

Abstract
The regulation of the synthesis of the endothelial-derived vasoconstrictor endothelin-1 (ET-1) is a complex process encompassing transcriptional as well as mRNA stability mechanisms. We have described recently the existence of a mechanism for the control of ET-1 expression based on the mRNA-destabilizing capacity of specific cytosolic proteins through interaction with AU-rich elements (AREs) present in the 3′ untranslated region of the gene. We now identify glyceraldehyde-3′-phosphate dehydrogenase (GAPDH) as a protein which binds to the AREs and is responsible for the destabilization of the mRNA. Oxidant stress alters the binding of GAPDH to the mRNA and its capacity to modulate ET-1 expression, a phenomenon occurring through specific S glutathionylation of the catalytically active residue Cys 152. Finally, we provide data consistent with a role for GAPDH in mRNA unwinding, yielding this molecule more prone to degradation. In contrast, S-thiolated GAPDH appears unable to modify mRNA unwinding, thus facilitating enhanced stability. Taken together, these results describe a novel, redox-based mechanism regulating mRNA stability and add a new facet to the panoply of GAPDH cellular homeostatic actions.
The endothelium contributes greatly to vascular homeostasis by functioning as a sensor of physical and chemical stimuli and responding to them with specific signals. Vascular tone and blood pressure are regulated by endothelial cells through a tight balance of the levels of vasodilator and vasoconstrictor substances, the most important being nitric oxide (NO) and endothelin-1 (ET-1), respectively. In addition to its constrictor action, ET-1 has been shown to be involved in normal tissue repair and also in the pathogenesis of fibrotic diseases (38). All of these ET-1 biological actions can lead to pathophysiological processes under certain circumstances. In this respect, elevated circulating and tissue ET-1 levels have been measured in patients suffering from certain vascular diseases, such as atherosclerosis, pulmonary hypertension, or skin and lung fibrosis (2, 32). Therefore, ET-1 production and secretion by vascular endothelial cells must be tightly regulated. Endothelial cells synthesize ET-1 as a propeptide of 212 residues, preproendothelin-1, which is then subjected to specific proteolysis to yield the bioactive ET-1 peptide of 21 amino acids. The regulation of the expression is a complex process that occurs mainly at the mRNA level and involves both transcriptional and posttranscriptional mechanisms (26, 27, 31, 45). Although the analysis of the transcription of the gene has attracted most of the attention, posttranscriptional mechanisms have recently been revealed as essential steps in the regulation of the biosynthesis (37, 43).
Generally speaking, many of the processes dealing with specific regulatory mechanisms at the posttranscriptional level take place within genetic elements present in the 3′ untranslated region (3′ UTR) of the genes (10, 17, 20). Adenine- and uridine-rich elements (AREs), which are often found in the 3′ UTR of cytokines, protooncogenes, and growth factors, constitute one of the most important cis-regulatory determinants of mRNA decay (12, 28). Several RNA binding proteins that interact specifically with AREs have been characterized. These include members of the Hu family, heterogeneous nuclear ribonucleoprotein D (hnRNPD/AUF-1), tristetraprolin (TTP), and KH-type splicing regulatory protein (KSRP) (21, 30, 35, 56). However, the current understanding of how ARE-mediated decay occurs in vivo is far from clear, a fact suggesting that the overall mechanism is rather complex and may involve ARE-binding proteins yet to be identified.
Very recently, we have described the existence of a mechanism for the control of ET-1 expression based on the mRNA-destabilizing capacity of specific cytosolic proteins through interaction with AREs present in the 3′ UTR of the ET-1 gene (43). Here, we report the identification of RNA binding proteins interacting with AREs of ET-1 mRNA from vascular endothelial cells and the mechanism by which the regulation of the expression of the gene is achieved. We show that the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) constitutes the major component interacting with functional AREs of the 3′ UTR of ET-1 mRNA and that this interaction is associated with reduced ET-1 expression by mRNA destabilization. We show also that oxidant stress alters the binding of GAPDH to the mRNA and its capacity to modulate ET-1 expression. This phenomenon occurs through specific posttranslational modification of the catalytically active GAPDH residue Cys 152 by the incorporation of glutathione, or S-glutathionylation. We suggest that this mechanism plays a critical role in the regulation of ET-1 mRNA expression by oxidative or nitrosative agents. Such a mechanism of control may be of importance in the vascular environment, where oxidative stress is able to promote cardiovascular disease by alteration of the synthesis of vasoactive factors.
MATERIALS AND METHODS
Cell culture.
The human umbilical vein endothelial cell-derived cell line EA.hy926 was obtained from C.-J. S. Edgell (University of North Carolina) (18) and maintained in Dulbecco modified Eagle medium (Invitrogen) containing 20% fetal calf serum, 4.5 g/liter glucose, 100 U/ml penicillin, 100 μg/ml streptomycin, and HAT supplement (0.1 mM hypoxanthine, 0.4 μM aminopterin, and 16 μM thymidine) at 37°C in a humidified atmosphere with 5% CO2. Cells were passed weekly at a 1:10 density and used for experiments at confluence. Cells for RNA experiments or cytoplasmic extract preparation were seeded onto 10-cm-diameter culture plates, whereas for transfection they were cultured on 6-, 12-, or 24-well plates depending on the particular experiment.
Tetracycline (Tet)-inducible HEK 293 cells were cultured in Dulbecco modified Eagle medium containing 10% fetal calf serum, 4.5 g/liter glucose, 100 U/ml penicillin, 100 μg/ml streptomycin, and 200 mg/ml hygromycin B.
Construction of reporter plasmids and DNA/RNA cell transfection.
Luciferase reporter plasmids containing the 3′ UTR sequence of the human ET-1 gene (EDN1) were generated as described previously (43). Briefly, a human ET-1 cDNA fragment containing the 3′ UTR sequence of the gene from the pUC13-full human ET-1 cDNA plasmid (kindly provided by Kenneth Bloch, Massachusetts General Hospital) was subcloned into pCR-Script to result in pCR-3′-UTR-human ET-1 [partial 3′ UTR corresponding to positions 272 to 1127 plus a poly(A) tract of 22 nucleotides (nt), numbered relative to the first nucleotide of the 3′ UTR (GenBank accession no. J05008) (26)]. This plasmid was used as the template for PCR performed with a set of primers to generate deletion fragments of the 3′ UTR fragment flanked by XbaI sites (Table 1) . The PCR products were digested by XbaI and cloned into the XbaI site of a luciferase reporter construct driven by a bp −650-to-+172 fragment of the human ET-1 promoter (−650bp-ppET-1 prom-luc) (45) to result in ET-1 prom-luc-3′-UTR constructs. A PCR product corresponding to the fragment from position 924 to position 1127 (fragment 924-1127) with specific mutations in AREs involved in mRNA stability (substitutions from AUUUA to GGGCC) was generated as described previously (43) and cloned into −650bp-ppET-1 prom-luc. The identities of the clones generated throughout this work were checked by sequencing.
TABLE 1.
Set of oligonucleotides used to generate constructs of the 3′ UTR of human ET-1 mRNA
| Primera | Sequenceb | Target |
|---|---|---|
| UTR-1 | GCTCTAGAAGGTCGACGGTATCGATAAG | Vector |
| UTR-2 | GCTCTAGAATGAGTGGCTGCAGGAATTC | 272-277 |
| UTR-3 | GCTCTAGAGACACAATGGTATAGGGTTG | 810-829 |
| UTR-4 | GCTCTAGACAACCCTATACCATTGTGTC | 829-810 |
| UTR-7 | GCTCTAGAGAATTTTGATGTACTTATTTTTTTATAG | 924-951 |
Primer pair UTR-2/UTR-1 was used to generate the full-length fragment (272-1127), primer pair UTR-3/UTR-1 was used to generate fragment 808-1127, primer pair UTR-2/UTR-4 was used to generate fragment 272-831, and primer pair UTR-7/UTR-1 was used to generate fragment 924-1127.
Nucleotides shown in bold carry specific sequences from the 3′ UTR. Oligonucleotides include an XbaI adapter to facilitate cloning (underlined).
Small interfering RNA (siRNA) technology was employed to reduce the content of endogenous GAPDH in EA.hy926 cells by using siRNA duplex oligonucleotides designed to target human GAPDH (silencer GAPDH, reference no. 4605; Ambion) or a siRNA negative control (reference no. 4611; Ambion).
Transient-transfection experiments were performed with EA.hy926 or HEK 293 cells as described previously (45). Reporter activity was estimated by luminometry using the pRL-CMV plasmid (Renilla luciferase under the control of the cytomegalovirus promoter) for normalization purposes (Promega) (45).
Preparation of cytosolic extracts and in vitro RNA binding assay.
Cytosolic extracts were prepared according to a procedure already described (43). Briefly, cell plates were washed once with ice-cold phosphate-buffered saline and scraped in the same solution. Upon centrifugation, they were resuspended in 10 mmol/liter HEPES, pH 7.9, 10 mmol/liter KCl, 0.1 mmol/liter EDTA, 0.1 mmol/liter EGTA, 1 mmol/liter dithiothreitol (DTT), and 1 μg/ml each of leupeptin, aprotinin, and pepstatin A (Sigma). After 15 min of incubation on ice, Nonidet P-40 was added to a final concentration of 0.5% and cells were vortexed for 10 s. Nuclei were then sedimented by centrifugation for 5 min at 14,000 × g, and the supernatant was stored at −80°C as a crude cytosolic fraction. Protein content was determined with a bicinchoninic acid (BCA) protein assay reagent kit (Pierce).
To determine in vitro RNA binding, RNA-electrophoretic mobility shift assays (RNA-EMSA) were performed using radiolabeled RNA probes. These probes were generated by in vitro transcription from pCR-II plasmids (Invitrogen) containing deletion fragments of the 3′ UTR of the human ET-1 gene using [α-32P]UTP (Amersham), as described previously (43). Transcripts for competition were generated by the same procedure but using unlabeled UTP. RNA binding experiments were carried out using 5 to 8 fmol of labeled RNA (about 60,000 cpm) and 4 μg of protein from cytosolic extracts in a final volume of 14 μl of binding buffer (50 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 5 mM DTT, 5% glycerol, and 10 mM Tris, pH 7.4, containing 0.2 mg/ml tRNA as the competitor for unspecific binding). After 15 min at room temperature, 6 U of RNase T1 (Roche) was added for 20 min to digest unbound RNA. RNA/protein complexes were resolved by nondenaturing 5% polyacrylamide gel electrophoresis in 0.25× Tris-borate-EDTA buffer and visualized by autoradiography. For competition experiments, cytosolic extracts were preincubated with the competitor at the concentrations indicated in the figure legends for 10 min prior to the addition of the radiolabeled probe.
For the nitrocellulose filter binding assay, reaction mixtures containing labeled RNA and protein in binding buffer without treatment with RNase T1 were filtered through nitrocellulose (BA85; Schleicher & Schuell). After the filter was washed five times with binding buffer, bound radioactivity was determined by scintillation counting.
Purification and identification of RNA binding proteins.
RNA binding proteins were purified by RNA-affinity chromatography. Briefly, cytosolic extracts from EA.hy926 cells seeded on to 15-cm-diameter plates (2 mg protein measured by the BCA assay) were incubated with 600 μg of 5′-biotinylated RNA (biotin-RNA) corresponding to the 3′ UTR fragment 939-991 (Curevac) in binding buffer including 1 U/μl RNasin (Promega) in a final volume of 1.2 ml for 30 min at room temperature. Afterwards, 2 ml of UItralink immobilized streptavidin (50% slurry equilibrated in binding buffer) (Pierce) was added and the mixture was incubated by rotation for 1 h at room temperature and then loaded on to a disposable 2.0-ml polystyrene column (Pierce). The packed column was washed extensively with binding buffer (20 to 25 ml), and bound proteins were eluted with 1 M NaCl in binding buffer and collected into 200-μl fractions. Eluates were analyzed for total protein by the Bradford assay (5a) and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). A control column without biotinylated RNA was processed in the same way in order to evaluate the nonspecific binding to the streptavidin beads.
