptoethanol.
Imaging mouse brains.
Anesthetized wild-type and Sept3−/− mice were transcardially perfused with heparinized saline followed by 10% formalin. Perfused brains were dissected and fixed further in 2% glutaraldehyde overnight at room temperature. Sections were cut sagittally and processed for hematoxylin and eosin staining by the Pathology Department at The Hospital for Sick Children (Toronto, ON, Canada).
Electron microscopy.
Mice were fixed with 10% formalin in heparinized saline by transcardial perfusion. Subsequently, the brains were dissected and postfixed in 2% glutaraldehyde in phosphate buffer overnight at room temperature. A 1-mm3 region of the CA1 region of the hippocampus was isolated using a vibratome. Thin sections (50 to 80 nm in thickness) were cut from the block and stained with 0.5% osmium tetroxide in phosphate buffer for 15 min and in 1% uranyl acetate in water for 15 min. After dehydration with increasing concentrations of ethanol, samples were embedded in Spurr resin (Canemco Inc., Montreal, QC, Canada). Sections were cut en face, stained with 2.0% uranyl acetate for 15 min and 0.2% lead citrate for 5 min, and then imaged with a Philips CM100 electron microscope.
Immunostaining hippocampal neurons.
Mouse hippocampal neurons were isolated from hippocampi from 17-day-old fetal mouse brains as previously described (8). After 3 or 10 days in culture, the neurons were fixed with 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and blocked with 5% horse serum in PBS for 1 h at room temperature. Primary antibodies were diluted in PBS containing 0.5% horse serum and incubated for 1 h at room temperature or overnight at 4°C. Fluorescence-labeled secondary antibodies were added for 1 h at room temperature and then washed with PBS.
Images of fluorescently stained hippocampal neurons mounted onto glass slides with Dako mounting medium (Glostrup, Denmark) were taken with a Zeiss LSM510 Multiphoton laser scanning confocal microscope (Zeiss, Thornwood, NY) and processed with Adobe Photoshop and Adobe Illustrator software. In order to quantitate axon outgrowth, digital images of hippocampal neurons were converted to GIF format with Adobe Photoshop software and then analyzed with ImageJ software using the NeuronJ plug-in.
Synaptic vesicle recycling with FM 1-43.
Primary mouse hippocampal neurons from wild-type or Sept3−/− brains were cultured for 10 days on glass coverslips. The coverslips were secured in an imaging chamber (RC-21BRFS and P-2 Series 20 platform; Warner Instruments, Hamden, CT) and mounted on the stage of a Leica DMIRB microscope (Richmond Hill, ON, Canada) using a stage adaptor (Series 20 Stage adaptor; Warner Instruments, Hamden, CT). Before each experiment, the growth medium was replaced with Tyrode's buffer (119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose and 25 mM HEPES, pH 7.4). Dye loading (a measure of endocytosis) was done in Tyrode's buffer containing 47 mM K+ (isotonic replacement of Na+ for K+) and 15 μM FM 1-43 (Invitrogen-Molecular Probes, Burlington, ON, Canada) for 90 s. The cells were washed by perfusion with Ca2+-free Tyrode's buffer for 10 min. Dye unloading (a measure of exocytosis) was elicited with five 1-min applications of Tyrode's buffer containing 90 mM K+ (separated by 1 min washes with Ca2+-free Tyrode's) to ensure maximal release of the dye. Fluorescence images of synapses (an average of three images) were collected on initial dye loading (Fpre) and after unloading (Fpost). The size of the recycling pool was calculated for each synapse as the change in fluorescence intensity: ΔF = Fpre − Fpost. All FM 1-43 experiments were performed at room temperature. AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDA (N-methyl-D-aspartate) glutamate receptors were blocked with CNQX (6-cyano-2,3-dihydroxy-7-nitro-quinoxaline; 10 μM) and APV (2-amino-5-phosphonovaleric acid; 50 μM), respectively, throughout the course of the experiments to prevent recurrent excitation and induction of synaptic plasticity. Fluorescence images were taken through a 40× (numerical aperture, 0.75) objective lens by exciting fluorescently labeled neurons at 488 nm using a Chroma fluorescein isothiocyanate filter cube (Rockingham, VT) and collecting the emitted light through a 535-nm emission filter with a Cascade II electron-multiplying charge-coupled-device camera (Tuscon, AZ). Fluorescent image acquisition, followed by a bright-field image using Nomarski optics, was controlled by Metafluor software, version 6.3R6 (Universal Imaging Corp., West Chester, PA), through a Lambda10-2 shutter controller (Sutter Instruments, Novato, CA). The data were calculated using Microsoft Excel software and plotted using GraphPad Prism, version 4. Statistical measurements between frequency histograms were made using the nonparametric Kolmogorov-Smirnov test (http://www.physics.csbsju.edu/stats/KS-test.html).
Calyx electrophysiology.
Brainstem slices were prepared from postnatal day 13 (P13) to P16 wild-type or Sept3−/− Sept5−/− mice as previously described for rats (18). Following decapitation with a small guillotine, brains were immediately immersed into semifrozen artificial cerebral spinal fluid (aCSF) containing 125 mM NaCl, 2.5 mM KCl, 10 mM glucose, 1.25 mM NaH2PO4, 2 mM Na-pyruvate, 2 mM myo-inositol, 0.5 mM ascorbic acid, 26 mM NaHCO3, 1 mM MgCl2, and 2 mM CaCl2 at a pH of 7.3 when oxygenated (95% O2 and 5% CO2), followed by rapid dissection. Transverse slices of the auditory brainstem containing the medial nucleus of the trapezoid body (MNTB) were cut at a thickness of 200 to 250 μm using a microtome (Leica VT1000S), followed by incubation at 37°C for 1 h prior to experimentation. All experiments were performed at room temperature (20 to 22°C).
For electrophysiological recordings, aCSF was supplemented with bicuculline (10 μM) and strychnine (1 μM) to block inhibitory inputs, tetrodotoxin (0.5 μM) to block Na+ channels, and 10 mM tetraethylammonium (TEA) and 0.3 mM 4-aminopyridine to block K+ channels. [Ca2+]outside was adjusted to 1 mM to improve voltage-clamp of presynaptic Ca2+ currents (ICa) (16, 68). Patch electrodes typically had resistances of 4 to 6 MΩ and 2.5 to 3 MΩ for presynaptic and postsynaptic recordings, respectively. For whole-cell voltage-clamp recordings, presynaptic and postsynaptic series resistances were 6 to 12 MΩ (<10 MΩ in the majority of recordings) and 4 to 8 MΩ, respectively, compensated to 90%, with cells showing higher resistances being omitted from analysis. Intracellular recording solution for presynaptic Ca2+ currents contained the following: 110 mM CsCl2, 40 mM HEPES, 0.5/10 mM EGTA, 1 mM MgCl2, 2 mM ATP, 0.5 mM GTP, 12 mM phosphocreatine, 20 mM TEA, and 3 mM K-glutamate at a pH of 7.3 after adjustment with CsOH. Intracellular solution for postsynaptic recordings was composed of the following: 97.5 mM K-gluconate, 32.5 mM CsCl2, 5 mM EGTA, 10 mM HEPES, 1 mM MgCl2, 30 mM TEA, and 3 mM lidocaine N-ethyl bromide (QX314) at a pH of 7.2. The holding potential was −80 mV and −60 mV for presynaptic and postsynaptic recordings, respectively. Presynaptic Ca2+ currents were evoked by various voltage command protocols indicated in the text, and leak subtraction was done with the online P/4 protocol. All recordings were made with a dual-channel amplifier (MultiClamp 700A; Molecular Devices, Sunnyvale, CA), and all reagents were purchased from Sigma (Oakville, ON, Canada), Tocris (Ellisville, MO), and Alomone Labs (Jerusalem, Israel).
Data were acquired online, filtered at 4 kHz, digitized at 50 kHz, and analyzed off-line using the pClamp9.2 software package (Molecular Devices, Sunnyvale, CA) and Microsoft Excel 2000. Excitatory postsynaptic current (EPSC) density (pA per ms) was used in all experiments as a measure of EPSC size. Measurement was limited to a 14-ms or 4-ms window for EPSCs generated from presynaptic voltage steps to 0 mV (10 ms) and 40 mV (4 ms), respectively, with the start of the detection window being placed on the EPSC onset. EPSC recovery was calculated as a percentage of the first EPSC current density for each sweep. Replenishment time constants were determined by fitting a biphasic standard exponential equation to the recovery curves and forcing the asymptote to 100%. In all cases a Levenberg-Marquardt search method, minimizing the sum of squared errors, was used in the Clampfit9.2 analysis package to evaluate equation parameters and their standard errors. Pooled data were expressed as the mean ± standard error from a population of synapses.
Hippocampus electrophysiology.
