

Abstract
Aberrant glycogen synthase kinase 3β (GSK-3β) activity is associated with the progression of several pathological conditions such as diabetes, Alzheimer's, and cancer. GSK-3β regulates cellular processes by directly phosphorylating metabolic enzymes and transcription factors. Here, we discovered a new target for GSK-3β phosphorylation: the human glucocorticoid receptor (GR). Glucocorticoid signaling is essential for life and regulates diverse biological functions from cell growth to metabolism to apoptosis. Specifically, we found hormone-dependent GR phosphorylation on serine 404 by GSK-3β. Cells expressing a GR that is incapable of GSK-3β phosphorylation had a redirection of the global transcriptional response to hormone, including the activation of additional signaling pathways, in part due to the altered ability of unphosphorylatable GR to recruit transcriptional cofactors CBP/p300 and the p65 (RelA) subunit of NF-κB. Furthermore, GSK-3β-mediated GR phosphorylation inhibited glucocorticoid-dependent NF-κB transrepression and attenuated the glucocorticoid-dependent cell death of osteoblasts. Collectively, our results describe a novel convergence point of the GSK-3β and the GR pathways, resulting in altered hormone-regulated signaling. Our results also provide a mechanism by which GSK-3β activity can dictate how cells will ultimately respond to glucocorticoids.
Glycogen synthase kinase 3 (GSK-3) is a serine/threonine kinase that acts on a wide variety of substrates, including glycogen synthase, β-catenin, NF-κB, c-Jun, c-Myc, cyclin D1, and eukaryotic initiation factor 2B (13, 31). Substrate phosphorylation by GSK-3 has been shown to be essential for such diverse biological responses as cell differentiation, cell survival, and metabolic processes (1). Unlike most kinases, GSK-3 is active in resting cells, and stimulation of cells by mitogens or growth factors leads to its inactivation. In addition, failure to inactivate GSK-3 kinase has been shown to have an important role in the progression of several disease states, such as cancer, diabetes, and Alzheimer's (3, 20, 33). Therefore, further exploration of the downstream targets of GSK-3 is essential for the better understanding of many pathological conditions.
Glucocorticoids (GCs) are a class of steroid hormones that are essential for human life. GCs have diverse, tissue-specific functions and regulate everything from cell growth and development to metabolism and even cell death. Drugs mimicking GC action are widely used to treat diseases such as cancer, inflammation, and autoimmune disorders (38). GCs function by binding and activating the GC receptor (GR), which then regulates the transcription of target genes. Depending on the cell type and promoter context, the GR can either up- or downregulate gene expression by recruiting cofactors and interacting with transcriptional machinery (24). Therefore, it is likely that transcriptional output will ultimately (i) determine how a cell will respond to hormone exposure and (ii) distinguish how GCs induce apoptosis in lymphocytes while inhibiting the same process in hepatocytes (10, 34, 41).
Since ligand binding to the GR enables its translocation from the cytoplasm to the nucleus of a cell, the transcriptional activity of the GR appears to be mainly governed by ligand binding. However, in addition to ligand binding, several recent studies provide evidence that cross talk from other signaling pathways are able to directly modulate the GR transcriptional responses by phosphorylating the GR (17). In fact, human GR phosphorylation by kinases, such as mitogen-activated protein kinases and cyclin-dependent kinases (CDKs), act to modulate the receptor protein stability, subcellular localization, protein interactions, and translational response (12, 26, 28, 32, 39, 44, 48). Interestingly, Rogatsky et al. have previously shown that GSK-3β-mediated phosphorylation of rat GR on threonine 171 can inhibit transcriptional activation (40). Thus, the GR phosphorylation status provides a potential mechanism to explain how the GR differentially regulates diverse subsets of genes in various cell types. Therefore, the control of receptor transcriptional activity via phosphorylation provides an increased array of regulatory inputs that, in addition to steroid hormones, can influence receptor function.
The human GR is phosphorylated on several sites, serines 203, 211, and 226 being the most thoroughly characterized at the molecular level (4, 14, 17, 23, 40, 47). With the utilization of mass spectrometry, we report here the discovery of a novel GR phosphorylation site, serine 404 (Ser404), and its important role in modulating GR function. We found that Ser404 was phosphorylated by GSK-3β in a hormone-dependent manner. Furthermore, cells lacking GR-Ser404 phosphorylation potential had a redirection of the transcriptional response to hormone and increased cell death. Conversely, cells with constitutive GSK-3β-mediated GR phosphorylation had decreased transcriptional responses and were more resistant to dexamethasone-dependent cell death. Our results suggest that Ser404 phosphorylation status is important for the ability of GR to signal within cells. Furthermore, our results also suggest that cellular conditions that result in altered GSK-3β activity will influence how cells ultimately respond to GC hormone stimulation.
MATERIALS AND METHODS
Reagents, antibodies, and plasmids.