Identification of proteins was performed by mass spectrometric analysis in the proteomics unit at Centro Nacional de Investigaciones Cardiovasculares, Madrid, Spain. Protein spots were excised manually and digested automatically using a Proteineer DP protein digestion station (Bruker Daltonics, Germany). For peptide mass fingerprinting and matrix-assisted laser desorption ionization (MALDI) Lift time of flight (TOF)/TOF mass spectrometer spectrum acquisition, an aliquot of α-cyano-4-hydroxycinnamic acid in 33% aqueous acetonitrile and 0.1% trifluoroacetic acid was mixed with an aliquot of the above-described digestion solution and the mixture was deposited onto an AnchorChip MALDI probe (Bruker Daltonics). Peptide mass fingerprint spectra were measured with an Ultraflex TOF/TOF MALDI mass spectrometer (Bruker Daltonics) in positive ion reflector mode. Mass measurements were performed automatically through fuzzy-logic-based software or manually. Each spectrum was calibrated internally with mass signals of trypsin autolysis ions to reach a typical mass measurement accuracy of ±25 ppm. The measured tryptic peptide masses were transferred through the MS BioTools program (Bruker Daltonics) as inputs to search the NCBInr database using Mascot software (Matrix Science, United Kingdom).
For RNA binding experiments, eluates from RNA affinity chromatography were concentrated and equilibrated in binding buffer by centrifugation in Centricon YM-10 filter devices (Millipore).
Protein studies.
For protein expression studies, EA.hy926 or HEK 293 cells were washed once with ice-cold phosphate-buffered saline and immediately lysed with NET-2 buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, and 0.05% NP-40, containing 1 μg/ml of each of the protease inhibitors leupeptin, aprotinin, and pepstatin A). Lysates were cleared from cell debris by centrifugation at 14,000 × g for 20 min at 4°C, and protein concentrations in the supernatants were determined by the BCA assay. Immunoprecipitation studies were done by incubation of total protein cell extracts with the corresponding antibody (anti-GAPDH, rabbit polyclonal [Abcam]; anti-green fluorescent protein [anti-GFP], mouse monoclonal [Roche]; or preimmune serum as a negative control) in NET-2 buffer for 1 h on ice in a final volume of 200 μl. Then, 25 μl of Ultralink immobilized protein A/G (50% slurry equilibrated in NET-2 buffer) (Pierce) was added and the mixture was incubated by rotation at 4°C for 2 h. Beads were washed five times with NET-2 buffer and pulled-down proteins were analyzed by Western blotting.
Immunoprecipitates and cytosolic or total cellular extracts were mixed with standard protein loading buffer, fractionated by SDS-PAGE, and transferred to Immobilon-P membranes (Millipore). As a control for even protein loading and transfer, membranes were stained with Ponceau S. Blots were probed with anti-GAPDH (1:2,000 dilution, mouse monoclonal 6C5; Santa Cruz), anti-GFP (1:1,000 dilution, mouse monoclonal; Roche), antiglutathione (1:500 dilution, mouse monoclonal; Virogen), or anti-β-actin (1:20,000 dilution, mouse monoclonal clone AC-15; Sigma), followed by horseradish peroxidase (HRP)-coupled secondary antibodies at a 1:2,000 dilution, and immunocomplexes were visualized using an ECL detection system from Amersham.
Incorporation of glutathione to proteins was analyzed by using biotin-labeled glutathione derivatives. In vitro experiments with purified proteins were done with biotin-labeled oxidized glutathione (GSSG). Biotin-GSSG was synthesized by coupling biotin to the primary amino groups of GSSG under mild alkaline conditions using sulfosuccinimidyl-6′-(biotinamido)-6-hexanamido hexanoate (Pierce) as the biotinylating reagent, as described previously (6). For in vivo experiments, the intracellular pool of glutathione was labeled by preincubation of cells with glutathione ethyl ester-biotin amide (bioGEE) (Invitrogen) for 1 h at 178 μM. Total cell extracts, immunoprecipitated GAPDH or GFP/GAPDH, or purified proteins were separated by SDS-PAGE and transferred to membranes as described above. The biotin label associated with proteins was revealed by incubation of the membranes with 1 μg/ml Immunopure streptavidin-HRP conjugate (Pierce).
Glutathione incorporation to purified rabbit muscle GAPDH was also estimated by derivatization of released thiols from GSSG-treated or S-nitrosoglutathione (GSNO)-treated protein by DTT reduction, coupled with detection by fluorescent high-pressure liquid chromatography (HPLC) as described previously (11). Briefly, 25 μg of protein was treated with GSSG or GSNO in RNA binding buffer, precipitated with trichloroacetic acid, and resuspended in 50 mM HEPES, pH 8.0, with 2% SDS and saturated sodium bicarbonate. Mixed disulfides were reduced with 2.5 mM DTT for 1 h at 40°C, released thiols were derivatized with 4 mM monobromobimane (Fluka), and protein was precipitated again. The supernatant was separated in a Supelco Ascentis C18 column using 0.25% acetic acid as solvent A, with a gradient ranging from 20% to 80% methanol (solvent B). Derivatized thiols were measured by fluorescence detection (excitation, 394 nm; emission, 480 nm), using reduced glutathione as a standard. Quantitative precipitation of GAPDH before mixed-disulfide reduction was assessed by SDS-PAGE.
Incorporation of glutathione to rabbit muscle GAPDH was confirmed by mass spectrometry analysis. Briefly, GAPDH was incubated with GSNO or GSSG and digested by trypsin, and the resulting tryptic peptides were injected onto a C18, reversed-phase nanocolumn (Discovery Bio Wide pore; Supelco) and analyzed in a continuous acetonitrile gradient. A flow rate of ca. 300 nl/min was used to elute peptides from the reversed-phase nanocolumn to an electrospray ion source coupled to a quadrupole ion trap mass spectrometer (Esquire HCT Ultra; Bruker Daltonics) for real-time ionization and fragmentation (proteomics unit, Centro Nacional de Investigaciones Cardiovasculares, Madrid, Spain).
RNA isolation and determination.
For RNA experiments, EA.hy926 or HEK 293 cells were washed with phosphate-buffered saline and processed for RNA isolation by guanidinium thiocyanate-phenol-chloroform extraction. To detect and quantify human ET-1 and GAPDH mRNA, RNase protection experiments were performed as described previously (45). For normalization purposes, 28S rRNA was also determined.
To generate a human ET-1 cDNA fragment, a PCR with reverse transcription products from EA.hy926 cells was performed with human ET-1 sense (CCAAGGAGCTCCAGAAACAG) and human ET-1 antisense (CTGTTGCCTTTGTGGGAAGT) primers. The 265-bp PCR fragment (positions 379 to 644 of human ET-1 mRNA, GenBank accession no. XM_004293) was TA cloned in pCRII vector (Invitrogen) to generate pCRII-human ET-1. A 148-bp NcoI fragment of pChug 20.1 (kindly provided by Michael Sirover, Temple University), corresponding to positions 243 to 391 of human GAPDH mRNA (GenBank accession no. NM_002046), was cloned into pXcm vector to generate pXcm-GAPDH. The plasmid pTRI-RNA-28S, containing a 115-bp cDNA fragment of the human 28S rRNA gene, was obtained from Ambion.
To generate radiolabeled human ET-1, human GAPDH, and 28S rRNA antisense probes for RNase protection assays, 0.5 μg of each of the corresponding linearized plasmids was in vitro transcribed using radiolabeled UTP. Densitometric analyses of resulting gels were performed using a FLA-3000 phosphorimager (Fuji). Antisense probes of human ET-1, human GAPDH, and 28S rRNA were 379, 241, and 153 nt, respectively. The corresponding protected fragments were 265, 148, and 115 nt.
Immunoprecipitation studies coupled with mRNA detection were performed as described above except that RNA bound to beads was isolated with an RNeasy mini kit according to the protocol recommended by the manufacturer (Qiagen). Luciferase and RNA polymerase 2A mRNAs were detected by Sybr green-based quantitative real-time PCR (qRT-PCR) in 25 μl in a 48-well thermal cycler (Mini-Opticon; Bio-Rad) with the following primers: luciferase sense (AAAAAGTTGCGCGGAGGAG), luciferase antisense (TTTTTCTTGCGTCGAGTTTTCC), RNA polymerase 2A sense (GCACCACGTCCAATGACAT), and RNA polymerase 2A antisense (GTGCGGCTGCTTCCATAA). Standard curves for each primer pair were done with serial dilutions of reverse transcription products in order to validate PCR conditions.
Expression and purification of recombinant GST fusion proteins.
The plasmid pGEX-2T (GE Healthcare) was used to generate recombinant glutathione S-transferase (GST)-GAPDH fusion proteins. Human GAPDH cDNA fragments covering the different domains of the protein were generated by PCR (using as a template the plasmid pChug 20.1) and were cloned into pGEX-2T to result in the set of GST-GAPDH plasmids used in this work (Table 2). Specific mutations in the GST-GAPDH full-length (2-335) Cys 152-to-Ser (TGC to TCC at codon 152) construct, as well as the GST-GAPDH 2-149/314-335 construct lacking the entire catalytic domain, were introduced by following the protocol of a QuikChange site-directed mutagenesis kit (Stratagene).
TABLE 2.
Sets of oligonucleotides used to generate GST-GAPDH fusion and mouse GAPDH/GFP C152S mutant constructs
| Primer function and namea | Sequenceb |
|---|---|
| GST-GAPDH fusion constructs | |
| GAP-1 | CCGGAATTCATGGGAAGGTGAAGGTCGG |
| GAP-2 | CCGGAATTCTTACTCCTTGGAGGCATG |
| GAP-3 | CCGGAATTCATTGCTGATGATCTTGAGGC |
| GAP-4 | CCGGAATTCATGCCTCCTGCACCACC |
| GAP-5 | CCGGAATTCCCAGGAAATGAGCTTGACAA |
| Mouse GAPDH/GFP C152S mutant constructs | |
| GAP-CYS | CATCAGCAATGCCTCCTcCACCACCAACTGCTTAGCACCC |
| GAP-delCAT | GCCTCAAGATCATCAGCAATTATGACAACGAATTTGGC |
| GAP-CYSm | TGTCAGCAATGCATCCTcCACCACCAACTGCTTAGCCC |
Primer pair GAP-1/GAP-2 was used to generate the full-length fragment (2-335), primer pair GAP-1/GAP-3 was used to generate fragment 2-149, primer pair GAP-4/GAP-5 was used to generate fragment 150-313, and primer pair GAP-4/GAP-2 was used to generate fragment 150-335. Oligonucleotides GAP-CYS and GAP-delCAT were used to generate the GST-GAPDH full-length C152S mutant (change from TGC to TCC at codon 152, indicated in lowercase italics) and the GST-GAPDH 2-149/314-335 construct lacking the entire catalytic domain, respectively. The GAPDH/GFP C152S mutant was generated with oligonucleotide GAP-CYSm (mouse) from plasmid pcDNA5/FRT/TO/GAPDH/GFP.
Nucleotides shown in bold carry specific sequences from human GAPDH targeted in the plasmid pChug 20.1. Oligonucleotides include an EcoRI adapter to facilitate cloning (underlined).
GST fusion proteins were expressed in Escherichia coli and purified on glutathione beads by use of a standard protocol described earlier (44). The purity of the recombinant proteins was verified by SDS-PAGE, and the concentration was determined with the Bradford assay (5a), using bovine serum albumin (BSA) as a standard.
Generation of stable transfectants of HEK 293 cells for in vivo RNA binding analysis and half-life mRNA studies.