Transverse slices of 3- to 8-week-old mouse hippocampus were cut at a thickness of 250 to 300 μm using a vibrating microtome (Leica VT 1000S; Wetzlar, Germany) in ice-cold cutting solution containing 81.2 mM NaCl, 23.4 mM NaHCO3, 69.9 mM sucrose, 23.3 mM glucose, 2.4 mM KCl, 1.4 mM Na2HPO4, 6.7 mM MgCl2, and 0.5 mM CaCl2 at a pH of 7.3 when bubbled in with 95% O2 and 5% CO2. The slices were incubated at 34°C for 30 min in the recovery solution followed by an additional 30 min at 34°C in a recording solution containing 125 mM NaCl, 2.5 mM KCl, 10 mM glucose, 1.25 mM NaH2PO4, 2 mM Na-pyruvate, 3 mM myo-inositol, 0.5 mM ascorbic acid, 26 mM NaHCO3, 1 mM MgCl2, and 2 mM CaCl2 at a pH of 7.3 when bubbled in with 95% O2 and 5% CO2. Subsequently, the temperature of the slices was brought down to room temperature. During whole-cell voltage clamp recordings, slices were superfused with recording solution, and to facilitate EPSC recording 10 μM bicuculline, a γ-aminobutyric acid receptor blocker was included. For synaptic experiments in CA1 pyramidal neurons, the pipette solution contained 97.5 mM K+-gluconate, 5 mM EGTA, 10 mM HEPES buffer, 1 mM MgCl2, 30 mM TEA, and 3 mM lidocaine N-ethyl bromide (QX314) (an intracellular blocker of Na+ currents) at a pH of 7.3. All reagents were purchased from Sigma (Oakville, ON, Canada), Tocris (Ellisville, MO), and Alomone Labs (Jerusalem, Israel).
Transverse slices were transferred to a continuously perfused recording chamber, with a flow rate set at approximately 1 ml/min, mounted on an Olympus microscope with Normarski optics and a 60× water immersion objective. CA1 pyramidal neurons were identified by morphology and their well-defined positions within the slice. Patch electrodes typically had resistances of 4 to 6 MΩ, and series resistance, compensated to 90% for whole-cell voltage-clamp recordings, was 6 to 10 MΩ. In voltage clamp mode, CA1 pyramidal neuron-evoked EPSCs were recorded at −60 mV, as were spontaneous EPSCs. Schaffer collaterals were stimulated in the stratum radiatum by a glass pipette placed close to the cell body layer. A cut was routinely made between area CA3 and CA1 to prevent recurrent excitation due to Schaffer collateral stimulation.
Data were acquired online using Patchmaster software and an EPC10 patch clamp amplifier (Heka Electronics Incorporated, Germany), filtered at 2 kHz, digitized at 10 to 50 kHz, and analyzed off-line with pClamp9.2 (Axon Instruments, Foster City, CA) and Mini Analysis software (Synaptosoft, Inc., Decater, GA). Results were compiled, and statistical analysis was performed using Microsoft Excel (Microsoft, Redmond, WA) and GraphPad (GraphPad Software Inc., San Diego, CA). All experiments were performed at room temperature. Values are given as means ± standard errors of the means (SEM), and confidence limits were determined using Student t tests.
Quantitative Western blot analysis of septin expression from wild-type and mutant mice.
Three brains from P30 wild-type, Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− mice were individually prepared as described in the Materials and Methods section (see “Tissue preparation and Western blot analysis”). For each gel, 30 μg of protein lysate from each genotype was loaded per lane. Two more sets of gels were created with the other brains (three independent sets). Following electrophoresis and gel transfer, each blot was first probed with a septin antibody (see “Tissue preparation and Western blot analysis”) and then with a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody to control for variability in gel loading. The results on autoradiographic film were digitized with a Dell A920 scanner to facilitate densitometry measurements using an ImageJ software package. Only protein bands that corresponded to known septin isoforms were analyzed. Changes in septin expression in mutant backgrounds were normalized to wild-type levels and are reported as percent expression levels (mutant/wild type × 100%). Similarly, GAPDH protein levels were measured and normalized to the wild-type level. These results were used to calibrate the percent expression level of septin from the corresponding blot. Measurements from all three sets were averaged, and significant differences, indicated by asterisks above the bar graphs, were determined using a Student's t test. The criterion for statistical significance was chosen to be a P value of <0.05.
Immunostaining vibratome slices.
Brain slices (200 μm in thickness) from the auditory cortex of P16 mice were cut and allowed to recover for 1 h in aCSF at 37°C. Subsequently, the brain slices were fixed, permeabilized, blocked, and stained using a Basic Vector Mouse-on-Mouse Immunodetection Kit according to the manufacturer's instructions (Vecta Laboratories, Burlingame, CA). SEPT3, SEPT5, and DNA were detected using a rabbit anti-SEPT3 antibody, a mouse SP20 antibody, and Hoechst 33342 stain (Invitrogen-Molecular Probes, Burlington, ON, Canada), respectively. See “Immunostaining hippocampal neurons” in Materials and Methods for the acquisition and processing of digital images of fluorescently labeled brain slices.
RESULTS
Generation of Sept3−/− and Sept3−/− Sept5−/− mice.
The function of SEPT3 was examined by disrupting the Sept3 gene locus in mice. A gene targeting vector was designed to replace a genomic region containing exons 2 to 5, which includes the P-loop consensus motif found in all GTPases (Fig. 1A). Out of the 167 antibiotic-resistant clones, four ES cell clones underwent homologous recombination with the targeting vector (Fig. 1B and C). Proper integration of the short and long arm of homology was confirmed for the ES cell clone 9F1, so it was used to generate chimeric founder mice, and the mutation was bred to homozygosity. Germ line transmission of the mutant allele was initially confirmed by Southern blot analysis (Fig. 1D) and then routinely determined by PCR (Fig. 1E). The absence of SEPT3 protein from brains of Sept3−/− mice was confirmed by Western blot analysis (Fig. 1F).
FIG. 1.

Gene targeting strategy, ES screening, and genotyping. (A) The mouse Sept3 gene was disrupted by replacing exon 2 to exon 5 with a Neor selection cassette. The cassette was targeted to the Sept3 locus by appending DNA fragments that were homologous to the Sept3 gene (targeting vector). A large piece of intron 1 and a small piece of exon 2 were used for the short arm of homology, whereas a piece of intron 5 through to the end of intron 8 was used for the large arm of homology. The endogenous and recombinant loci are shown above and below the targeting vector, respectively. For perspective, the location of relevant protein domains in relation to the encoding exons is shown at the top (PRO is the proline-rich domain, G1 is the P-loop motif, and G3 and G4 represent the other GTP binding features). Screening for recombination events was performed by Southern blotting using the 5′ and 3′ probes or by PCR using the Ex5-Ex6 and Ex2-Neo primer pairs. The sizes of DNA fragments that each probe is expected to detect by Southern blotting or that each primer pair is expected to amplify during PCR are shown on the right. The locations of only the relevant restriction enzyme cut sites are indicated for simplicity. (B) Screening results for proper homologous recombination of the short arm of homology in ES cells. Among the three ES cell clones shown, only clone 9F1 was targeted correctly. (C) Screening results for proper homologous recombination of the long arm of homology in ES cells. Out of six ES cell clones shown, only clones 9F1, 2A2, 11E2, and 12E2 were targeted correctly. (D) Genotype determined by Southern blot analysis of mouse tails from an F2 generation. (E) Genotype determined by PCR analysis of mouse tails from an F2 generation. (F) Western blot analysis of 20 μg of brain tissue from wild-type and Sept3−/− mice using an antibody to SEPT3.
Sept3−/− mice are viable at birth and live as long as their wild-type counterparts. Breeding Sept3+/− mice produced Sept3−/− pups at the expected Mendelian ratio (data not shown). We then determined if Sept3−/− mice were fertile since the lack of SEPT4 causes male sterility in mice due to loss of annuli, ring-like structures important for sperm motility, from sperm tails (26, 36). Although no SEPT3 protein was detected in testes by Western blot analysis, putative Sept3 transcripts were found in the testes by Northern blot analysis (71, 74), raising the possibility that the protein may have a specialized role in this tissue. However, Sept3−/− male mice are still fertile (data not shown). Sept3−/− female mice were also tested for fertility because the expression of SEPT3 in female reproductive organs was not determined previously. Similar to Sept3−/− male mice, Sept3−/− female mice were also fertile (data not shown). These results indicate no primary functional role for SEPT3 in germ cells. Sept3−/− mice were subsequently bred with Sept5−/− mice to generate Sept3−/− Sept5−/− mice. Like Sept3−/− mice, Sept3−/− Sept5−/− mice were viable and lived as long as their wild-type counterparts. In addition, both male and female double knockout mice were fertile.
SEPT5 complex in the absence of SEPT3.
In Drosophila embryos, Sep2 but not Sep1 failed to accumulate at the cellularization front in the absence of Pnut (1). It was postulated from this observation that septin complex assembly might occur in a hierarchical manner. We therefore determined whether the lack of SEPT3 affected the ability of SEPT5 to associate with other septins. SEPT5 was found to coimmunoprecipitate with SEPT2, SEPT4, SEPT6, SEPT7, SEPT8, SEPT9, and SEPT11 (Fig. 2). SEPT1 was not found in this complex but has been observed in another brain septin complex (34). This result does not distinguish between a single complex and multiple complexes containing SEPT5. Nevertheless, the association of other septins with SEPT5 was unaffected by the lack of SEPT3. Therefore, the absence of SEPT3 did not prevent or enhance the interaction of any other septin with SEPT5.
FIG. 2.

Lack of SEPT3 does not alter the association of other septins with SEPT5. Detergent-solubilized brain lysates from wild-type and Sept3−/− mice are shown in the first and second lanes, respectively. After the SEPT5 monoclonal antibody (SP20) was used to immunoprecipitate SEPT5 from wild-type or from Sept3−/− brain lysates, the coimmunoprecipitating septins were detected by immunoblotting (lanes 4 and 5). Mouse serum (MS) was used as a negative control for the immunoprecipitation (IP) procedure (lane 3). The input brain lysate together with the immunoprecipitates were separated by SDS-polyacrylamide gel electrophoresis, transferred onto PVDF membrane, and probed with the indicated septin antibodies.
Septin expression in the absence of SEPT3 and SEPT5.
The lack of one protein can affect the stability of other proteins that are associated with it in a complex. For example, the targeted disruption of amphiphysin I results in complete loss of its partner amphiphysin II by proteolysis (12). Likewise for septin complexes, knockdown of SEPT2 by RNA interference causes the level of SEPT7 to decrease and vice versa (34). Moreover in Sept5−/− mouse brains, there is a significant change in the expression of SEPT2 and SEPT7 (47). Therefore, the expression levels of other septins were compared by Western blot analysis between wild-type, Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− mouse brains at 0, 2, and 4 weeks of age (Fig. 3). Quantitation of septin levels at 4 weeks of age revealed several differences in the mutant backgrounds (Fig. 4). In the absence of SEPT3, there was a significant decrease in the expression of SEPT2, SEPT6, and the largest form of SEPT8. In the absence of SEPT5, there was a significant increase in the levels of SEPT2 and a significant decrease in the levels of SEPT7, which is consistent with previous results (47). In addition to SEPT7, a significant decrease in the levels of SEPT3, SEPT6, the largest isoform of SEPT8, and SEPT11 was also observed. In the absence of both SEPT3 and SEPT5, there was a significant decrease in the levels of SEPT6, SEPT7, both forms of SEPT8, and SEPT11, whereas one isoform of SEPT4 increased. Although the expression level of SEPT12 was not tested in either Sept3−/− or Sept5−/− mouse brains, its expression level was not detected in the brains of Sept3−/− Sept5−/− mice. This septin is predominantly expressed in the testes and closely related to SEPT3 because it lacks a predicted coiled-coil domain.
FIG. 3.

Levels of septin expression in the absence of SEPT3, SEPT5, and both septins. Brains from wild-type, Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− mice at P0, P14, and P28 were homogenized and then solubilized in SDS gel sample buffer. In each lane, 20 μg of protein sample was separated by SDS-polyacrylamide gel electrophoresis, transferred onto PVDF membrane, and probed with the indicated septin antibodies. A number of the septins undergo alterative splicing to produce multiple isoforms of different sizes on gels.
FIG. 4.

Quantitation of septin expression in the absence of SEPT3, SEPT5, and both septins. P30 brains from wild-type, Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− mice were homogenized and solubilized in SDS gel sample buffer. In each lane, 30 μg of protein sample was separated by SDS-polyacrylamide gel electrophoresis, transferred onto a PVDF membrane, and probed with the indicated septin antibody followed by the GAPDH antibody. The relative level of each septin after calibration was measured in three independent experiments and averaged. Any significant deviation in septin expression from wild-type levels is indicated by an asterisk.
It should be noted that extra bands of unconfirmed identity were observed for SEPT1, SEPT9, and SEPT11. The appearance of these bands may be associated with the mixed genetic background of the single and double knockout mice. Furthermore, the levels of SEPT10, SEPT13, and SEPT14 were not tested in this study due to the lack of specific antibodies for these septins.
Axon outgrowth and synapse formation.
Septins are known to associate with the exocyst complex (24), which is a group of proteins implicated in neurite outgrowth and synapse formation. Thus, the roles of SEPT3 and SEPT5 were examined in the development of isolated neurons. Primary hippocampal neurons were cultured from wild-type and Sept3−/− mouse brains and tested for their ability to polarize, form axons, and make synapses. Primary hippocampal neurons lacking SEPT3 exhibited normal growth rates and acquired characteristic markers of polarity after 3 days in vitro (Fig. 5A). Staining for neurofilament heavy chain and F-actin, which marked emerging axons and growth cones, respectively, revealed normal development of these structures. When the loss of SEPT3 was compounded with the loss of SEPT5, these neurons were still able to polarize, which can be seen by the asymmetric distribution of axonal (neurofilament-H) and dendritic (MAP2) markers in their processes (Fig. 5A). Similarly, no significant difference in axonal growth rates was observed when neurofilament-H-positive, MAP2-negative processes of stage III (13) wild-type and Sept3−/− Sept5−/− hippocampal neurons were measured after 2 days in vitro (67.7 ± 1.6 μm and 69.2 ± 1.8 μm, respectively [SEM]; P = 0.52; total number of neurons analyzed from three wild-type litters, 357; total number of neurons analyzed from four Sept3−/− Sept5−/− litters, 438).
FIG. 5.

Loss of SEPT3 or SEPT3 and SEPT5 does not affect the development of primary hippocampal neurons in vitro. Primary mouse hippocampal neurons from wild-type, Sept3−/−, and Sept3−/− Sept5−/− brains were cultured for 3 days (A) or 10 days (B). Wild-type and Sept3−/− neurons cultured for 3 days were stained for SEPT3, F-actin, and neurofilament-H (green, red, and blue, respectively, in the merged panel). In neurons lacking SEPT3, processes containing the axon-specific protein neurofilament-H still form growth cones at their tips (white arrow). Sept3−/− Sept5−/− neurons cultured for 3 days were stained in the top series of panels for SEPT3, F-actin, and neurofilament-H (green, red, and blue, respectively, in the merged panel) or in the bottom series of panels for SEPT5, F-actin, and MAP2 (green, red, and blue, respectively, in the merged panel). Like Sept3−/− neurons, F-actin-rich growth cones at the tips of axons (arrow) are still present in the absence of SEPT3 and SEPT5. Wild-type and Sept3−/− neurons cultured for 10 days were stained for SEPT3 and synaptophysin. Sept3−/− Sept5−/− neurons cultured for 10 days were stained in the left series of panels for SEPT3 and synaptophysin or in the right series of panels for SEPT5 and VAMP2. A digitally magnified image of the region highlighted by the hatched box is shown directly below. The arrows point to presynaptic terminals enriched with synaptophysin. In neurons lacking SEPT3 or SEPT3 and SEPT5, many punctate regions enriched with synaptophysin or VAMP2 still form (white arrows).
After 10 days in vitro, primary hippocampal neurons form a large number of synapses onto neighboring dendrites or cell bodies. At this stage, presynaptic terminals are enriched with the synaptic vesicle protein synaptophysin and SEPT3 (Fig. 5B). When neurons isolated from Sept3−/− mice were grown for 10 days in culture, they were still able to form synapses (Fig. 5B). The accumulation of synaptophysin-positive rich areas along the processes of these neurons revealed no obvious differences in either the distribution or abundance of synaptic contacts with dendrites or cell bodies. SEPT5 is also enriched in presynaptic terminals of hippocampal neurons, where it colocalizes with the synaptic vesicle protein VAMP2 (47). Even without SEPT3 and SEPT5, hippocampal neurons made synaptic connections with each other after 10 days in vitro (Fig. 5B).
Hippocampal architecture is normal in the absence of SEPT3 and SEPT5.
Although axon outgrowth and synapse formation appeared normal in vitro, the correct development of complex brain structures may be more sensitive to the absence of critical proteins such as SEPT3 and SEPT5. The anatomy of the hippocampus was examined in detail because both SEPT3 and SEPT5 are expressed in this region of the brain (73) and could be affected by their absence. Gross defects in the hippocampus are easy to identify because the neurons in this area have a distinct topographical organization. Nevertheless, no major abnormalities were detected in the architecture of the hippocampus viewed sagittally from Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− brains (Fig. 6A).
FIG. 6.

Hippocampal anatomy in wild-type, Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− brains. (A) A series of magnified images of the hippocampus from wild-type, Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− mice. Pictures were taken from sagittal sections of formalin-fixed, P30 brains that were stained with hematoxylin and eosin. The bubbles and large spaces around each hippocampus were a result of artifacts that were introduced during tissue preparation. (B) A series of electron transmission micrographs that were taken of nerve terminals from the CA1 region of the hippocampus (boxed area illustrated in panel A). Two representative examples from each genotype are shown.
Nerve terminal ultrastructure.
Although hematoxylin- and eosin-stained brain sections are suitable for making qualitative assessments about tissue architecture, we turned to transmission electron microscopy, which provides better resolution and contrast to address subcellular structures. Since SEPT3 and SEPT5 are associated with synaptic vesicles and concentrated in synapses (32, 73), the ultrastructure of nerve terminals from the CA1 region of the hippocampus were compared between wild-type, Sept3−/−, Sept5−/−, and Sept3−/− Sept5−/− mice (Fig. 6B). Presynaptic terminals from mouse hippocampal neurons had no obvious changes in terminal area, synaptic vesicle size, or synaptic vesicle density in the absence of SEPT3 or SEPT5. Likewise, when the loss of SEPT3 was compounded with the loss of SEPT5, the morphology of these terminals was similar to that of the wild type.
Synaptic vesicle protein expression in the absence of SEPT3 and SEPT5.