To study in vivo binding of the ET-1 mRNA 3′ UTR and GAPDH, we generated Tet-inducible HEK 293 cells stably expressing a mouse GAPDH/GFP fusion protein and GFP only as the control. A blunt-ended BglII/HincII mouse GAPDH/GFP fragment generated from the pGAPDH/GFP plasmid (kindly provided by J.-L. Dreyer, University of Fribourg, Switzerland) was cloned into pcDNA5/FRT/TO vector (Invitrogen) cut with EcoRV to result in pcDNA5/FRT/TO/GAPDH/GFP. A similar cloning strategy was used to generate pcDNA5/FRT/TO/GFP. These constructs were then cotransfected with the Flp recombinase expression plasmid pOG44 into the Flp-In T-Rex 293 cell line (Invitrogen). Cells of this line stably express the Tet repressor and contain a single integrated Flp recombination target (FRT) site. Flp recombinase expression from the pOG44 vector mediates insertion of GFP-based constructs into the genome at the integrated FRT site through site-specific DNA recombination. Cells were selected with hygromycin B after 48 h, and clones appeared after 10 to 15 days. Isogenic pooled clones were expanded and used for experiments of induction by the Tet analogue doxycycline (Dox). A specific mutation in GFP/GAPDH, Cys 152 to Ser, was introduced by site-directed mutagenesis (Table 2) in order to generate HEK 293 cells overexpressing a GFP/GAPDH C152S mutant fusion protein. Immunofluorescence analysis was done with HEK 293 cells seeded on 10-mm-diameter glass coverslips in 24-well plates. Cells were fixed with 3.5% paraformaldehyde and permeabilized with 70% methanol. GAPDH expression was analyzed with a primary anti-GAPDH antibody (1:50 dilution, monoclonal antibody; Santa Cruz) and the corresponding Alexa 488-coupled secondary antibody. Nuclear staining was performed with DAPI (4′,6-diamidino-2-phenylindole) (Sigma). Cell fluorescence from GFP, Alexa 488, and DAPI was visualized by confocal microscopy (Leica TCS-SP2-AOBS).
To estimate the half-lives of mRNAs in a Tet-inducible expression system, luciferase cassettes fused to ET-1 3′ UTR fragments were cloned into the vector pcDNA5/FRT/TO to obtain the corresponding pcDNA5/FRT/TO/luc-3′-UTR constructs (43). These plasmids were used to generate HEK 293 cells overexpressing luciferase-3′ UTR-derived mRNAs, as described before (43). Rates of decay of 3′ UTR mRNA species in Tet-regulated, stably transfected HEK 293 cells were estimated by measuring luciferase activity before and after induction of cells with Dox from 2 to 72 h. Luciferase induction kinetics for these cell lines were fitted to the equation 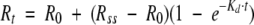 , where t (time) and Rt (luciferase activity at a given time) are the variables, R0 is the initial value, Rss is the value at the stationary state, and Kd is the decay constant. Kd values for the Rss that gave the best fit were chosen with GraphPad Prism 4 (GraphPad Software) and are given per hour (±standard errors). These decay constants were used to calculate the apparent luciferase protein half-life for these induction kinetics (t1/2 = 0.693/Kd). As levels of luciferase protein turnover were identical in all of the cell lines (3 h by cycloheximide experiments [data not shown]), mRNA half-lives for the corresponding transcripts were estimated by subtracting this 3-h value from the protein half-life value (43).
, where t (time) and Rt (luciferase activity at a given time) are the variables, R0 is the initial value, Rss is the value at the stationary state, and Kd is the decay constant. Kd values for the Rss that gave the best fit were chosen with GraphPad Prism 4 (GraphPad Software) and are given per hour (±standard errors). These decay constants were used to calculate the apparent luciferase protein half-life for these induction kinetics (t1/2 = 0.693/Kd). As levels of luciferase protein turnover were identical in all of the cell lines (3 h by cycloheximide experiments [data not shown]), mRNA half-lives for the corresponding transcripts were estimated by subtracting this 3-h value from the protein half-life value (43).
RNA degradation experiments.
RNA degradation assays were performed by exposing the radiolabeled, 5′-capped, 3′-polyadenylated 924-1127 3′ UTR RNA probe to cytosolic extracts from EA.hy926 cells. In order to generate a plasmid for in vitro transcription of a polyadenylated RNA probe, an EcoRI 3′ UTR 924-1127 fragment was cloned into the EcoRI site of the pSD3 plasmid [a pSP65 derivative with an 85-bp poly(A) tract downstream from the cloning site (kindly provided by Peter Good, University of Wisconsin)] to generate pSD3/924-1127-poly(A). A stable radiolabeled RNA probe was also included as an internal control. For that purpose, a 105-nt fragment of the 3′ UTR of Xenopus laevis β-globin (corresponding to positions 1647 to 1748 of the X. laevis major β-globin gene, GenBank accession no. J00978) followed by a poly(A) tract of 100 nt was cloned into the plasmid pCR-Script to generate pCR-3′-UTR-β-globin-poly(A). In vitro transcription was done following the protocol described above, including an m7g(5′)ppp(5′)g cap (Ambion) as the substrate. RNAs were incubated at room temperature with cytosolic extracts (20 μg of protein), and at various time points, aliquots were removed, protein was phenol extracted, and the remaining intact RNA was measured by denaturing urea-polyacrylamide gel electrophoresis coupled with autoradiography.
Fluorometric RNA unwinding assay.
RNA unwinding by GAPDH was analyzed by following a fluorometric method based on the displacement of a fluorescent dye from double-stranded RNA upon unwinding, as previously described (19). The standard reaction mixture consisted of a solution of 900 μl of 1 μM DAPI in binding buffer, 58 nM of 3′ UTR RNA probe, and various concentrations of rabbit muscle GAPDH or BSA as controls (Sigma). Fluorescence measurements were carried out at room temperature using a cuvette with agitation on a Fluorolog-3 spectrofluorometer (Horiba), with excitation and emission wavelengths for DAPI of 345 and 467 nm, respectively. A baseline was recorded with DAPI before the addition of RNA and/or protein. RNA unwinding by GAPDH was estimated as the reduction in the initial fluorescence of DAPI bound to RNA.
Other experimental procedures and reagents.
ET-1 peptide determination was performed by an enzyme-linked immunosorbent assay (ELISA) (endothelin-1 Biotrak ELISA system; Amersham), as described previously (8). GAPDH activities from cell extracts and purified proteins were measured spectrophotometrically as an increase in the absorption at 340 nm resulting from the reduction of NAD+ by using an assay system including dl-glyceraldehyde-3-phosphate (Sigma) and NAD+ in pyrophosphate-arsenate buffer, as described previously (29).
Statistical analysis.
Experimental data were analyzed by unpaired Student's t test in the case of normal distribution of data or by nonparametric tests as appropriate. The P values obtained are indicated in the figure legends when statistically significant. Apparent dissociation constants for the binding of GAPDH to RNA from filter nitrocellulose binding assays were estimated using GraphPad Prism 4 statistical software.
RESULTS
AREs in the 3′ UTR mediate destabilization of human ET-1 mRNA.
The 3′ UTR of human ET-1 mRNA contains a number of AU-rich repeats of the type AUUUA, which are cis-acting sequences critical for the regulation of mRNA stability. AREs in the 3′ UTR of human ET-1 mRNA are located predominantly in the distal portion, with five runs of the form AUnA (n = 3, 5, or 7) within a short region spanning positions 940 to 1035 (Fig. 1A). We have recently described that three AREs (ARE3, ARE4, and ARE5) contained within that region (952-991) exert a strong repressional control over ET-1 gene expression, as observed in experiments performed with luciferase reporters in bovine aortic endothelial cells (43). By generating a Tet-inducible system for the expression of ET-1 3′ UTR-containing mRNA species in HEK 293 cells, we observed that a reduced expression of the luciferase reporter occurs by increased mRNA destabilization, with decay constants of ARE-containing mRNAs doubling those without them (decay constants were, for ARE-containing mRNAs, 0.058 ± 0.004 h−1 for the 3′ UTR construct and 0.061 ± 0.004 h−1 for the 808-1127 construct and, for non-ARE-containing mRNAs, 0.035 ± 0.002 h−1 for the construct lacking the 3′ UTR and 0.031 ± 0.001 h−1 for the 272-831 construct [see Materials and Methods for details]). We also observed that AREs involved in the destabilization were the target for the binding of cytosolic proteins (43). In order to identify and characterize the protein(s) interacting with these control elements, we used the established human umbilical vein endothelial cell-derived cell line EA.hy926. First, we checked whether the repressional control mediated by the AREs in the 3′ UTR was also operating in human endothelial cells. Transfection of EA.hy926 cells with luciferase cassettes driven by the human ET-1 promoter fused to different fragments of the 3′ UTR showed that the full-length 3′ UTR (272-1127) promoted reduced luciferase expression compared to that of a construct without this sequence (ΔUTR) (Fig. 1B). This reduction in luciferase expression was mimicked only by 3′ UTR-derived constructs including distal AREs (808-1127 and 924-1127) and was abolished by the specific mutation of the three AREs within the 952-991 segment (the 924-1127 mutant). Luciferase mRNA level measurements confirmed these observations (data not shown). As previously shown with bovine aortic endothelial cells, reduced reporter expression correlates with the binding of cytosolic proteins. Figure 1C shows that binding of cellular proteins to the radiolabeled 924-1127 probe was competed with excess unlabeled full-length 3′ UTR, by 808-1127 and by itself but not by the non-ARE probe 272-831, indicating that the target sequence for the interaction with proteins lies in positions 924 to 1127. Consistently with the reporter studies, radiolabeled 924-1127 with specific mutations disrupting AREs in segment 952-991 showed reduced binding capacity compared to that of a wild-type probe (Fig. 1D). Therefore, human endothelial cells recapitulate the mechanism of mRNA destabilization observed in the bovine endothelium and they constitute a good model to characterize the identity of the protein(s) responsible for the effect.
FIG. 1.

AREs in the 3′ UTR control ET-1 expression through the interaction of specific cytosolic proteins. (A) Schematic representations of the 3′ UTR of the human ET-1 gene and constructs used in this study. Black bars represent the region containing AREs involved in mRNA destabilization. Sequences of wild-type and ARE mutant constructs are also shown. prom, promoter; luc, luciferase. (B) Analysis of the luciferase expression of ET-1 promoter/luciferase/3′ UTR-derived constructs in transiently transfected EA.hy926 cells. Values are expressed as percentages of luciferase activity for the ΔUTR construct after normalization with a Renilla control plasmid (means ± standard deviations, n = 6) (*, P < 0.01 compared with the ΔUTR value). (C and D) Interactions of 3′ UTR-derived RNA probes with cytoplasmic proteins extracted from EA.hy926 cells. Competition of specific binding to the 924-1127 probe was assayed with a molar excess (1, 10, and 100 times) of unlabeled 3′ UTR, 272-831, 808-1127, and 924-1127 fragments (C). Binding to radiolabeled wild-type and ARE mutant 924-1127 probes is also shown (D). The autoradiograms shown correspond to representative RNA-EMSA performed twice with two independent preparations. The position of the detected protein complexes is indicated (arrowhead).
Purification and identification of ET-1 mRNA 3′ UTR-binding protein(s).
As a capture aid to isolate ET-1 mRNA 3′ UTR-binding proteins, we used a 5′-biotinylated 939-991 probe in RNA affinity chromatography experiments. Validation of the biotinylated probe was performed by competition of binding of radiolabeled 924-1127 probe to proteins from cytosolic extracts via RNA-EMSA. As shown in Fig. 2A, increasing concentrations of the biotinylated probe reduced the presence of the radiolabeled RNA-protein complex. RNA chromatography was performed with the biotinylated probe bound to streptavidin beads and cytosolic extracts from EA.hy926 cells. A column without RNA was packed with streptavidin beads only as a control, assuming that differential binding to the biotin-RNA column compared to the no-RNA column may come from both selective and nonselective interaction with RNA. After several washing steps, the RNA binding proteins were eluted by passing 1 M NaCl and collected into 200 μl-fractions. Analysis of the eluates for total protein content by the Bradford assay (5a) (Fig. 2B) and SDS-PAGE (Fig. 2C, left) showed that numerous proteins were eluted from the RNA-loaded beads (“Biotin-RNA” column, fractions 2 to 4), whereas only few proteins bound to unloaded resin (“No RNA” column, fractions 2 to 4). Attention was paid to a major component of about 40 kDa, which was eluted only from the RNA-loaded column. The stained protein band was cut out, and its identity was determined by peptide mass fingerprinting. This analysis resulted, strikingly, in the identification of the glycolytic enzyme GAPDH as a putative protein interacting with the 3′ UTR of the ET-1 mRNA. The identity of the protein was further confirmed by immunoblotting with a specific anti-GAPDH antibody (Fig. 2C, right). Proteins from the biotin-RNA and no-RNA columns were concentrated and equilibrated in binding buffer, and their capacities to interact with radiolabeled 924-1127 probe were analyzed by RNA-EMSA. Figure 2D, left, shows similar RNA-protein complex patterns for EA.hy926 cytosolic extracts and eluates from the RNA-loaded column, whereas the control resin gave no signal. Again, transfer of proteins to a membrane and further immunoblot analysis with an anti-GAPDH antibody revealed a signal coincidence between cytosolic extracts and the eluted fraction from the RNA-loaded column (Fig. 2D, right).