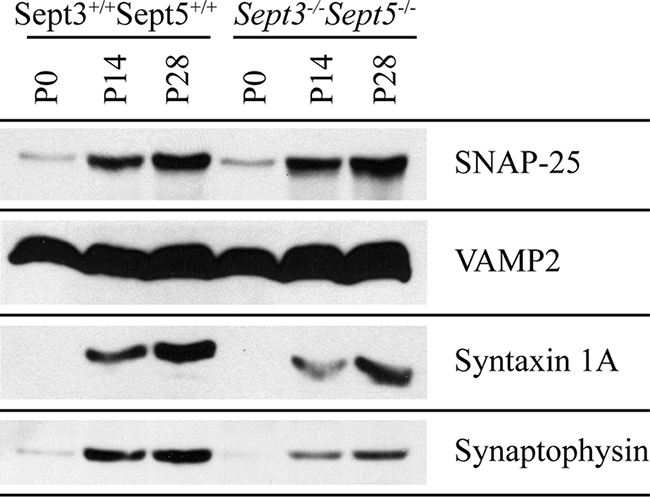
Although the architecture of Sept3−/− and Sept3−/− Sept5−/− neurons looks similar to wild-type neurons, there could be differences in their function. Since septins are thought to be important for secretion, we examined the levels of synaptic vesicle fusion proteins in the absence of SEPT3 and SEPT5 (Fig. 7). In Sept3−/− Sept5−/− brain tissue, the levels of syntaxin 1 and synaptophysin were 68% and 65% of that in wild-type brain tissue, whereas the levels of VAMP2 and SNAP-25 were 98% and 108% of that in wild-type tissue, respectively. These observations are consistent with previous reports showing that SEPT5 associates with synaptophysin and syntaxin (4). Loss of SEPT5 might disrupt the stability of these two proteins in nerve terminals, thereby impairing the process of synaptic vesicle fusion. Therefore, we turned to FM 1-43 imaging and electrophysiology on single and double knockout neurons to address this question.
FIG. 7.

Expression of synaptic vesicle proteins in the absence of SEPT3 and SEPT5. Brains from wild-type and Sept3−/− Sept5−/− mice at P0, P14, and P28 were homogenized and then solubilized in SDS gel sample buffer. In each lane, 20 μg of protein sample was separated by SDS-polyacrylamide gel electrophoresis, transferred onto PVDF membrane, and probed with the indicated antibodies.
Properties of synaptic transmission in the absence of SEPT3 and SEPT5.
Genetic mutations of presynaptic proteins can alter certain properties of synaptic transmission in the absence of any obvious morphological defects. In the absence of SEPT5, no differences from wild-type mice were observed in several synaptic transmission parameters at the hippocampal CA3-CA1 synapse (47). The parameters used to examine presynaptic physiology were revisited in mice lacking SEPT3 and SEPT5 to determine if a phenotype would manifest with a compound genetic deletion.
Paired-pulse facilitation is a form of short-term plasticity seen at many synapses and is characterized by an increase in neurotransmitter release evoked by a second stimulus during twin-pulse stimulation. When the latency between the first and second pulse is shorter than the time it takes to extrude Ca2+ out from the nerve terminal, the residual intracellular Ca2+ left over from the first stimulus contributes to a greater Ca2+ rise, and hence greater evoked release, during the second stimulus. Thus, changes in paired-pulse facilitation can indicate changes in the sensitivity of the presynaptic release apparatus to Ca2+. Paired-pulse facilitation is taken as the ratio between the postsynaptic response amplitude evoked by the second stimulus and the response amplitude evoked by the first. When the paired-pulse ratio (PRR) was measured with an interpulse interval of 150 ms (Fig. 8A, B, and E), no difference between wild-type (PPR 0.77 ± 0.05; n = 5 cells) and Sept3−/− Sept5−/− (PPR 0.78 ± 0.02; n = 12 cells) mice was observed (P = 0.95).
FIG. 8.
Loss of SEPT3 does not affect paired-pulse facilitation or the amplitude and frequency of spontaneous EPSCs in hippocampal CA1 pyramidal neurons. (A) The top trace represents superimposed AMPA receptor-mediated EPSCs from a wild-type mouse evoked by paired stimuli (150 ms apart). The bottom trace represents an average of 15 consecutive traces. (B) The top trace represents superimposed AMPA receptor-mediated EPSCs from a Sept3−/− Sept5−/− mouse evoked by paired stimuli (150 ms apart). The bottom trace represents an average of 15 consecutive traces. (C) A representative 4-s segment of spontaneous EPSCs recorded from a wild-type mouse. A blow-up of the postsynaptic currents is shown in the inset (inset scale, 10 pA/6 ms). (D) A representative 4-s segment of spontaneous EPSCs recorded from a Sept3−/− Sept5−/− mouse. A blow-up of the postsynaptic currents is shown in the inset (inset scale, 10 pA/6 ms). (E) Pooled data showing the PPR of EPSCs for wild-type and Sept3−/− Sept5−/− mice. (F) Pooled data showing frequency of spontaneous EPSCs for wild-type and Sept3−/− Sept5−/− mice. (G) Pooled data showing average amplitude for spontaneous EPSCs for wild-type and Sept3−/− Sept5−/− mice. Error bars in panels E, F, and G show SEMs.
Measurements of both size and frequency of spontaneous EPSCs can also give some information about changes at the synapse. A change in the former is often indicative of a change in postsynaptic sensitivity, whereas a change in the latter is indicative of a change in presynaptic sensitivity. When the amplitudes of spontaneous EPSCs were measured from wild-type and Sept3−/− Sept5−/− mice (Fig. 8C, D, and G), no significant difference was observed between them (38.4 ± 6.1 pA [n = 10 cells] versus 44.0 ± 5.0 pA [n = 10 cells], respectively; P = 0.49). When the frequencies of spontaneous EPSCs were measured from the same mice (Fig. 8C, D, and F), no significant difference was observed (0.44 ± 0.09 Hz for wild-type and 0.47 ± 0.08 Hz for Sept3−/− Sept5−/− mice; n = 10 cells for each genotype; P = 0.84). The analyses of spontaneous EPSCs indicate that in the absence of both SEPT3 and SEPT5, there appears to be no change in the synaptic vesicle release probability or postsynaptic receptor responsiveness.
The synaptic vesicle recycling pool.
Primary hippocampal neurons typically contain 100 to 200 synaptic vesicles per terminal. The recycling pool size is about 10 to 20% of the total (22) and is defined as the population of synaptic vesicles that maintain neurotransmitter release during moderate (physiological) stimulation (48). To obtain a quantitative measurement of the functional synaptic vesicle pool size, the fluorescent indicator dye FM 1-43 was used to measure the size of the recycling pool. When added to the bath containing high K+ (dye loading), some FM 1-43 inserted into the plasma membrane is internalized into recycling synaptic vesicles, providing one index of synaptic vesicle endocytosis (37). Destaining of synaptic vesicles by exocytosis (dye unloading) is triggered by a second K+ depolarization. Wild-type nerve terminals from 10-day hippocampal neuronal cultures were depolarized by bath application of 47 mM K+ in the presence of 15 μM FM 1-43 for 90 s (Fig. 9A, steps 1 to 5) to completely label the recycling pool of synaptic vesicles (22). After a 10-min wash in Ca2+-free buffer, a punctate distribution of dye-loaded nerve terminals was clearly evident (Fig. 9B, middle panel). When this procedure was repeated for Sept3−/− nerve terminals, the total amount of dye uptake was not different from wild-type terminals (data not shown). This suggests that SEPT3 is not essential for synaptic vesicle endocytosis and that its absence does not affect the amount of recycling that occurs in response to high K+ for 90 s. Next, dye-loaded synaptic vesicles were depolarized with 90 mM K+ to elicit exocytosis (dye unloading) (Fig. 9A, steps 5 to 7). Following the depolarizing stimulus, wild-type nerve terminals were completely destained (Fig. 9B, right panel), and the same was true for knockout nerve terminals (data not shown). The overall size of the synaptic vesicle recycling pool was quantitatively determined (ΔF = Fpre − Fpost) for nerve terminals in the presence and absence of SEPT3 and displayed as a frequency distribution (Fig. 9C). The distribution of recycling pool sizes was found to be broad but overlapping for both genotypes. Using the data sets from the experiment shown in Fig. 9C, a cumulative frequency curve was constructed for each genotype (Fig. 9D) so that the differences between genotypes could be examined by a Kolmogorov-Smirnov test. This test, which measures the statistical difference between two data sets without making any assumptions about their distributions, confirmed that there was no change in the size of the recycling pool in the absence of SEPT3 (P = 0.362; n = 532 terminals from seven wild-type pups and n = 555 terminals from six Sept3−/− pups). The results demonstrate there was no overall effect on synaptic vesicle fusion and recycling in Sept3−/− mice.
FIG. 9.

Synaptic vesicle recycling pool size in Sept3−/− neurons. (A) FM 1-43 loading and unloading protocol used to measure the size of the synaptic vesicle recycling pool. The diagram outlines the steps involved in loading and unloading presynaptic terminals of FM 1-43. (B) Differential interference contrast (DIC) image of a wild-type mouse primary hippocampal neuron cultured for 10 days from a wild-type mouse (left panel). A fluorescence image of the same wild-type neuron studded with FM 1-43-loaded nerve terminals is shown in the middle panel. A fluorescence image of the same wild-type neuron after unloading nerve terminals of FM 1-43 by K+ depolarization is shown in the right panel. A magnified view of the region highlighted by the hatched box is shown directly below. (C) The size of the recycling pool was measured individually from each synapse, and the data were plotted as a frequency distribution. The distributions of the recycling pool size from wild-type and Sept3−/− nerve terminals is represented by the open bars and the closed bars, respectively (n = 532 terminals from seven wild-type pups and n = 555 terminals from six Sept3−/− pups). (D) A cumulative frequency curve was constructed for statistical analysis from the distribution of recycling pool sizes in panel C. Data from wild-type nerve terminals are plotted with squares, whereas data from Sept3−/− nerve terminals are plotted with triangles. A.F.U., arbitrary fluorescence units.
SEPT3 and SEPT5 are not crucial for the replenishment of the immediately releasable pool of synaptic vesicles.
In order to determine the importance of SEPT3 and SEPT5 in the replenishment of the readily releasable pool (RRP) of synaptic vesicles, we turned to the calyx of Held nerve terminal which makes a mono-synaptic connection with the MNTB. Like hippocampal boutons, the calyx of Held expresses both SEPT3 and SEPT5, and these two septins show a strong degree of colocalization throughout the terminal (Fig. 10). The large size of the calyx of Held terminal makes it feasible to do paired whole-cell voltage clamp recordings (Fig. 11A). Furthermore, direct voltage clamping of the presynaptic terminal allows for precise control of the ionic currents triggering release of synaptic vesicles, which can be measured from the current response of the postsynaptic MNTB neuron (6, 18). These characteristics make the calyx of Held-MNTB synapse a unique model in which to examine aspects of synaptic transmission and dissect the components of the RRP.
FIG. 10.

SEPT3 and SEPT5 are expressed in the calyx of Held nerve terminal. (A) A brain slice from the auditory cortex of a P16 mouse was stained for SEPT3 (green), SEPT5 (red), and DNA (Hoechst; blue). A digitally magnified image of the region highlighted by the hatched box is shown in panel B below.
FIG. 11.

Loss of SEPT3 and SEPT5 does not affect the replenishment of the immediately available pool of synaptic vesicles. (A) Schematic illustration of the paired voltage clamp recording arrangement from the calyx of Held-MNTB synapse. (B) Presynaptic voltage clamp command paradigm used to measure recovery of the portion of the RRP of synaptic vesicles available in response to a single action potential. Two voltage steps (−80 to 40 mV; 4 ms), separated by increasing Δt ([Dt] intervals ranging from 5 ms to 10 s), were used to evoke ICa and IEPSC from voltage-clamped presynaptic calyces and their corresponding postsynaptic MNTB neurons. (C) Presynaptic ICa (top) and postsynaptic IEPSC (bottom) recorded from wild-type (left) and Sept3−/− Sept5−/− (right) synapses and superimposed at selected interstep intervals (Dt) in response to the presynaptic voltage command shown in panel A. (D) Pooled data for wild-type (closed circles) and Sept3−/− Sept5−/− (open diamonds) plotting recovery of the IEPSC area against the interstep interval (Dt). Error bars show SEMs.
The pool of synaptic vesicles can be subdivided into two main components: those that are releasable under conditions of typical activity (RRP) and those that refill the RRP, or the reserve pool (44). The RRP can be further subdivided into an immediately releasable pool (IM-RRP) and an intermediate pool (IT-RRP). The IM-RRP pool consists of those synaptic vesicles that are available for release in response to the Ca2+ current (ICa) evoked from a single presynaptic action potential and are most likely those synaptic vesicles in close proximity to voltage-gated Ca2+ channels (VGCCs) and active zones. The IT-RRP pool consists of synaptic vesicles that are recruited into release during sustained elevations in intraterminal [Ca2+], which occurs during repetitive high-frequency activity or sustained presynaptic influxes of Ca2+ (51, 53).
To assay whether loss of SEPT3 and SEPT5 impacted the ability of the calyx of Held to replenish the IM-RRP, we depolarized voltage-clamped presynaptic terminals from −80 mV to 40 mV for 4 ms, which triggered a large inward Ca2+ current through VGCCs (Fig. 11B and C). During the step, a majority of VGCCs enter an open state; however, they do not flux Ca2+ ions as 40 mV is past the effective reversal potential for Ca2+. Upon repolarization, a steep electrochemical gradient drives Ca2+ ions into the terminal, during the brief period before VGCCs close, producing a large tail current (Fig. 11C). The Ca2+ flux through individual VGCCs creates [Ca2+] domains which are analogous to those resulting from the innervation of a single action potential into the nerve terminal (7). However, under these conditions, all VGCCs contribute to the ICa rather than the relatively small fraction activated during an action potential, especially at physiological temperatures (75a). The resulting synaptic output contains all of the synaptic vesicles that would be available during any single action potential, or the IM-RRP.
By separating two such depolarizations by an interstep interval (Δt) of variable duration, we determined the portion of the IM-RRP that recovered for a range of Δt (5 ms to 10 s) in both wild-type and Sept3−/− Sept5−/− synapses (Fig. 11D). This protocol failed to completely deplete the IM-RRP even at the shortest Δt possible (5 ms). At this short time interval, recovery was 54% ± 4% and 40% ± 8% for wild-type and Sept3−/− Sept5−/− synapses, respectively, which were not significantly different (unpaired t test, P > 0.05). The recovered portion of the IM-RRP continued to increase with increasing Δt and followed a biphasic time course. Fast recovery time constants were 39 ± 13 ms and 43 ± 7 ms for wild-type and Sept3−/− Sept5−/− synapses, respectively, while slow time constants were 2,500 ± 600 ms and 2,400 ± 600 ms, respectively. Given that no significant differences were observed between wild-type and Sept3−/− Sept5−/− synapses in the rates of recovery of their IM-RRP (Fig. 11C and D), we concluded that SEPT3 and SEPT5 are not essential for the trafficking of synaptic vesicles to the active zone and their positioning into the immediately available pool.
SEPT3 and SEPT5 are not crucial for the replenishment of the readily releasable pool of synaptic vesicles.
Although SEPT3 and SEPT5 are not essential for replenishment of the IM-RRP, they may be involved in trafficking synaptic vesicles from the reserve pool of synaptic vesicles to the broader pool of available synaptic vesicles recruited primarily during repetitive activity (IT-RRP and IM-RRP) (67). To investigate whether SEPT3 and SEPT5 are involved in the replenishment of the RRP, we depolarized voltage-clamped presynaptic calyces from −80 mV to 0 mV for 10 ms (Fig. 12A). At 0 mV, the maximal current flux occurs through P/Q-type VGCCs, which are the exclusive type mediating release of synaptic vesicles at this developmental stage in the calyx of Held (28). During voltage steps, a large sustained ICa was recorded, which smears the terminal with Ca2+ and results in release of a large number of synaptic vesicles (Fig. 12B). In response, postsynaptic MNTB neurons registered a large inward current that decayed to near baseline, suggesting that the RRP had been largely depleted (Fig. 12B). Voltage steps of this type are thought to trigger release of a pool of synaptic vesicles larger than those depleted during high-frequency activity at the calyx of Held (53).
FIG. 12.

Loss of SEPT3 and SEPT5 does not affect replenishment of the readily releasable pool of synaptic vesicles. (A) Presynaptic voltage-clamp command paradigm used to measure recovery of the readily releasable pool of synaptic vesicles. Two voltage steps (−80 to 0 mV; 10 ms), separated by increasing Δt ([Dt] intervals ranging from 20 ms to 20 s), were used to evoke ICa and IEPSC from voltage-clamped presynaptic calyces and their corresponding postsynaptic MNTB neurons. (B) Presynaptic ICa (top) and postsynaptic IEPSC (bottom), recorded from wild-type (left) and Sept3−/− Sept5−/− (right) synapses and superimposed, at selected interstep intervals (Dt) in response to the presynaptic voltage command shown in panel B. (C) Pooled data for wild-type (closed circles) and Sept3−/− Sept5−/− (open diamonds) plotting recovery of IEPSC area against interstep interval (Δt). Error bars show SEMs.
To assay the recovery of the RRP, we separated two depolarizing steps by varying the Δt (20 ms to 20 s) and analyzed the recovery of the RRP in wild-type and Sept3−/− Sept5−/− synapses (Fig. 12B). This protocol effectively depleted the RRP at a short Δt (20 ms) to ∼10% in wild-type and Sept3−/− Sept5−/− synapses. Here also, a biphasic exponential function described the recovery of the RRP well for both experimental groups. Fast time constants were 156 ± 6 ms and 148 ± 6 ms for wild-type and Sept3−/− Sept5−/− synapses, respectively, while slow time constants were 9,100 ± 600 ms and 7,200 ± 600 ms, respectively, in line with estimates from this and other central synapses (59, 66, 67). Despite the sizable difference in the slow time constant between experimental groups, the results were not statistically different (P > 0.05). As observed for the IM-RRP, recovery kinetics of the RRP (IM-RRP plus IT-RRP) showed no significant difference with or without SEPT3 and SEPT5, implying that these two septins are not crucial for the replenishment of the entire RRP or the IM-RRP subset (Fig. 12C).
DISCUSSION
SEPT3 is the only septin exclusively expressed in neurons and is enriched in presynaptic nerve terminals, which suggests that it might have unique functions in synapse formation or maturation or play a role in exocytosis or endocytosis. The successful generation of Sept3−/− mice revealed that SEPT3 is not essential for mouse development, fertility, or viability. In particular, the architecture and fine structure of the hippocampus appear unperturbed despite the fact that SEPT3 is found in presynaptic terminals. This finding is in line with a very recent study showing that genetic ablation of SEPT3 in mice does not affect the cerebellum and cerebral cortex in addition to the hippocampus (20). Furthermore, this group also verified that SEPT3 does not play a significant role in neuronal development and synapse formation. Consistent with the lack of gross anatomical changes in presynaptic nerve terminals, no changes in the physiology of synaptic vesicle pool size, synaptic vesicle fusion, and synaptic vesicle recycling were observed in the absence of SEPT3.