FIG. 2.

Purification of GAPDH and identification of GAPDH as an ET-1 mRNA 3′ UTR-binding protein. (A) Validation of the 5′-biotinylated 939-991 RNA probe used in RNA affinity chromatography experiments by competition of specific binding of EA.hy926 cytosolic proteins to the radiolabeled 924-1127 probe. A molar excess from 0.001 to 100 times was assayed by RNA-EMSA. (B and C) Enrichment of ET-1 mRNA 3′ UTR-binding proteins by RNA affinity chromatography. After extensive washing of columns packed with or without biotin-RNA, the total protein content from 1 M NaCl-eluted fractions was assayed (B). Coomassie staining pattern of proteins from fractions 2 to 4 bound to biotin-RNA and no-RNA columns. The arrow indicates the position of the major 40-kDa component, further identified by peptide mass fingerprinting as GAPDH (C, left). A GAPDH-specific antibody was used in immunoblotting of eluted fractions (C, right). The sequence of human GAPDH is also shown, with peptides covered by matrix-assisted laser desorption ionization-time of flight analysis indicated shown in red (aa, amino acids) (C, bottom). (D) RNA binding capacity of proteins eluted from RNA affinity chromatography columns. Proteins eluted in fraction 2 from biotin-RNA and no-RNA columns were concentrated, equilibrated in binding buffer, and assayed by RNA-EMSA with the radiolabeled 924-1127 probe (left). In a similar EMSA, proteins were transferred to a membrane and assayed with an anti-GAPDH antibody (right). Cytosolic extracts from EA.hy926 cells were also included in this study. (E) RNA binding capacity of commercial preparations of rabbit muscle GAPDH. Various amounts (1, 2, and 5 μg) of rabbit muscle GAPDH or BSA were assayed by RNA-EMSA with radiolabeled 924-1127. Cytosolic extracts from EA.hy926 cells were also included in this study. (F) Effect of mutation of AREs on RNA binding capacity of rabbit muscle GAPDH and eluted proteins from fraction 2 (biotin-RNA). RNA-EMSA were performed with wild-type and ARE mutant 924-1127 probes. The autoradiograms shown correspond to representative RNA-EMSA performed twice with two independent preparations. The position of the detected protein complexes is indicated (arrowhead). (G) Estimation of apparent dissociation constant (KDapp) by nitrocellulose filter binding assay. Radiolabeled RNA was incubated with the indicated concentrations of rabbit muscle GAPDH in binding buffer. Following incubation for 15 min at room temperature, samples were filtered through nitrocellulose and washed extensively. Bound radioactivity was determined by scintillation counting. Values are shown as percentages of the maximum and represent experiments performed four times with essentially the same results.
GAPDH is a key glycolytic enzyme catalyzing the oxidative phosphorylation of glyceraldehyde 3-phosphate (G3P) to 1,3-diphosphoglycerate. Different functions beyond the glycolytic activity have been postulated for GAPDH, including roles in apoptosis, DNA replication and repair, endocytosis, and mRNA regulation. Actually, early investigations reported the interaction of GAPDH with RNA and more specifically with AU-rich RNA, involving this protein in the control of mRNA decay (5, 14, 41, 47). In order to validate the specificity of GAPDH as an ET-1 mRNA 3′ UTR-binding protein, we performed RNA-EMSA with commercial preparations of GAPDH from rabbit muscle and radiolabeled 924-1127 probe. Figure 2E shows that increasing amounts of rabbit GAPDH resulted in robust signals from RNA-protein complexes. Equivalent amounts of the control protein BSA did not give any significant signal. Whether GAPDH interacts specifically with AREs within the 3′ UTR of the human ET-1 mRNA was also checked by RNA-EMSA. Both the rabbit muscle GAPDH and the eluate from the biotin-RNA resin were able to interact with the radiolabeled ARE wild-type probe, in the same way as the cytosolic extracts from EA.hy926 cells. In contrast, the ARE mutant probe gave no significant signal (Fig. 2F). A quantitative analysis of RNA binding of rabbit muscle GAPDH performed by a nitrocellulose filter binding assay yielded an apparent dissociation constant of 0.98 μM (Fig. 2G).
To determine whether GAPDH binds to ET-1 mRNA in vivo, we performed immunoprecipitation experiments with specific antibodies, and the presence of ET-1 mRNA in the purified complex was investigated by qRT-PCR. EA.hy926 cells produce little ET-1 mRNA and peptide, so we found it difficult to see enrichment of ET-1 mRNA in immunocomplexes pulled down by an anti-GAPDH antibody over the background level observed with a control immunoglobulin G antibody (data not shown). In order to circumvent this problem and to permit accurate normalization for antibody incubation, we generated stable transfectants of HEK 293 cells expressing mouse GAPDH fused to GFP or GFP only, under the control of a Tet-coupled switch (see Materials and Methods for details). Figure 3A (top) shows that the incubation with the Tet analog Dox induced the overexpression of GFP and GAPDH/GFP from the corresponding cell types, as observed by Western blotting. Immunoprecipitation with a specific anti-GFP antibody gave robust signals, as also shown in Fig. 3A (bottom). Under confocal microscopy, GAPDH/GFP displayed a cytosolic localization, in the same way as endogenous GAPDH. In contrast, control GFP distributed throughout the cell (data not shown). This observation, together with the fact that GAPDH/GFP associates with endogenous GAPDH, as it was also immunoprecipitated by the anti-GFP antibody, indicates that the GFP moiety does not significantly alter the behavior of the protein. We further transfected these cells transiently with luciferase constructs with or without the 3′ UTR of the ET-1 gene. As shown in Fig. 3B, reduced luciferase reporter expression was detected in the 3′ UTR-containing construct compared to the level for the control without this sequence (ΔUTR), in both GAPDH/GFP- and GFP-expressing cells. This result indicates that the mechanism for the control of mRNA expression by the 3′ UTR is also operative in HEK 293 cells and that the overexpression of GAPDH/GFP does not modify this effect, probably due to a high level of endogenous GAPDH expression (Fig. 3A, top right). Under these conditions, the detection of luciferase mRNA in the anti-GFP immunoprecipitates showed a significant enrichment of this RNA in GAPDH/GFP-overexpressing cells transfected with the 3′ UTR-containing construct, with little or no signal in cells transfected with luciferase-ΔUTR or in cells expressing GFP (Fig. 3C), a result which is compatible with the intracellular association between GAPDH and the 3′ UTR.
FIG. 3.

GAPDH binds to the 3′ UTR of ET-1 mRNA in vivo. (A) HEK 293 cells expressing mouse GAPDH fused to GFP and GFP only in a Tet-sensitive way were generated by stable transfection. GFP and GAPDH antigens were detected by immunoblotting of GFP- and GAPDH/GFP-expressing cells in the absence or presence of the Tet analog Dox (Input). Immunoprecipitation with a specific anti-GFP antibody and further detection of GFP and GAPDH by immunoblotting are also shown (IP-GFP). The positions of GAPDH/GFP, GFP only, and endogenous GAPDH are indicated at right. (B and C) Analysis of the luciferase (luc) mRNA expression of ET-1 promoter/luciferase/3′ UTR constructs in GAPDH/GFP- and GFP-expressing HEK 293 cells. Dox-treated cells were transiently transfected with ΔUTR and 3′ UTR luciferase reporters, and luciferase mRNA was detected by qRT-PCR. Values are expressed as percentages of luciferase mRNA for the ΔUTR construct for GFP-expressing cells normalized after detection of RNA polymerase 2A mRNA (means ± standard deviations, n = 6) (*, P < 0.01 compared with the ΔUTR value) (B). Immunoprecipitation with an anti-GFP antibody coupled with qRT-PCR detection of luciferase mRNA. Under these conditions, RNA polymerase 2A mRNA was not significantly detected over background levels. Luciferase mRNA expression is expressed as enrichment over control GFP-expressing cells transfected with luciferase-ΔUTR (means ± standard deviations, n = 6) (*, P < 0.05 compared with the corresponding ΔUTR value) (C). NT, not transfected.
The above-described data demonstrate that GAPDH is a major component interacting with the 3′ UTR of human ET-1 mRNA both in vitro and in vivo. This interaction occurs through specific binding to the AREs involved in the mRNA destabilization, which suggests a role for GAPDH in the control of ET-1 mRNA decay.
Endogenous GAPDH mediates ET-1 mRNA 3′ UTR-dependent downregulation of ET-1 expression.
In order to analyze whether GAPDH may play a role in the control of ET-1 expression, we manipulated the levels of the protein in EA.hy926 cells. Since GAPDH is an abundant protein, we opted to downregulate its endogenous levels by siRNA technology. We transfected EA.hy926 cells with specific GAPDH siRNA duplexes and compared their effects with results for a siRNA control. The efficiency of the endogenous GAPDH knockdown was checked by immunoblotting, as shown in Fig. 4A (a maximal reduction of 50% with 10 pmol siRNA per well). No effect on the expression of the unrelated protein β-actin was observed. Figure 4B and C also show that siRNA GAPDH transfection results in a similar reduction rate of GAPDH mRNA, as determined by RNase protection experiments. Under these conditions, we observed a marked increase in ET-1 mRNA expression levels (158.75 ± 24.98 for GAPDH siRNA, as a percentage of control siRNA), a result with is also seen at the level of the ET-1 peptide (Fig. 4D). Downregulation of GAPDH expression correlates with a moderate but significant reduction in the capacity of cytosolic extracts from EA.hy926 cells to bind radiolabeled RNA in gel shift experiments (62.35 ± 2.48 for GAPDH siRNA, as a percentage of control siRNA) (Fig. 4E and F). Cotransfection of GAPDH and control siRNAs together with luciferase constructs including the 3′ UTR of the human ET-1 mRNA revealed an increased reporter activity in GAPDH-silenced cells, an effect that was not observed with cells cotransfected with the same luciferase construct but without the 3′ UTR (ΔUTR) (Fig. 4G).
FIG. 4.

Selective downregulation of endogenous GAPDH enhances ET-1 expression by a 3′ UTR-dependent mechanism. (A) Immunoblot analysis of GAPDH expression in EA.hy926 cells silenced with siRNA GAPDH or control or untransfected (None) cells. β-Actin expression was also detected for the loading control. Values at the top show the results of densitometric quantification of GAPDH-immunoreactive bands. (B and C) Detection of ET-1 and GAPDH mRNA by RNase protection studies with siRNA-silenced cells. The positions of protected ET-1 and GAPDH mRNAs and 28S rRNA used for normalization purposes are indicated. tRNA (negative control), antisense probes, and a molecular size marker are also included. The autoradiogram shown corresponds to a representative RNase protection gel experiment performed twice with two independent preparations (B). Results of densitometric analyses of RNase protection studies (means ± standard deviations, n = 4) (*, P < 0.05 compared with the corresponding control) (C). (D) Effect of selective downregulation of GAPDH on ET-1 peptide released by EA.hy926 cells. Supernatants of cells transfected with siRNA GAPDH and control were assayed for the ET-1 peptide by specific ELISA (means ± standard deviations, n = 4) (*, P < 0.01 compared with the corresponding control). (E and F) RNA binding activity of cytosolic extracts from cells transfected with siRNA GAPDH or control to the 924-1127 probe. The RNA-EMSA gel shown corresponds to a representative experiment performed twice with two independent preparations (E). Results of densitometric analyses of RNA-EMSA studies (means ± standard deviations, n = 4) (*, P < 0.05 compared with the corresponding control) (F). (G) Analysis of the levels of luciferase expression of ET-1 promoter/luciferase/3′ UTR constructs in cells transfected with siRNA GAPDH and control. Values are expressed as percentages of luciferase activity for the corresponding ΔUTR construct/siRNA control after normalization with a Renilla control plasmid (means ± standard deviations, n = 6) (*, P < 0.01 compared with the corresponding ΔUTR value).