SEPT5 is another septin that is predominantly expressed in the nervous system, but it is also expressed in blood cells (75). Like SEPT3, generation of Sept5−/− mice revealed that lack of SEPT5 was not essential for mouse development, fertility, or viability, and no irregularities were found in synaptic transmission. However, secretion of large, dense core granules from platelets was enhanced (11), supporting the idea that SEPT5 is a negative regulator of exocytosis (4). The discrepancy in secretion between nerve terminals and platelets lacking SEPT5 was thought to be due to functional redundancy because SEPT3 is not expressed in platelets (data not shown). Therefore, we tested the hypothesis that either SEPT3 or SEPT5 is sufficient for synaptic transmission by creating Sept3−/− Sept5−/− mice. Once again, though, loss of both septins did not produce any changes beyond what was observed for either single knockout mouse line, ruling out the possibility that SEPT3 and SEPT5 functionally compensate for the loss of the other.
Mouse knockouts for two other septins, Sept4−/− and Sept6−/−, were originally reported not to have severe neurological impairments (26, 36, 45). Targeted deletion of SEPT4 resulted in male mice that were sterile, due to a structural defect in the sperm tail annulus which impaired sperm motility. In the brains of Sept4−/− mice, no gross structural abnormalities were detected, but a reduction in the number of Purkinje cells in the cerebellum was observed with no obvious motor deficiencies. Sept6−/− mice are fertile, and they do not have any gross structural abnormalities in the cerebellum or impaired neurological function, as assessed by motor performance on a rotor rod task (45). Generation of compound Sept4−/− Sept6−/− mice did not produce defects beyond those already observed in Sept4−/− and Sept6−/− mice (45). In an exciting new finding, it was shown that Sept4−/− mice exhibit reduced dopaminergic synaptic transmission and are susceptible to the aggregation of α-synuclein (27), which are hallmark symptoms associated with Parkinson's disease.
In addition to mouse knockouts, knockdown of SEPT7 was examined in hippocampal neurons by two independent groups (61, 70). Both groups found that lowering SEPT7 protein levels resulted in neurons with impaired dendritic branching and increased density of elongated spines/immature protrusions. This phenotype is consistent with the localization of SEPT7 to dendritic branch points and the base of filopodia. In Sept3−/− Sept5−/− mice, the level of SEPT7 is significantly reduced from wild-type levels. However, the extent to which this reduction has an effect on dendrite morphology is likely to be minimal since synaptic transmission in the hippocampus of Sept3−/− Sept5−/− mice appears to be normal. A greater than 55% reduction in SEPT7 levels may be necessary to see a phenotype in mice.
Several reasons might potentially explain the absence of an observable phenotype in Sept3−/−, Sept5−/−, or Sept3−/− Sept5−/− mice. One possibility is that another related septin from a similar class is compensating for the loss of these septins during development or within nerve terminals. SEPT3, SEPT9, and SEPT12 belong to a group of septins that lack a predicted coiled-coil domain in their C-terminal extension (65). However, the levels of three SEPT9 isoforms did not change dramatically in the brains of Sept3−/− or Sept3−/− Sept5−/− mice. This result does not exclude the possibility of significant changes in small, isolated regions of the brain where SEPT3 function is most important. The other member of this class, SEPT12, appears to be specifically expressed in the testes (58) and does not get upregulated in brain tissue in the absence of SEPT3 and SEPT5. In contrast to SEPT3, SEPT5 belongs to a separate class with three other septins (SEPT1, SEPT2, and SEPT4) based on sequence similarity. In the absence of SEPT5, the levels of SEPT1 appeared unaltered whereas SEPT2 levels appeared to be upregulated. SEPT2 is required for septin filament assembly in vitro and depletion of this septin in cell lines leads to an increased incidence of binucleated cells (35, 54). However, immunofluorescence analysis of SEPT2 in primary rat hippocampal neurons isolated from Sept5−/− mice did not reveal any upregulation of SEPT2 in these neurons (data not shown). Once again, the changes in SEPT2 levels could be occurring in small, isolated regions of the brain where SEPT5 function is most important. However, it remains to be demonstrated whether septins in the same class are functionally interchangeable. Surprisingly, in the absence of both SEPT3 and SEPT5, very little upregulation was observed in the expression of the other septins, with the exception of one isoform of SEPT4. Instead, the protein levels of several septins were significantly reduced. However, it has not been clearly demonstrated whether our antibodies can detect all septin isoforms in the brain. Furthermore, the levels of SEPT10, SEPT13, and SEPT14 remain to be examined.
A second explanation for the absence of an observable phenotype in Sept3−/−, Sept5−/−, or Sept3−/− Sept5−/− mice is that functional redundancy could occur at the level of more distantly related septins. Immunoprecipitation of SEPT3 or SEPT5 from brain lysates pulls down many other septins. Since the lack of SEPT3 had little impact on the composition of septin complexes isolated from neurons, the overall function of septin complexes may not be impaired. Functionally redundant septins are present in budding yeast and fruit flies. For example, S. cerevisiae lacking Shs1/Sep7 grow as well as the wild type, suggesting that Cdc3, Cdc10, Cdc11, and Cdc12 are sufficient on their own for cell division (64). Even a complex of Cdc3, Cdc11, and Cdc12, without Cdc10, is sufficient for growth at suboptimal growth temperatures under nutrient-rich conditions (19). In Drosophila, depletion of the septin peanut did not inhibit cytokinesis during embryogenesis until the pupal stage even without maternal contribution of peanut protein (1). The absence of peanut and Sep1 did not result in gross abnormalities in the developing fly central nervous system (14). It is thought that the continued presence of Sep2 is sufficient to allow embryonic cell divisions and neuronal development to proceed normally in the absence of peanut and Sep1 (1). Interestingly, in C. elegans and D. melanogaster, there are no septin family members like SEPT3 that lack a predicted C-terminal coiled-coil domain. This suggests that these septins may be involved in highly specialized functions that are not apparent.
In the absence of certain septins, septin filaments may be structurally unstable but still retain sufficient function during neuronal development and synaptic transmission to produce a normal phenotype. While the lack of Cdc10 does not impair cytokinesis at suboptimal growth temperatures, it does affect the position of the remaining septins at the mother-bud neck junction (19). Instead of accumulating evenly at this junction, septins accumulate more on the bud side, as judged by immunofluorescence microscopy. Electron microscope images of budding yeast lacking Cdc10 show no discernible filaments where wild-type septin filaments are normally observed. When purified S. cerevisiae Cdc3, Cdc11, and Cdc12 are combined in vitro, they make filaments without Cdc10; however, the Cdc3/Cdc11/Cdc12 (Cdc3/11/12) filaments are short, unstable, and curved and do not make lateral associations (64). This contrasts with the wild-type filaments which are long, rigid, straight, and invariably paired. The mammalian SEPT2/6/7 complex is presumed to be the functional equivalent of the budding yeast Cdc3/11/12 complex. The SEPT2/6/7 complex makes filaments in vitro that are 10 nm in diameter but are curved and unstable like Cdc3/11/12 filaments (34, 54). This suggests the possibility that mammalian septin filaments lacking SEPT3, like yeast septin filaments lacking Cdc10, may retain enough structural integrity to sufficiently function in neurons. Although yeast lacking Cdc11 exhibits more severe growth defects than yeast lacking Cdc10, these mutants are still capable of cell division at 22°C. In addition, the remaining septins are able to form filaments, albeit they are structurally similar to the Cdc10-deficient filaments. Therefore, septin filaments lacking SEPT5 may retain sufficient integrity to function properly in nerve terminals. However, it remains to be demonstrated whether Cdc10 and SEPT3 or Cdc11 and SEPT5 share homologous functions.
It is possible that that septins may not be required for neural development, synapse formation, maturation, or synaptic transmission. Therefore, SEPT3 and SEPT5 may play entirely unrelated roles or else their roles in these functions are regulatory rather than obligatory. In Schizosaccharomyces pombe, deletion of all mitotic septins only delays cell division, resulting in a chain-of-cells phenotype (5, 62). In this case it was suggested that septins may function to enhance the efficiency of cytokinesis. Similarly, SEPT3 and SEPT5 in nerve terminals may function to enhance the efficiency of synaptic vesicle fusion or recycling. Such a role would not be detectable as an overall block or enhancement of synaptic vesicle exocytosis or endocytosis unless the appropriate stimulation or conditions were found.
Although the molecular details of septin function in the brain remain elusive, the neural importance of septins is reflected in their association with many neurological disorders. Moreover, the association of septins with the synaptic vesicle trafficking machinery suggests that they are likely to be involved in neuronal growth and synaptic transmission in some way that is yet to be defined. Although the lack of an individual septin such as SEPT3, SEPT4, SEPT5, or SEPT6 does not seem to have a large impact on synaptic transmission, the generation of combination knockouts along with detailed electrophysiological measurements to dissect out septin function in intact brain circuits promises to be an exciting platform for future studies of septin function.
Acknowledgments
C.W.T. was supported by a Doctoral Research Award from the Canadian Institutes of Health Research, the RESTRCOMP Trainee Award from the Hospital for Sick Children, and the Joe A. Connolly Memorial Award from the Faculty of Medicine at the University of Toronto (Toronto, ON, Canada). W.S.T. and L.Y.W. are the recipients of Canada Research Chairs and are supported by the Canadian Institutes of Health Research (FRN 13465 and FRN 143867, respectively). P.J.R. is supported by grants from the Australian National Health and Medical Research Council.