Taken together, these results indicate a significant role of endogenous GAPDH in the control of the expression of the ET-1 gene in EA.hy926 cells through a mechanism mediated by the 3′ UTR sequence of the ET-1 mRNA.
S glutathionylation of the catalytic cysteine residue (Cys 152) controls binding of GAPDH to AREs within the 3′ UTR of ET-1 mRNA: a redox-sensitive mechanism for the regulation of ET-1 mRNA stability by GAPDH.
We further examined the requirements for the binding of GAPDH to 3′ UTR mRNA. For that purpose, we generated deletion mutant constructs of GAPDH covering catalytic and NAD+-binding domains fused to GST (Fig. 5A and B) and tested their capacities to interact with radiolabeled RNA in gel shift experiments. Figure 5A shows that only the full-length construct (GST fused to GAPDH 2-335) bound to the RNA probe. Neither the catalytic domain nor the NAD+-binding domain on its own was able to show any significant binding capacity, and the same negative result was observed with the GST partner alone. The fact that a full-length configuration of the protein is required for the binding to RNA suggests the participation of both catalytic and NAD+-binding domains at the active site of the enzyme. In order to verify this hypothesis, we performed RNA-EMSA with GST-GAPDH full-length, radiolabeled RNA and increasing concentrations of the substrate G3P and the cofactor NAD+. Figure 5C shows efficient inhibition of binding to RNA by both G3P and NAD+ at the millimolar or submillimolar range, comparable to Km values for both molecules in most mammalian GAPDH enzymes. The fact that these natural substrates are able to compete efficiently with RNA binding suggests that the interaction takes place within the active site of the protein.
FIG. 5.

Requirements for the binding of GAPDH to the 3′ UTR of ET-1 mRNA, and effects of GSNO and GSSG on the RNA binding capacity. (A) Full-length (2-335) and deletion mutant constructs of GAPDH covering catalytic and NAD+-binding domains fused to GST were generated, and the RNA binding capacities of 0.5 and 1 μg of the corresponding recombinant protein were assayed by RNA-EMSA with the radiolabeled 924-1127 probe. (B) Schematic representation of GAPDH constructs used in this study. Black bars indicate the catalytic domain, and white bars indicate the NAD+-binding domain. The position of the catalytically active residue Cys 152 changed to serine in the C152S mutant is shown by the red X. (C) Competition of the RNA binding capacity of GST-GAPDH (2-335) by the substrate G3P and the cofactor NAD+. The effect of increasing concentrations of G3P and NAD+ was tested by RNA-EMSA with the 924-1127 probe. The results of a densitometric analysis of RNA-EMSA gels performed with the radiolabeled 924-1127 probe are shown. (D to G) RNA binding capacities of wild-type and C152S mutant GST-GAPDH treated with increasing concentrations of GSNO (D and E) or GSSG (F and G). Representative autoradiograms show the effects of GSNO (D) and GSSG (F) on the binding of wild-type and mutant GST-GAPDH and of rabbit muscle GAPDH to radiolabeled 924-1127 probe, by RNA-EMSA. Results of densitometric analysis of RNA-EMSA gels were used to determine the effects of GSNO (E) and GSSG (G). Values are shown as percentages of the control in the absence of competitor or pretreatment and represent densitometric analysis of experiments performed twice with two independent preparations giving essentially the same results (means ± standard deviations, n = 4). (H) GAPDH activities associated with the GST-GAPDH wild type and C152S mutant, and effects of increasing concentrations of GSSG and GSNO on GAPDH activity from the GST-GAPDH wild type. Values are shown as percentages of the control without treatment from experiments performed with 1 μg of protein (means ± standard deviations, n = 4).
GAPDH is comprised of four identical 37-kDa subunits. Each subunit contains four cysteines; two of them (Cys 152 and Cys 156) are located in the catalytic site of each GAPDH subunit. The catalytically active residue Cys 152 interacts with a histidine to form a highly reactive thiolate group (Cys-S−), which is required for GAPDH activity and, interestingly, is the target for specific posttranslational modifications by oxidants. To examine whether a potential modification of the catalytic cysteine may influence the ability of GAPDH to bind AREs within the 3′ UTR of ET-1 mRNA, we generated a mutant construct of full-length GST-GAPDH, replacing cysteine with serine at position 152 (Fig. 5B). This change prevents the thiol modification by oxidants from occurring, without significantly impairing the protein structure. Both the wild type and the C152S mutant bind equally to RNA, suggesting that the serine residue accommodates the structure without affecting the binding to RNA (Fig. 5D and F). Accumulating evidence suggests that NO may play a role in the redox control of cellular activities through the specific modification of redox-sensitive thiols (36). The low-molecular-weight nitrosothiol GSNO is well known to mimic the actions of NO as a reactive species and has been described to modify GAPDH by S nitrosylation and S glutathionylation. We analyzed the RNA binding capacities of both wild-type and C152S mutant GAPDH after treatment with increasing concentrations of GSNO. Figure 5D and E show that the wild-type enzyme was particularly sensitive to the action of GSNO, much in the same manner as rabbit muscle GAPDH, whose interaction with RNA was abrogated at concentrations of ≥7 mM. On the contrary, C152S mutant binding was not significantly altered. We performed the same experiments with GSSG, which allows the incorporation of glutathione to cysteine but does not induce S nitrosylation. As found with GSNO, wild-type GAPDH was more sensitive to the action of GSSG than the C152S mutant (Fig. 5F and G). Binding to RNA of rabbit muscle GAPDH was also inhibited by GSSG. The chemical alteration of the cysteine residue was further corroborated by direct measurement of the enzymatic activity (conversion of G3P to 1,3-diphosphoglycerate in the presence of NAD+ and phosphate). GSNO and GSSG caused a concentration-dependent inhibition of GAPDH activity in the wild-type enzyme (Fig. 5H). The C152S mutant showed residual activity compared to that of the wild type, as this cysteine residue is essential for enzymatic activity. Taken together, these results suggest that both compounds may alter the interaction to RNA by covalent incorporation of glutathione to Cys 152. Considering that binding experiments were performed in the presence of 5 mM DTT and that GST fusion proteins were used as crude preparations from reduced glutathione (GSH)-resin eluates (containing about 1 mM reduced GSH in the binding buffer), concentrations of GSNO or GSSG required to see binding impairment (above 7 mM) are within the range of the physiological concentration of the redox pair GSH/GSSG present in mammalian cells (1 to 10 mM) and may represent conditions of oxidative stress, with the equilibrium clearly displaced to GSSG.
In agreement with published observations, we further confirmed that under our experimental conditions GSSG induced the formation of a mixed disulfide to the enzyme in a redox-sensitive manner. Figure 6A shows that treatment of rabbit muscle GAPDH with biotin-labeled GSSG resulted in a covalent, DTT-sensitive incorporation of biotin to the protein, as visualized with streptavidin-HRP. Both wild-type and C152S mutant GST-GAPDH were labeled after treatment with biotin-GSSG. The overall labeling of wild-type GST-GAPDH was higher than that of the C152S mutant, a result that suggests that specific incorporation to Cys 152 occurs and that additional cysteines of the molecule are also targets for the modification (Fig. 6B). The extent of glutathionylation induced by GSNO and GSSG was also estimated by monobromobimane derivatization and HPLC coupled with fluorescence detection (11). Both GSNO and GSSG were able to modify rabbit muscle GAPDH at the concentration range used in binding experiments (Fig. 6C), an observation that was also confirmed by trypsin digestion and mass spectrometry using a quadrupole ion trap (data not shown).
FIG. 6.

In vitro and in vivo S glutathionylation of GAPDH. (A and B) In vitro treatment of rabbit muscle GAPDH (5 μg) with increasing concentrations of biotin-GSSG (from 2 μM to 2 mM) in the presence and absence of 5 mM DTT (A, top). The effect of 2 mM biotin-GSSG on wild-type and C152S GST-GAPDH (5 μg) is also shown (B, top). Biotin labeling was revealed with streptavidin-HRP. Numbers in parentheses refer to the relative intensities of the bands after densitometric analysis. Aliquots of the same set of protein samples were analyzed by Coomassie staining for control loading purposes (A and B, bottom). (C) S glutathionylation of rabbit muscle GAPDH by GSNO and GSSG, analyzed by monobromobimane derivatization and HPLC coupled with fluorescence detection (means ± standard deviations, n = 6). prot, protein. (D) Analysis of S glutathionylation of GAPDH in vivo. The intracellular pool of GSH was labeled by incubation of EA.hy926 cells with bioGEE before incubation with 60 μM H2O2. Total cell extracts or immunoprecipitated (IP) GAPDH was analyzed by immunoblotting with anti-GAPDH antibody and streptavidin-HRP. The effect of the preincubation with the antioxidant N-acetylcysteine (NAC) (10 mM) is also shown. The blots shown correspond to representative experiments performed twice with two independent preparations. The positions of the GAPDH and GST-GAPDH proteins are indicated by arrowheads in panels A, B, and D.
S glutathionylation can be induced by incubation with H2O2 in cellular systems. We examined whether the incubation of EA.hy926 cells with H2O2 results in the incorporation of glutathione to GAPDH under our experimental conditions. For that purpose, we labeled the intracellular pool of GSH by preincubation of cells with bioGEE (1 h at 178 μM). Then, cells were treated with 60 μM H2O2 for 2 h, and endogenous GAPDH was immunoprecipitated with an anti-GAPDH antibody. Immunoprecipitates were then separated by SDS-PAGE, transferred to blots, and probed with streptavidin-HRP. As shown in Fig. 6D, basal labeling of endogenous GAPDH was greatly increased upon incubation of cells with H2O2. Both basal labeling and H2O2-induced labeling of GAPDH were inhibited by preincubation of cells with the antioxidant N-acetylcysteine. In another set of similar experiments, immunoprecipitates were probed with a specific antibody against GSH, and essentially the same results were found (data not shown). These results indicate that treatment of EA.hy926 cells with H2O2 results in specific S glutathionylation of GAPDH in vivo.
Considering that in vitro S glutathionylation affects binding to AREs within the 3′ UTR of ET-1 mRNA and that incubation of cells with H2O2 induces thiolation of endogenous GAPDH, we then examined whether treatment with H2O2 may affect ET-1 mRNA expression by such a mechanism. First, we checked whether oxidant stress with H2O2 affects ET-1 mRNA expression. Figure 7A and B show that H2O2 incubation induces time- and concentration-dependent increases in ET-1 mRNA levels (2- to 2.5-fold increases at 2 h and 60 to 100 μM). We also analyzed the effect of the treatment with H2O2 on the reporter expression from constructs containing the 3′ UTR of human ET-1 mRNA. As shown in Fig. 7C, H2O2 dose-dependently increased the luciferase expression from a construct including the 3′ UTR, without significantly affecting the expression from the ΔUTR construct, a result suggesting the impairment by H2O2 of the capacity of the 3′ UTR to regulate mRNA expression. In order to confirm that the effect of H2O2 occurs through the modulation of the mRNA stability, we estimated decay constants of luciferase reporters fused to 3′ UTR or ΔUTR in Tet-regulated, stably transfected HEK 293 cells by a method developed by our group and recently published (43). Luciferase activity and mRNA were measured from luciferase-3′ UTR and luciferase-ΔUTR stable clones before and after induction with Dox for time periods ranging from 2 h to 72 h. Decay constants of 3′ UTR and ΔUTR mRNA species in cells treated with 100 μM H2O2 or under basal conditions can be estimated from the Dox induction kinetics shown in Fig. 7D (see Materials and Methods for details). Careful analysis of these parameters clearly showed that the half-life values for ΔUTR mRNA were not substantially altered by oxidative conditions, whereas those for the intact 3′ UTR increased significantly after treatment with H2O2.
FIG. 7.

ET-1 mRNA expression is regulated at the level of transcript stability by oxidant stress in a 3′ UTR-dependent manner. (A and B) Time- and concentration-dependent effects of H2O2 on ET-1 mRNA expression in EA.hy926 cells. Results of densitometric analyses of RNase protection studies are shown (means ± standard deviations, n = 4) (*, P < 0.01 compared with the corresponding control in the absence of H2O2). (C) Analysis of the luciferase expression of ET-1 promoter/luciferase/3′ UTR constructs in cells treated with increasing concentrations of H2O2. Values are expressed as percentages of luciferase activity for the corresponding ΔUTR construct without treatment after normalization with a Renilla control plasmid (means ± standard deviations, n = 6) (*, P < 0.01 compared with the corresponding ΔUTR value). (D) Estimation of luciferase-3′ UTR and -ΔUTR mRNA half-lives in Tet-inducible HEK 293 cells. Stable isogenic transfectants of HEK 293 cells exhibiting Tet-inducible expression of luciferase reporters fused to the ET-1 3′ UTR fragment or ΔUTR were generated as described in Materials and Methods. The time course of luciferase induction after addition of Dox to the cultures was determined by luminometry with cells pretreated with 100 μM H2O2 for 2 h. Activity is expressed as normalized units (n.u.) (means ± standard deviations, n = 4; normalization with total protein determination). Induction kinetics were fit to an exponential function to estimate the decay constants shown at the bottom. These decay constants allow calculation of the corresponding apparent half-lives of luciferase protein after subtracting the 3-h value from these values of the mRNA half-lives (see Materials and Methods and Results for details). (E) Establishment of a stable HEK 293 cell line expressing mouse C152S mutant GAPDH/GFP in a Tet-regulated way. Levels of GAPDH/GFP and endogenous GAPDH expression from wild-type and mutant (mut) cell lines were analyzed by immunoblotting in the absence or presence of Dox (and H2O2). S glutathionylation of GAPDH/GFP fusion proteins and endogenous GAPDH was studied by labeling of the intracellular pool of GSH with bioGEE and immunoprecipitation with an anti-GFP antibody. Note that, as shown in Fig. 3, the anti-GFP antibody is able to pull down GAPDH/GFP fusion proteins and the associated endogenous GAPDH. Biotin-GSH labeling was revealed with streptavidin-HRP (top right, representative blot; bottom right, densitometric analysis of biotin-GSH incorporation [means ± standard deviations, n = 3] [*, P < 0.01 compared with the corresponding control in the absence of H2O2]). (F) Analysis of levels of luciferase expression of transiently transfected luciferase-3′ UTR and -ΔUTR constructs in wild-type and C152S mutant HEK 293 cells incubated in the absence or presence of 100 μM H2O2. Values are expressed as percentages of luciferase activity for the corresponding ΔUTR construct without treatment after normalization with a Renilla control plasmid (means ± standard deviations, n = 6) (*, P < 0.01 compared with the corresponding ΔUTR value).
We also investigated whether this effect of H2O2 takes place through a mechanism eventually involving the S thiolation of Cys 152 of GAPDH. For that purpose, we also generated stable transfectants of HEK 293 cells expressing a mouse GAPDH/GFP fusion protein in which cysteine was replaced by serine at position 152, and we compared the capacities of H2O2 to induce S glutathionylation of GAPDH and to modulate mRNA stability in HEK 293 cells overexpressing wild-type versus C152S mutant GAPDH/GFP. Figure 7E (left) shows that both cell lines display similar GAPDH/GFP levels after induction with Dox and that this expression is not altered by treatment with H2O2. S thiolation of GAPDH forms was estimated by labeling the intracellular pool of GSH by preincubation of cells with bioGEE and further immunoprecipitation of GAPDH/GFP with an anti-GFP antibody, followed by detection of biotin-glutathione incorporation with streptavidin-HRP. As also shown in Fig. 7E (right, Western blot and densitometric analyses), basal labeling of GAPDH/GFP was significantly increased in H2O2-treated cells expressing wild-type GAPDH/GFP but not in those expressing the C152S mutant. In contrast, endogenous GAPDH, which was coimmunoprecipitated with the anti-GFP antibody, showed similar labeling increments in both the wild-type and mutant cell lines. By transfection of luciferase-3′ UTR and ΔUTR constructs, we also studied whether the impairment by H2O2 of the capacity of the 3′ UTR to regulate mRNA stability is altered in cells overexpressing mutant GAPDH/GFP. As shown in Fig. 7F, whereas the treatment with H2O2 induced an increase in the expression of luciferase-3′ UTR in wild-type cells, the C152S mutant cell line was refractory to the effect of H2O2. No alteration in the expression of luciferase-ΔUTR was observed.
Therefore, our results indicate that oxidant stress increases ET-1 mRNA expression in a 3′ UTR-dependent manner through a mechanism which implies specific S glutathionylation of GAPDH Cys 152 and alteration of binding to AREs in the 3′ UTR of the ET-1 gene.
RNA unwinding as a potential mechanism for GAPDH-mediated RNA destabilization.
To our knowledge, there is no previous report describing RNase activity associated with the GAPDH enzyme, and our preliminary experiments also confirmed this point (data not shown). Therefore, it is of interest to analyze how this protein may induce destabilization of ET-1 mRNA. In order to study the contribution of GAPDH to the degradation rate of this RNA, we exposed the radiolabeled, 5′-capped, 3′-polyadenylated 924-1127 RNA probe to cytosolic extracts from EA.hy926 cells transfected with siRNA GAPDH and control. The integrity of the remaining radiolabeled RNA probe was assessed by denaturing electrophoresis coupled with autoradiography after normalization of the signals with a stable β-globin RNA included in the assay as an internal control. Figure 8A and B show that cytosolic extracts from siRNA GAPDH-silenced cells degrade the RNA probe at a significantly lower rate than do cells transfected with the siRNA control. These results indicate a significant role for endogenous GAPDH in the regulation of mRNA stability in this cell assay. Additionally, we analyzed whether the pretreatment of EA.hy926 cytosolic extracts with GSSG alters ET-1 mRNA decay. As shown in Fig. 8C and D, in vitro GSSG treatment drastically reduced the capacity of the extracts to degrade ET-1 mRNA. In order to analyze whether GAPDH is involved in this effect of GSSG, RNA degradation experiments were performed with cytosolic extracts from HEK 293 cells overexpressing wild-type or C152S mutant GAPDH/GFP. Figure 8E and F show that GSSG pretreatment inhibited the degradation of ET-1 mRNA in extracts from wild-type cells but not from C152S mutant cells, an observation demonstrating the role of GAPDH in the control of the mRNA stability by a redox-sensitive mechanism.
FIG. 8.


RNA unwinding as a potential mechanism for GAPDH-mediated mRNA destabilization. (A and B) The radiolabeled, 5′-capped, 3′-polyadenylated 924-1127 RNA probe was exposed to cytosolic extracts from EA.hy926 cells transfected with siRNA GAPDH or control. After the indicated times, reaction volumes were deproteinized and the integrity of the remaining radiolabeled RNA was assayed by denaturing electrophoresis and autoradiography. Values shown in panel B are percentages of the remaining RNA estimated by densitometric analysis after normalization of the signals with a stable β-globin RNA (globin) included in the assay as an internal control (means ± standard deviations, n = 3) (*, P < 0.01 compared with the corresponding value for the siRNA control). (C and D) RNA degradation assays performed with EA.hy926 cytosolic extracts pretreated in vitro in the absence or presence of 2 mM GSSG. After the indicated times, the remaining radiolabeled RNA was assayed as described above (C, representative gel; D, densitometric analyses [means ± standard deviations, n = 3] [*, P < 0.01 compared with the corresponding control in the absence of GSSG]). (E and F) RNA degradation assays performed with wild-type and C152S mutant HEK 293 cytosolic extracts pretreated in vitro in the absence or presence of 2 mM GSSG. After the indicated times, the remaining radiolabeled RNA was assayed as described above (E, representative gels; F, densitometric analyses [means ± standard deviations, n = 3] [*, P < 0.01 compared with the corresponding control in the absence of GSSG]). (G) Secondary structure predicted for the region of the 3′ UTR from 932-1120. The multiple alignment of 3′ UTR ET-1 sequences from human, mouse, pig, cattle, and dog was computed with ClustalW (left), and the predicted structure was obtained by running the aligned sequences with the Alifold algorithm, which considers the phylogenetic information as well as the thermodynamic stability of a set of aligned RNA molecules (right). Consistent mutations are shown by red circles around the varying positions. Inconsistent mutants are indicated by blue instead of black lettering. The positions of the AREs shown to participate in the binding to GAPDH are also indicated. (H, I, and J) Fluorometric RNA unwinding assays performed with the RNA probe and increasing concentrations of rabbit muscle GAPDH. Real-time fluorescence signals from DAPI were recorded before the addition of RNA and GAPDH (arrows) (a.u., arbitrary units). The effect of GAPDH on the intrinsic fluorescence of DAPI in the absence of RNA was also studied (bottom set of lines) (H). The fluorescence decrease was estimated as the fraction of initial fluorescence corresponding to the addition of RNA and is represented as a function of the concentration of GAPDH (I). The RNA unwinding capacity of GAPDH treated with increasing concentrations of GSSG is also shown (J). The values shown correspond to representative experiments performed twice with two independent preparations giving essentially the same results. (K) Model for the redox-sensitive regulation of mRNA stability by GAPDH. In the absence of an oxidative insult, native GAPDH interacts with the ET-1 mRNA through certain AREs in the 3′ UTR and disrupts the structure of the 3′ UTR mRNA, contributing to accelerated degradation. When Cys 152 of GAPDH is S glutathionylated, binding to ET-1 mRNA cannot occur, and this is associated with stabilization of the mRNA.
Previous reports of GAPDH binding to AU-rich RNAs have described rearrangements of the RNA structure, with the loss of double-stranded-RNA regions upon interaction (15, 47). It is therefore plausible to hypothesize that GAPDH interaction with ET-1 mRNA facilitates the degradation of the mRNA by modification of the RNA structure at the 3′ UTR, making it more susceptible to attack by cytosolic ribonucleases. The distal portion of the ET-1 3′ UTR is highly conserved among different species, including human, mouse, pig, cattle, and dog, as shown in the ClustalW alignment depicted in Fig. 8G (left), a fact suggesting that this sequence may adopt a characteristic RNA secondary structure that is conserved in evolution. Computer-assisted RNA folding utilities such as the Alifold program, which combine the thermodynamic stability with the phylogenetic information contained in the sequence covariations of a collection of RNA molecules, are particularly interesting to use to search for a conserved secondary structure(s) (25). By applying the Alifold algorithm to this collection of ET-1 3′ UTR sequences, an evolutionary conserved and thermodynamically stable RNA secondary structure was predicted, with a high content of the double-stranded helical region (Fig. 8G, right). In order to determine whether this region actually may contain a significant percentage of duplex RNA and whether GAPDH may eventually disrupt it, we performed a fluorometric assay based on the displacement of a fluorescent dye from double-stranded RNA upon unwinding (19). DAPI, a well-characterized fluorescent probe for nucleic acids, exhibits significant fluorescence enhancement when bound to duplex RNA. As shown in Fig. 8H, the addition of an RNA probe (58 nM) to a solution of DAPI (1 μM) resulted in a strong increase in fluorescence, thus indicating a significant portion of double-stranded RNA in the structure. Interestingly, increasing amounts of rabbit muscle GAPDH induced a significant reduction in the fluorescence from DAPI, likely coming from the displacement of the dye by local RNA unwinding (Fig. 8H and I). This effect on DAPI fluorescence was observed only when RNA was present and was not observed with similar amounts of a control protein, such as BSA (data not shown). Additionally, we studied the effect of pretreatment of the protein with GSSG on the capacity of GAPDH to unwind RNA. As shown in Fig. 8J, the incubation of the protein with increasing concentrations of GSSG reversed the reduction of fluorescence observed with intact GAPDH, an effect which is compatible with the covalent modification of Cys 152 and the alteration of the binding to RNA. Overall, the above-described data are consistent with GAPDH inducing the conversion of helical segments to unpaired strands. Hence, GAPDH binding to RNA may be coupled with the facilitation of RNA degradation by cellular ribonucleases.