We thank Audrey Lam (The Hospital for Sick Children) for growing, electroporation, and selection of ES clones; Linda Wei (The Hospital for Sick Children) for aggregation and animal breeding during the generation of the Sept3 knockout; Nancy Simpson (The University Health Network, Toronto, ON, Canada) for help with animal husbandry; Robert Temkin at the Advanced Bioimaging Center (The Hospital for Sick Children) for assistance with electron microscopy; and Yanchun Wang (Toronto Centre for Phenogenomics) and Bonnie Welsh (The Hospital for Sick Children) for their assistance with perfusion-fixation of mouse brains. We also thank Mathew Estey, Xiao-Rong Peng, Carol Froese, and Laura Pritzker for technical assistance and critical reading of the manuscript; Sergio Grinstein for the use of the digital image acquisition system; and especially Doris L. Fortin and Richard H. Kramer for their support during the revision of the manuscript.
Footnotes
Published ahead of print on 22 September 2008.
REFERENCES
- 1.Adam, J. C., J. R. Pringle, and M. Peifer. 2000. Evidence for functional differentiation among Drosophila septins in cytokinesis and cellularization. Mol. Biol. Cell 113123-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahuja, P., E. Perriard, W. Trimble, J. C. Perriard, and E. Ehler. 2006. Probing the role of septins in cardiomyocytes. Exp. Cell Res. 3121598-1609. [DOI] [PubMed] [Google Scholar]
- 3.Barr, A. M., C. E. Young, K. Sawada, W. S. Trimble, A. G. Phillips, and W. G. Honer. 2004. Abnormalities of presynaptic protein CDCrel-1 in striatum of rats reared in social isolation: relevance to neural connectivity in schizophrenia. Eur. J. Neurosci. 20303-307. [DOI] [PubMed] [Google Scholar]
- 4.Beites, C. L., H. Xie, R. Bowser, and W. S. Trimble. 1999. The septin CDCrel-1 binds syntaxin and inhibits exocytosis. Nat. Neurosci. 2434-439. [DOI] [PubMed] [Google Scholar]
- 5.Berlin, A., A. Paoletti, and F. Chang. 2003. Mid2p stabilizes septin rings during cytokinesis in fission yeast. J. Cell Biol. 1601083-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borst, J. G., F. Helmchen, and B. Sakmann. 1995. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J. Physiol. 489:825-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borst, J. G., and B. Sakmann. 1998. Calcium current during a single action potential in a large presynaptic terminal of the rat brainstem. J. Physiol. 506:143-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brewer, G. J., J. R. Torricelli, E. K. Evege, and P. J. Price. 1993. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35567-576. [DOI] [PubMed] [Google Scholar]
- 9.Byers, B., and L. Goetsch. 1976. A highly ordered ring of membrane-associated filaments in budding yeast. J. Cell Biol. 69717-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casamayor, A., and M. Snyder. 2003. Molecular dissection of a yeast septin: distinct domains are required for septin interaction, localization, and function. Mol. Cell. Biol. 232762-2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dent., J., K. Kato, X. R. Peng, C. Martinez, M. Cattaneo, C. Poujol, P. Nurden, A. Nurden, W. S. Trimble, and J. Ware. 2002. A prototypic platelet septin and its participation in secretion. Proc. Natl. Acad. Sci. USA 993064-3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Paolo, G., S. Sankaranarayanan, M. R. Wenk, L. Daniell, E. Perucco, B. J. Caldarone, R. Flavell, M. R. Picciotto, T. A. Ryan, O. Cremona, and P. De Camilli. 2002. Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron 33789-804. [DOI] [PubMed] [Google Scholar]
- 13.Dotti, C. G., C. A. Sullivan, and G. A. Banker. 1988. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 81454-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fares, H., M. Peifer, and J. R. Pringle. 1995. Localization and possible functions of Drosophila septins. Mol. Biol. Cell 61843-1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farkasovsky, M., P. Herter, B. Voss, and A. Wittinghofer. 2005. Nucleotide binding and filament assembly of recombinant yeast septin complexes. Biol. Chem. 386643-656. [DOI] [PubMed] [Google Scholar]
- 16.Fedchyshyn, M. J., and L. Y. Wang. 2005. Developmental transformation of the release modality at the calyx of Held synapse. J. Neurosci. 254131-4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Field, C. M., O. al-Awar, J. Rosenblatt, M. L. Wong, B. Alberts, and T. J. Mitchison. 1996. A purified Drosophila septin complex forms filaments and exhibits GTPase activity. J. Cell Biol. 133605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forsythe, I. D. 1994. Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J. Physiol. 479381-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frazier, J. A., M. L. Wong, M. S. Longtine, J. R. Pringle, M. Mann, T. J. Mitchison, and C. Field. 1998. Polymerization of purified yeast septins: evidence that organized filament arrays may not be required for septin function. J. Cell Biol. 143737-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujishima, K., H. Kiyonari, J. Kurisu, T. Hirano, and M. Kengaku. 2007. Targeted disruption of Sept3, a heteromeric assembly partner of Sept5 and Sept7 in axons, has no effect on developing CNS neurons. J. Neurochem. 10277-92. [DOI] [PubMed] [Google Scholar]
- 21.Hall, P. A., K. Jung, K. J. Hillan, and S. E. Russell. 2005. Expression profiling the human septin gene family. J. Pathol. 206269-278. [DOI] [PubMed] [Google Scholar]
- 22.Harata, N., J. L. Pyle, A. M. Aravanis, M. Mozhayeva, E. T. Kavalali, and R. W. Tsien. 2001. Limited numbers of recycling vesicles in small CNS nerve terminals: implications for neural signaling and vesicular cycling. Trends Neurosci. 24637-643. [DOI] [PubMed] [Google Scholar]
- 23.Hartwell, L. H. 1971. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp. Cell Res. 69265-276. [DOI] [PubMed] [Google Scholar]
- 24.Hsu, S. C., C. D. Hazuka, R. Roth, D. L. Foletti, J. Heuser, and R. H. Scheller. 1998. Subunit composition, protein interactions, and structures of the mammalian brain Sec6/8 complex and septin filaments. Neuron 201111-1122. [DOI] [PubMed] [Google Scholar]
- 25.Huang, Y. W., M. C. Surka, D. Reynaud, C. Pace-Asciak, and W. S. Trimble. 2006. GTP binding and hydrolysis kinetics of human septin 2. FEBS J. 2733248-3260. [DOI] [PubMed] [Google Scholar]
- 26.Ihara, M., A. Kinoshita, S. Yamada, H. Tanaka, A. Tanigaki, A. Kitano, M. Goto, K. Okubo, H. Nishiyama, O. Ogawa, C. Takahashi, S. Itohara, Y. Nishimune, M. Noda, and M. Kinoshita. 2005. Cortical organization by the septin cytoskeleton is essential for structural and mechanical integrity of mammalian spermatozoa. Dev. Cell 8343-352. [DOI] [PubMed] [Google Scholar]
- 27.Ihara, M., N. Yamasaki, A. Hagiwara, A. Tanigaki, A. Kitano, R. Hikawa, H. Tomimoto, M. Noda, M. Takanashi, H. Mori, N. Hattori, T. Miyakawa, and M. Kinoshita. 2007. Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of alpha-synuclein neurotoxicity. Neuron 53519-533. [DOI] [PubMed] [Google Scholar]
- 28.Iwasaki, S., and T. Takahashi. 1998. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J. Physiol. 509419-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.John, C. M., R. K. Hite, C. S. Weirich, D. J. Fitzgerald, H. Jawhari, M. Faty, D. Schlapfer, R. Kroschewski, F. K. Winkler, T. Walz, Y. Barral, and M. O. Steinmetz. 2007. The Caenorhabditis elegans septin complex is nonpolar. EMBO J. 263296-3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joo, E., C. W. Tsang, and W. S. Trimble. 2005. Septins: traffic control at the cytokinesis intersection. Traffic 6626-634. [DOI] [PubMed] [Google Scholar]
- 31.Kinoshita, A., M. Kinoshita, H. Akiyama, H. Tomimoto, I. Akiguchi, S. Kumar, M. Noda, and J. Kimura. 1998. Identification of septins in neurofibrillary tangles in Alzheimer's disease. Am. J. Pathol. 1531551-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinoshita, A., M. Noda, and M. Kinoshita. 2000. Differential localization of septins in the mouse brain. J. Comp. Neurol. 428223-239. [DOI] [PubMed] [Google Scholar]
- 33.Kinoshita, M. 2006. Diversity of septin scaffolds. Curr. Opin. Cell Biol. 1854-60. [DOI] [PubMed] [Google Scholar]
- 34.Kinoshita, M., C. M. Field, M. L. Coughlin, A. F. Straight, and T. J. Mitchison. 2002. Self- and actin-templated assembly of mammalian septins. Dev. Cell 3791-802. [DOI] [PubMed] [Google Scholar]
- 35.Kinoshita, M., S. Kumar, A. Mizoguchi, C. Ide, A. Kinoshita, T. Haraguchi, Y. Hiraoka, and M. Noda. 1997. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes Dev. 111535-1547. [DOI] [PubMed] [Google Scholar]
- 36.Kissel, H., M. M. Georgescu, S. Larisch, K. Manova, G. R. Hunnicutt, and H. Steller. 2005. The Sept4 septin locus is required for sperm terminal differentiation in mice. Dev. Cell 8353-364. [DOI] [PubMed] [Google Scholar]
- 37.Klingauf, J., E. T. Kavalali, and R. W. Tsien. 1998. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature 394581-585. [DOI] [PubMed] [Google Scholar]