DISCUSSION
In an earlier study, we showed that endothelial ET-1 expression is under the control of a mechanism based on the mRNA-destabilizing capacity of specific cytosolic proteins through interaction with AREs present in the 3′ UTR of the ET-1 gene (43). Our current work has identified by means of RNA affinity chromatography the glycolytic enzyme GAPDH as the major component interacting with AREs of the 3′ UTR of ET-1 mRNA. The results presented here also show that GAPDH plays an essential role in the control of ET-1 mRNA decay. (i) RNA binding experiments and immunoprecipitation with mRNA coupled with qRT-PCR show that GAPDH binds to ET-1 mRNA both in vitro and in vivo. Quantitative analysis of RNA binding gave an apparent dissociation constant of 0.98 μM, which is within the range of the cellular concentration of GAPDH (1 to 5 μM), as described previously (52). This interaction occurs through specific binding to AREs previously reported to control the expression of the gene. (ii) siRNA-based downregulation of endogenous GAPDH results in an increase in ET-1 gene expression, and this occurs through a 3′ UTR-dependent mechanism by mRNA stabilization. ET-1 expression in the endothelium is regulated through both transcriptional and posttranscriptional mechanisms (26, 27, 31, 37, 43, 45). GAPDH-mediated destabilization of mRNA expression might serve as a mechanism conferring to the ET-1 gene the capacity to respond rapidly to changes in transcriptional activity. This is important in the context of vascular pathophysiology, as the endothelium senses a variety of stimuli and needs to be able to activate specific responses to them, such as fine regulation of ET-1 biosynthesis.
For many years, GAPDH has been regarded only as the enzyme catalyzing the sixth step in glycolysis, the process converting glucose into pyruvate. However, a number of independent studies have recently reported a variety of diverse biological properties for this protein, including playing roles in apoptosis, DNA replication and repair, endocytosis, and mRNA regulation (52). Concerning RNA metabolism, earlier studies identified GAPDH as a specific RNA binding protein. RNA targets for this activity include AREs in the 3′ UTRs of lymphokine gamma interferon, c-myc, granulocyte-macrophage colony-stimulating factor (41), and colony-stimulating factor 1 (5) mRNAs; 5′ UTR and 3′ UTR stem-loops of hepatitis A virus (47); the 3′ genome sequence element of human parainfluenza I virus type 3 (14); and, very recently, the negative strand of tomato bushy stunt virus (55). These observations prompted authors to hypothesize that GAPDH could regulate mRNA or viral RNA turnover. Our work extends these observations to the ET-1 gene transcript and unveils a clear role for GAPDH in the control of mRNA stability.
GAPDH is a particularly interesting protein as it represents the archetypal example of covalent modification by oxidants at its highly reactive Cys 152 residue. The protein undergoes S nitrosylation by NO (22, 40), NAD+ covalent linkage upon S nitrosylation (40), nitroalkylation by nitrated fatty acids (3), S glutathionylation by glutathione or even by NO (39), and extensive oxidation by peroxynitrite or H2O2 (33, 53). Among these modifications, S glutathionylation, the formation of mixed disulfides between glutathione and the cysteine of some proteins, has attracted great interest, as it has recently emerged as a potential mechanism for the regulation of signaling and metabolic pathways under both physiological and pathological conditions (13, 36). We describe herein that the in vitro treatment of GAPDH with the NO donor GSNO or with GSSG impaired the capacity of the protein to interact with RNA. Whereas other covalent modifications may also be responsible for the observed effect, S glutathionylation seems the most likely. Results supporting this statement have been obtained both in vitro and in vivo. (i) Treatment of purified proteins with a biotin-labeled glutathione derivative, biotin-GSSG, resulted in DTT-sensitive biotin incorporation. Using an alternative approach based on monobromobimane derivatization and HPLC coupled with fluorescence detection, we observed specific S glutathionylation by GSSG and GSNO at the same range of concentrations able to alter RNA binding. This result was also confirmed by trypsin digestion and mass spectrometry. (ii) Oxidative challenge of cells with H2O2 has previously been described to induce S glutathionylation of intracellular proteins (42, 49). In the work presented here, biotin labeling of the intracellular pool of GSH and incubation with H2O2 resulted in specific biotin incorporation of numerous proteins, among them GAPDH. Interestingly, S glutathionylation correlates with an impaired RNA binding capacity and a 3′ UTR-dependent increase in ET-1 mRNA expression. Therefore, our results are compatible with a mechanism for the control of ET-1 expression based on the capacity of GAPDH to destabilize the mRNA. This mechanism is redox sensitive and involves specific S glutathionylation of Cys 152. Replacement of this cysteine residue with serine prevents the modification and, therefore, makes the protein insensitive to the redox regulation. The existence of this mechanism of control may shed light on previous observations of upregulated ET-1 expression upon exposure of endothelial cells to oxidants (46, 51).
The regulation of the mRNA-destabilizing capacity of GAPDH by oxidative and nitrosative stress presents an extremely interesting scenario regarding the endothelium, where the contribution of NO and oxidants to the vascular pathophysiology is important (24, 48). Indeed, it has long been described that GAPDH functions as an NO and oxidant stress sensor (23). Very recently, new unpredicted roles for GAPDH, including the control of cell death by apoptosis in different cell systems, have been reported (1, 9, 16, 22). According to these reports, GAPDH may couple the cellular energy status with the capacity of the cells to survive. NO-mediated S nitrosylation of GAPDH has been described to promote apoptotic cell death (22, 50). Our results add a new level of complexity to the cellular roles of GAPDH, as they also suggest the existence of a functional link between the regulation of the biosynthesis of vasoactive factors like ET-1 and the redox balance and, in light of the above-mentioned reports, cell energy homeostasis and survival. Actually, in our study, the fact that the GAPDH substrates G3P and NAD+ effectively inhibit binding to RNA suggests that the fraction of protein engaged in RNA interaction is unable to catalyze enzymatic activity and vice versa. Therefore, cell energy metabolism may indirectly influence ET-1 expression by controlling the amount of GAPDH capable of interacting with mRNA. In this respect, previous reports have described that the binding to AU-rich RNA and the glycolytic action of GAPDH occur in the same region of the protein, the Rossman fold, where the catalytic residue Cys 152 is located (7, 41), and that reactive oxygen species induce the release of GAPDH from AU-rich RNA binding sites (4). The model presented here may therefore explain how oxidant stress uncouples the capacity of the protein to interact with RNA.
Concerning the molecular mechanism by which GAPDH binding to RNA controls the stability of the transcript, our experiments show that the protein is unable to hydrolyze RNA per se but enhances the rate of degradation by cytosolic extracts. Therefore, it is likely that the GAPDH interaction with ET-1 mRNA facilitates the degradation of the mRNA by cytosolic ribonucleases. Computer-assisted structural analysis of the distal portion of the 3′ UTR of ET-1 mRNA using a set of aligned RNA sequences from different species predicts a significant content of double-stranded helical regions, which, according to our fluorometric experiments, become disturbed upon binding to GAPDH. This effect was abolished when the protein was pretreated with oxidant GSSG, a condition shown to covalently modify Cys 152. Published works have already reported that binding of GAPDH to AU-rich stem-loop structures results in the loss of double-stranded-RNA regions (15, 47). We hypothesize that GAPDH may function as an RNA anti-chaperone protein, promoting by RNA unwinding and hence facilitating the degradation of RNA by additional cellular components. In addition, this capacity of the protein is regulated by the redox state through covalent modification of the catalytic cysteine (Fig. 8K). According to current knowledge, the regulation of expression of ARE-containing mRNAs involves the participation of two well-defined degradative pathways. On one hand, the exosome, a large multiprotein complex first described for Saccharomyces cerevisiae, is responsible for the 3′-to-5′ exonucleolytic degradation of ARE-containing RNA (34). An alternative pathway of ARE-containing mRNA degradation occurs at processing bodies, cytoplasmic foci that contain decapping enzymes, and 5′-to-3′ exonucleases (54). How the GAPDH-based mechanism described here can be integrated into these degradative pathways remains unknown and is an objective of our current research.
Acknowledgments
This work was supported by grants SAF2006-02410 and Consolider CSD-2007-0020, Network of Excellence for the Research on Oxidative Stress in Spain (NEROSS), from Plan Nacional de I+D+I, CARDIOVREP, from Comunidad de Madrid to S.L.; by a grant from the Sociedad Española de Nefrología to S.L.; and by a grant from Fundación de Investigación Médica Mutua Madrileña to F.R.-P. Fernando Rodríguez-Pascual is the holder of a Ramon y Cajal contract from the Ministerio de Educación y Ciencia.
We thank J. Fernando Díaz (Centro de Investigaciones Biológicas, Madrid, Spain) and Magdalena Torres (Universidad Complutense de Madrid, Spain) for their technical assistance.
Footnotes
Published ahead of print on 22 September 2008.
REFERENCES
- 1.Almeida, B., S. Buttner, S. Ohlmeier, A. Silva, A. Mesquita, B. Sampaio-Marques, N. S. Osorio, A. Kollau, B. Mayer, C. Leao, J. Laranjinha, F. Rodrigues, F. Madeo, and P. Ludovico. 2007. NO-mediated apoptosis in yeast. J. Cell Sci. 1203279-3288. [DOI] [PubMed] [Google Scholar]
- 2.Arendt, R. M., U. Wilbert-Lampen, L. Heucke, M. Schmoeckel, K. Suhler, and W. O. Richter. 1993. Increased endothelin plasma concentrations in patients with coronary artery disease or hyperlipoproteinemia without coronary events. Res. Exp. Med. 193225-230. [DOI] [PubMed] [Google Scholar]
- 3.Batthyany, C., F. J. Schopfer, P. R. Baker, R. Duran, L. M. Baker, Y. Huang, C. Cervenansky, B. P. Branchaud, and B. A. Freeman. 2006. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 28120450-20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berry, M. D. 2004. Glyceraldehyde-3-phosphate dehydrogenase as a target for small-molecule disease-modifying therapies in human neurodegenerative disorders. J. Psychiatr. Neurosci. 29337-345. [PMC free article] [PubMed] [Google Scholar]
- 5.Bonafe, N., M. Gilmore-Hebert, N. L. Folk, M. Azodi, Y. Zhou, and S. K. Chambers. 2005. Glyceraldehyde-3-phosphate dehydrogenase binds to the AU-rich 3′ untranslated region of colony-stimulating factor-1 (CSF-1) messenger RNA in human ovarian cancer cells: possible role in CSF-1 posttranscriptional regulation and tumor phenotype. Cancer Res. 653762-3771. [DOI] [PubMed] [Google Scholar]
- 5a.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72248-254. [DOI] [PubMed] [Google Scholar]
- 6.Brennan, J. P., J. I. Miller, W. Fuller, R. Wait, S. Begum, M. J. Dunn, and P. Eaton. 2006. The utility of N,N-biotinyl glutathione disulfide in the study of protein S-glutathiolation. Mol. Cell. Proteomics 5215-225. [DOI] [PubMed] [Google Scholar]
- 7.Carlile, G. W., R. M. Chalmers-Redman, N. A. Tatton, A. Pong, K. E. Borden, and W. G. Tatton. 2000. Reduced apoptosis after nerve growth factor and serum withdrawal: conversion of tetrameric glyceraldehyde-3-phosphate dehydrogenase to a dimer. Mol. Pharmacol. 572-12. [PubMed] [Google Scholar]
- 8.Castañares, C., M. Redondo-Horcajo, N. Magan-Marchal, P. ten Dijke, S. Lamas, and F. Rodriguez-Pascual. 2007. Signaling by ALK5 mediates TGF-beta-induced ET-1 expression in endothelial cells: a role for migration and proliferation. J. Cell Sci. 1201256-1266. [DOI] [PubMed] [Google Scholar]
- 9.Colell, A., J. E. Ricci, S. Tait, S. Milasta, U. Maurer, L. Bouchier-Hayes, P. Fitzgerald, A. Guio-Carrion, N. J. Waterhouse, C. W. Li, B. Mari, P. Barbry, D. D. Newmeyer, H. M. Beere, and D. R. Green. 2007. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 129983-997. [DOI] [PubMed] [Google Scholar]
- 10.Conne, B., A. Stutz, and J. D. Vassalli. 2000. The 3′ untranslated region of messenger RNA: a molecular ‘hotspot’ for pathology? Nat. Med. 6637-641. [DOI] [PubMed] [Google Scholar]
- 11.Cotgreave, I. A., and P. Moldeus. 1986. Methodologies for the application of monobromobimane to the simultaneous analysis of soluble and protein thiol components of biological systems. J. Biochem. Biophys. Methods 13231-249. [DOI] [PubMed] [Google Scholar]
- 12.Chen, C. Y., and A. B. Shyu. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20465-470. [DOI] [PubMed] [Google Scholar]
- 13.Dalle-Donne, I., R. Rossi, D. Giustarini, R. Colombo, and A. Milzani. 2007. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 43883-898. [DOI] [PubMed] [Google Scholar]
- 14.De, B. P., S. Gupta, H. Zhao, J. A. Drazba, and A. K. Banerjee. 1996. Specific interaction in vitro and in vivo of glyceraldehyde-3-phosphate dehydrogenase and LA protein with cis-acting RNAs of human parainfluenza virus type 3. J. Biol. Chem. 27124728-24735. [DOI] [PubMed] [Google Scholar]
- 15.Dollenmaier, G., and M. Weitz. 2003. Interaction of glyceraldehyde-3-phosphate dehydrogenase with secondary and tertiary RNA structural elements of the hepatitis A virus 3′ translated and non-translated regions. J. Gen. Virol. 84403-414. [DOI] [PubMed] [Google Scholar]
- 16.Du, Z. X., H. Q. Wang, H. Y. Zhang, and D. X. Gao. 2007. Involvement of glyceraldehyde-3-phosphate dehydrogenase in tumor necrosis factor-related apoptosis-inducing ligand-mediated death of thyroid cancer cells. Endocrinology 1484352-4361. [DOI] [PubMed] [Google Scholar]
- 17.Eberhardt, W., A. Doller, E.-S. Akool, and J. Pfeilschifter. 2007. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 11456-73. [DOI] [PubMed] [Google Scholar]
- 18.Edgell, C. J., C. C. McDonald, and J. B. Graham. 1983. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc. Natl. Acad. Sci. USA 803734-3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eggleston, A. K., N. A. Rahim, and S. C. Kowalczykowski. 1996. A helicase assay based on the displacement of fluorescent, nucleic acid-binding ligands. Nucleic Acids Res. 241179-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garneau, N. L., J. Wilusz, and C. J. Wilusz. 2007. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8113-126. [DOI] [PubMed] [Google Scholar]
- 21.Gherzi, R., K. Y. Lee, P. Briata, D. Wegmuller, C. Moroni, M. Karin, and C. Y. Chen. 2004. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell 14571-583. [DOI] [PubMed] [Google Scholar]
- 22.Hara, M. R., N. Agrawal, S. F. Kim, M. B. Cascio, M. Fujimuro, Y. Ozeki, M. Takahashi, J. H. Cheah, S. K. Tankou, L. D. Hester, C. D. Ferris, S. D. Hayward, S. H. Snyder, and A. Sawa. 2005. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7665-674. [DOI] [PubMed] [Google Scholar]
- 23.Hara, M. R., M. B. Cascio, and A. Sawa. 2006. GAPDH as a sensor of NO stress. Biochim. Biophys. Acta 1762502-509. [DOI] [PubMed] [Google Scholar]
- 24.Hare, J. M., and J. S. Stamler. 2005. NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Investig. 115509-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofacker, I. L., M. Fekete, and P. F. Stadler. 2002. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 3191059-1066. [DOI] [PubMed] [Google Scholar]
- 26.Inoue, A., M. Yanagisawa, Y. Takuwa, Y. Mitsui, M. Kobayashi, and T. Masaki. 1989. The human preproendothelin-1 gene. Complete nucleotide sequence and regulation of expression. J. Biol. Chem. 26414954-14959. [PubMed] [Google Scholar]
- 27.Kawana, M., M. E. Lee, E. E. Quertermous, and T. Quertermous. 1995. Cooperative interaction of GATA-2 and AP1 regulates transcription of the endothelin-1 gene. Mol. Cell. Biol. 154225-4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khabar, K. S. 2005. The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J. Interferon Cytokine Res. 251-10. [DOI] [PubMed] [Google Scholar]
- 29.Krebs, E. G. 1955. Glyceraldehyde-3-phosphate dehydrogenase from yeast, p. 407-411. In S. P. Colowick and N. O. Kaplan (ed.), Methods in enzymology, vol. 1. Academic Press, New York, NY. [Google Scholar]
- 30.Lai, W. S., E. Carballo, J. R. Strum, E. A. Kennington, R. S. Phillips, and P. J. Blackshear. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 194311-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee, M. E., M. S. Dhadly, D. H. Temizer, J. A. Clifford, M. Yoshizumi, and T. Quertermous. 1991. Regulation of endothelin-1 gene expression by Fos and Jun. J. Biol. Chem. 26619034-19039. [PubMed] [Google Scholar]
- 32.Lerman, A., B. S. Edwards, J. W. Hallett, D. M. Heublein, S. M. Sandberg, and J. C. Burnett, Jr. 1991. Circulating and tissue endothelin immunoreactivity in advanced atherosclerosis. N. Engl. J. Med. 325997-1001. [DOI] [PubMed] [Google Scholar]
- 33.Little, C., and P. J. O'Brien. 1969. Mechanism of peroxide-inactivation of the sulphydryl enzyme glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 10533-538. [DOI] [PubMed] [Google Scholar]
- 34.Liu, Q., J. C. Greimann, and C. D. Lima. 2006. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 1271223-1237. [DOI] [PubMed] [Google Scholar]
- 35.Ma, W. J., S. Cheng, C. Campbell, A. Wright, and H. Furneaux. 1996. Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem. 2718144-8151. [DOI] [PubMed] [Google Scholar]
- 36.Martinez-Ruiz, A., and S. Lamas. 2007. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc. Res. 75220-228. [DOI] [PubMed] [Google Scholar]
- 37.Mawji, I. A., G. B. Robb, S. C. Tai, and P. A. Marsden. 2004. Role of the 3′-untranslated region of human endothelin-1 in vascular endothelial cells. Contribution to transcript lability and the cellular heat shock response. J. Biol. Chem. 2798655-8667. [DOI] [PubMed] [Google Scholar]
- 38.Miyauchi, T., and T. Masaki. 1999. Pathophysiology of endothelin in the cardiovascular system. Annu. Rev. Physiol. 61391-415. [DOI] [PubMed] [Google Scholar]
- 39.Mohr, S., H. Hallak, A. de Boitte, E. G. Lapetina, and B. Brune. 1999. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 2749427-9430. [DOI] [PubMed] [Google Scholar]
- 40.Mohr, S., J. S. Stamler, and B. Brune. 1996. Posttranslational modification of glyceraldehyde-3-phosphate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J. Biol. Chem. 2714209-4214. [DOI] [PubMed] [Google Scholar]
- 41.Nagy, E., and W. F. Rigby. 1995. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold). J. Biol. Chem. 2702755-2763. [DOI] [PubMed] [Google Scholar]
- 42.Niture, S. K., C. S. Velu, N. I. Bailey, and K. S. Srivenugopal. 2005. S-thiolation mimicry: quantitative and kinetic analysis of redox status of protein cysteines by glutathione-affinity chromatography. Arch. Biochem. Biophys. 444174-184. [DOI] [PubMed] [Google Scholar]
- 43.Reimunde, F. M., C. Castañares, M. Redondo-Horcajo, S. Lamas, and F. Rodriguez-Pascual. 2005. Endothelin-1 expression is strongly repressed by AU-rich elements in the 3′-untranslated region of the gene. Biochem. J. 387763-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-Pascual, F., M. Hausding, I. Ihrig-Biedert, H. Furneaux, A. P. Levy, U. Forstermann, and H. Kleinert. 2000. Complex contribution of the 3′-untranslated region to the expressional regulation of the human inducible nitric-oxide synthase gene. Involvement of the RNA-binding protein HuR. J. Biol. Chem. 27526040-26049. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Pascual, F., M. Redondo-Horcajo, and S. Lamas. 2003. Functional cooperation between Smad proteins and activator protein-1 regulates transforming growth factor-beta-mediated induction of endothelin-1 expression. Circ. Res. 921288-1295. [DOI] [PubMed] [Google Scholar]
- 46.Ruef, J., M. Moser, W. Kubler, and C. Bode. 2001. Induction of endothelin-1 expression by oxidative stress in vascular smooth muscle cells. Cardiovasc. Pathol. 10311-315. [DOI] [PubMed] [Google Scholar]
- 47.Schultz, D. E., C. C. Hardin, and S. M. Lemon. 1996. Specific interaction of glyceraldehyde 3-phosphate dehydrogenase with the 5′-nontranslated RNA of hepatitis A virus. J. Biol. Chem. 27114134-14142. [DOI] [PubMed] [Google Scholar]
- 48.Schulze, P. C., and R. T. Lee. 2005. Oxidative stress and atherosclerosis. Curr. Atheroscler. Rep. 7242-248. [DOI] [PubMed] [Google Scholar]
- 49.Schuppe-Koistinen, I., P. Moldeus, T. Bergman, and I. A. Cotgreave. 1994. S-thiolation of human endothelial cell glyceraldehyde-3-phosphate dehydrogenase after hydrogen peroxide treatment. Eur. J. Biochem. 2211033-1037. [DOI] [PubMed] [Google Scholar]
- 50.Sen, N., M. R. Hara, M. D. Kornberg, M. B. Cascio, B.-I. Bae, N. Shahani, B. Thomas, T. M. Dawson, V. L. Dawson, S. H. Snyder, and A. Sawa. 2008. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 10866-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sethi, A. S., D. M. Lees, J. A. Douthwaite, A. B. Dawnay, and R. Corder. 2006. Homocysteine-induced endothelin-1 release is dependent on hyperglycaemia and reactive oxygen species production in bovine aortic endothelial cells. J. Vasc. Res. 43175-183. [DOI] [PubMed] [Google Scholar]
- 52.Sirover, M. A. 1999. New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim. Biophys. Acta 1432159-184. [DOI] [PubMed] [Google Scholar]
- 53.Souza, J. M., and R. Radi. 1998. Glyceraldehyde-3-phosphate dehydrogenase inactivation by peroxynitrite. Arch. Biochem. Biophys. 360187-194. [DOI] [PubMed] [Google Scholar]
- 54.Stoecklin, G., T. Mayo, and P. Anderson. 2006. ARE-mRNA degradation requires the 5′-3′ decay pathway. EMBO Rep. 772-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang, R. Y.-L., and P. D. Nagy. 2008. Tomato bushy stunt virus co-opts the RNA-binding function of a host metabolic enzyme for viral genomic RNA synthesis. Cell Host Microbe 3178-187. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, W., B. J. Wagner, K. Ehrenman, A. W. Schaefer, C. T. DeMaria, D. Crater, K. DeHaven, L. Long, and G. Brewer. 1993. Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol. Cell. Biol. 137652-7665. [DOI] [PMC free article] [PubMed] [Google Scholar]