- 38.Kuhlenbaumer, G., M. C. Hannibal, E. Nelis, A. Schirmacher, N. Verpoorten, J. Meuleman, G. D. Watts, E. De Vriendt, P. Young, F. Stogbauer, H. Halfter, J. Irobi, D. Goossens, J. Del-Favero, B. G. Betz, H. Hor, G. Kurlemann, T. D. Bird, E. Airaksinen, T. Mononen, A. P. Serradell, J. M. Prats, C. Van Broeckhoven, P. De Jonghe, V. Timmerman, E. B. Ringelstein, and P. F. Chance. 2005. Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat. Genet. 371044-1046. [DOI] [PubMed] [Google Scholar]
- 39.Martinez, C., J. Corral, J. A. Dent., L. Sesma, V. Vicente, and J. Ware. 2006. Platelet septin complexes form rings and associate with the microtubular network. J. Thromb. Haemost. 41388-1395. [DOI] [PubMed] [Google Scholar]
- 40.Martinez, C., M. A. Sanjuan, J. A. Dent, L. Karlsson, and J. Ware. 2004. Human septin-septin interactions as a prerequisite for targeting septin complexes in the cytosol. Biochem. J. 382783-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez, C., and J. Ware. 2004. Mammalian septin function in hemostasis and beyond. Exp. Biol. Med. 2291111-1119. [DOI] [PubMed] [Google Scholar]
- 42.Mendoza, M., A. A. Hyman, and M. Glotzer. 2002. GTP binding induces filament assembly of a recombinant septin. Curr. Biol. 121858-1863. [DOI] [PubMed] [Google Scholar]
- 43.Nagata, K., T. Asano, Y. Nozawa, and M. Inagaki. 2004. Biochemical and cell biological analyses of a mammalian septin complex, Sept7/9b/11. J. Biol. Chem. 27955895-55904. [DOI] [PubMed] [Google Scholar]
- 44.Neher, E. 1998. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20389-399. [DOI] [PubMed] [Google Scholar]
- 45.Ono, R., M. Ihara, H. Nakajima, K. Ozaki, Y. Kataoka-Fujiwara, T. Taki, K. Nagata, M. Inagaki, N. Yoshida, T. Kitamura, Y. Hayashi, M. Kinoshita, and T. Nosaka. 2005. Disruption of Sept6, a fusion partner gene of MLL, does not affect ontogeny, leukemogenesis induced by MLL-SEPT6, or phenotype induced by the loss of Sept4. Mol. Cell. Biol. 2510965-10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan, F., R. L. Malmberg, and M. Momany. 2007. Analysis of septins across kingdoms reveals orthology and new motifs. BMC Evol. Biol. 7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng, X. R., Z. Jia, Y. Zhang, J. Ware, and W. S. Trimble. 2002. The septin CDCrel-1 is dispensable for normal development and neurotransmitter release. Mol. Cell. Biol. 22378-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rizzoli, S. O., and W. J. Betz. 2005. Synaptic vesicle pools. Nat. Rev. Neurosci. 657-69. [DOI] [PubMed] [Google Scholar]
- 49.Rodal, A. A., L. Kozubowski, B. L. Goode, D. G. Drubin, and J. H. Hartwig. 2005. Actin and septin ultrastructures at the budding yeast cell cortex. Mol. Biol. Cell 16372-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodriguez-Escudero, I., F. M. Roelants, J. Thorner, C. Nombela, M. Molina, and V. J. Cid. 2005. Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast. Biochem. J. 390613-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakaba, T., and E. Neher. 2001. Quantitative relationship between transmitter release and calcium current at the calyx of Held synapse. J. Neurosci. 21462-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saraste, M., P. R. Sibbald, and A. Wittinghofer. 1990. The P-loop-a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 15430-434. [DOI] [PubMed] [Google Scholar]
- 53.Schneggenburger, R., A. C. Meyer, and E. Neher. 1999. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23399-409. [DOI] [PubMed] [Google Scholar]
- 54.Sheffield, P. J., C. J. Oliver, B. E. Kremer, S. Sheng, Z. Shao, and I. G. Macara. 2003. Borg/septin interactions and the assembly of mammalian septin heterodimers, trimers, and filaments. J. Biol. Chem. 2783483-3488. [DOI] [PubMed] [Google Scholar]
- 55.Sirajuddin, M., M. Farkasovsky, F. Hauer, D. Kuhlmann, I. G. Macara, M. Weyand, H. Stark, and A. Wittinghofer. 2007. Structural insight into filament formation by mammalian septins. Nature 449311-315. [DOI] [PubMed] [Google Scholar]
- 56.Son, J. H., H. Kawamata, M. S. Yoo, D. J. Kim, Y. K. Lee, S. Kim, T. M. Dawson, H. Zhang, D. Sulzer, L. Yang, M. F. Beal, L. A. Degiorgio, H. S. Chun, H. Baker, and C. Peng. 2005. Neurotoxicity and behavioral deficits associated with Septin 5 accumulation in dopaminergic neurons. J. Neurochem. 941040-1053. [DOI] [PubMed] [Google Scholar]
- 57.Spiliotis, E. T., and W. J. Nelson. 2006. Here come the septins: novel polymers that coordinate intracellular functions and organization. J. Cell Sci. 1194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steels, J. D., M. P. Estey, C. D. Froese, D. Reynaud, C. Pace-Asciak, and W. S. Trimble. 2007. Sept12 is a component of the mammalian sperm tail annulus. Cell Motil. Cytoskeleton 64794-807. [DOI] [PubMed] [Google Scholar]
- 59.Stevens, C. F., and T. Tsujimoto. 1995. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc. Natl. Acad. Sci. USA 92846-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Surka, M. C., C. W. Tsang, and W. S. Trimble. 2002. The mammalian septin MSF localizes with microtubules and is required for completion of cytokinesis. Mol. Biol. Cell 133532-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tada, T., A. Simonetta, M. Batterton, M. Kinoshita, D. Edbauer, and M. Sheng. 2007. Role of septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr. Biol. 171752-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tasto, J. J., J. L. Morrell, and K. L. Gould. 2003. An anillin homologue, Mid2p, acts during fission yeast cytokinesis to organize the septin ring and promote cell separation. J. Cell Biol. 1601093-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tybulewicz, V. L., C. E. Crawford, P. K. Jackson, R. T. Bronson, and R. C. Mulligan. 1991. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 651153-1163. [DOI] [PubMed] [Google Scholar]
- 64.Versele, M., B. Gullbrand, M. J. Shulewitz, V. J. Cid, S. Bahmanyar, R. E. Chen, P. Barth, T. Alber, and J. Thorner. 2004. Protein-protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol. Biol. Cell 154568-4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Versele, M., and J. Thorner. 2005. Some assembly required: yeast septins provide the instruction manual. Trends Cell Biol. 15414-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang, L. Y., and L. K. Kaczmarek. 1998. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394384-388. [DOI] [PubMed] [Google Scholar]
- 67.Weis, S., R. Schneggenburger, and E. Neher. 1999. Properties of a model of Ca++-dependent vesicle pool dynamics and short term synaptic depression. Biophys. J. 772418-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu, L. G., R. E. Westenbroek, J. G. Borst, W. A. Catterall, and B. Sakmann. 1999. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J. Neurosci. 19726-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie, H., M. Surka, J. Howard, and W. S. Trimble. 1999. Characterization of the mammalian septin H5: distinct patterns of cytoskeletal and membrane association from other septin proteins. Cell Motil. Cytoskeleton 4352-62. [DOI] [PubMed] [Google Scholar]
- 70.Xie, Y., J. P. Vessey, A. Konecna, R. Dahm, P. Macchi, and M. A. Kiebler. 2007. The GTP-binding protein Septin 7 is critical for dendrite branching and dendritic-spine morphology. Curr. Biol. 171746-1751. [DOI] [PubMed] [Google Scholar]
- 71.Xiong, J. W., A. Leahy, and H. Stuhlmann. 1999. Retroviral promoter-trap insertion into a novel mammalian septin gene expressed during mouse neuronal development. Mech. Dev. 86183-191. [DOI] [PubMed] [Google Scholar]
- 72.Xue, J., P. J. Milburn, B. T. Hanna, M. E. Graham, J. A. Rostas, and P. J. Robinson. 2004. Phosphorylation of septin 3 on Ser-91 by cGMP-dependent protein kinase-I in nerve terminals. Biochem. J. 381753-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xue, J., C. W. Tsang, W. P. Gai, C. S. Malladi, W. S. Trimble, J. A. Rostas, and P. J. Robinson. 2004. Septin 3 (G-septin) is a developmentally regulated phosphoprotein enriched in presynaptic nerve terminals. J. Neurochem. 91579-590. [DOI] [PubMed] [Google Scholar]
- 74.Xue, J., X. Wang, C. S. Malladi, M. Kinoshita, P. J. Milburn, I. Lengyel, J. A. Rostas, and P. J. Robinson. 2000. Phosphorylation of a new brain-specific septin, G-septin, by cGMP-dependent protein kinase. J. Biol. Chem. 27510047-10056. [DOI] [PubMed] [Google Scholar]
- 75.Yagi, M., B. Zieger, G. J. Roth, and J. Ware. 1998. Structure and expression of the human septin gene HCDCREL-1. Gene 212229-236. [DOI] [PubMed] [Google Scholar]
- 75a.Yang, Y. M., and L. Y. Wang. 2006. Amplitude and kinetics of action potential-evoked Ca2+ current and its efficacy in triggering transmitter release at the developing calyx of held synapse. J. Neurosci. 265698-5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang, J., C. Kong, H. Xie, P. S. McPherson, S. Grinstein, and W. S. Trimble. 1999. Phosphatidylinositol polyphosphate binding to the mammalian septin H5 is modulated by GTP. Curr. Biol. 91458-1467. [DOI] [PubMed] [Google Scholar]