Dexamethasone (Dex; 1,4-pregnadien-9-fluoro-16-methyl-11β,17,21-triol-3,20-dione) was purchased from Steraloids (Newport, RI). Doxycycline was purchased from Sigma (St. Louis, MO). The GSK-3α/β inhibitor 6-bromoindirubin-3′-oxime (BIO) was purchased from Calbiochem (San Diego, CA), and SB415286, lithium chloride, and AR-A014418 were purchased from Sigma. Rabbit anti-phospho-GR antibodies were produced by using peptides made by AnaSpec (San Jose, CA), and the antisera were produced by Covance (Denver, PA). The anti-phospho-GR antibodies purified in our laboratory were epitope purified and characterized in collaboration with Paul Housley (University of South Carolina). pGSK-3β-Flag (WT, S9A, and Y216F) were generously provided by Xiao-Fan Wang (Duke University). pSuper-shScramble and pSuper-shGSK-3 constructs were kindly provided by William Snider (University of North Carolina, Chapel Hill). Reporter plasmids pGRE2-Luc, pGL2-3XMHCLuc, and pGL3-hRL were described previously (29, 30). pTRE-hGRα-S404A and pTRE-hGRα-S404D were generated by site-directed mutagenesis of pTRE-hGRα (29) using a QuikChange kit (Stratagene, La Jolla, CA). N-terminal Flag-tagged GRα (pcDNA-hGRα-Flag and pcDNA-hGRα-S404A-Flag) were constructed by PCR and inserted into pcDNA3.1/Zeo(+) (Invitrogen, Carlsbad, CA). All cloning and mutagenesis products were verified by DNA sequencing at the DNA Sequencing Core at the National Institute of Environmental Health Sciences.
Cell culture and stable cell line production.
U-2 OS, MG-63, Hep-G2, and HeLa cells (American Type Culture Collection, Manassas, VA) were maintained in Dulbecco modified Eagle medium-F-12 medium supplemented with 10% fetal calf serum. Wild-type (WT) and GSK-3β-null mouse embryonic fibroblasts (MEFs) were generously provided by Jim Woodgett (Mount Sinai Hospital-SLRI) and cultured as previously described (21). Geneticin (500 μg/ml; Invitrogen) and hygromyocin (200 μg/ml; Invitrogen) were used to establish U-2 OS cell lines stably expressing WT-, S404A-, and S404D-hGRα, similar to that described previously (29). Briefly, U-2 OS cells were transfected with pTET-OFF and clonally selected with Geneticin to produce the parental U-2 OS cell line (null). Parental U-2 OS cells were then transfected with pTRE2 vector containing WT-, S404A-, or S404D-GR and clonally selected with Geneticin and hygromycin. Receptor levels were compared by Western blot analysis, and clones expressing comparable receptor levels were used. All results were validated with the use of several independent clones for each mutant of the GR. FuGENE reagent (Roche, Indianapolis, IN) was used at 3 μl per 1 μg of DNA for transfection and transient expression of hGRα-Flag, GSK-3β, and shGSK. All Dex treatments were performed in growth medium supplemented with 10% charcoal-dextran-stripped fetal calf serum.
Western blot analysis and immunoprecipitations.
Following treatments as indicated, the cells were washed in ice-cold phosphate-buffered saline (PBS) and lysed for 60 min on ice in Triton X-100 lysis buffer (50 mM Tris, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100 [pH 7.4]) supplemented with phosphatase inhibitor cocktail set II (Calbiochem) and protease inhibitor cocktail tablets (Roche). The resulting detergent-solubilized whole-cell extracts were clarified by microcentrifugation and quantitated using a BCA protein quantitation kit (Pierce, Rockford, IL). Immunoprecipitations were carried out by adding anti-Flag (Sigma), anti-GR 57 (11), or anti-GR (BD Biosciences, San Jose, CA) antibodies to 500 μg of whole-cell extracts, followed by incubation for 16 h at 4°C with slow rotation. The resulting immunocomplexes were collected by microcentrifugation and washed several times with PBS. For in vitro kinase assays, GR immunocomplexes were incubated with active GSK-3β (3 ng; Chemicon), 20 mM MOPS (morpholinepropanesulfonic acid), 10 mM magnesium acetate, 100 μM ATP, and 1 μCi of γ-32P for 10 min at 30°C. Kinase reactions were stopped with the addition of 3× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer and boiled for 5 min. All immunocomplexes were then resolved on 4 to 20% ReadyGel Tris-Gly gels (Bio-Rad, Hercules, CA). For Western blot analysis, 20 to 50 μg of protein from whole-cell extracts was similarly resolved. Nuclear and/or cytoplasmic fractionation was performed by using a Biovision kit (Biovision, Mountain View, CA) according to the manufacturer's directions. All proteins were electrophoretically immobilized to nitrocellulose membranes, which were subsequently probed with anti-GR antibodies (1:1,000; BD Biosciences), anti-GR 57 antibodies (1:500), anti-pSer404 GR antibodies (1:1,000), anti-pSer211 GR antibodies (1:2,000), anti-β-actin antibodies (1:2,500; Chemicon, Billerica, MA), anti-Bcl-xL antibodies (1:1,000; Chemicon), anti-GSK-3β antibodies (1:500; Cell Signaling, Danvers, MA), anti-GSK-3α antibodies (1:500; Chemicon), anti-p65 (RelA) antibodies (1:500; Cell Signaling), anti-CBP/p300 antibodies (1:500; Santa Cruz Biotechnologies, Santa Cruz, CA), anti-GRIP1 antibodies (1:500; Chemicon), anti-cIAP antibodies (1:1,000; Cell Signaling), anti-GAPDH antibodies (1:500; Cell Signaling), anti-nucleolin antibodies (1:1,000; Santa Cruz Biotechnologies), or anti-survivin antibodies (1:1,000; Stressgen, Ann Arbor, MI). Densitometry and quantitation of three independent experiments was performed by using NIH ImageJ software and normalized to β-actin expression.
Identification of GR phosphorylation sites.
Flag-tagged human GRα was transiently expressed in U-2 OS cells for 48 h and then immunoprecipitated with anti-Flag antibodies conjugated to agarose beads (Sigma). Immunoprecipitates were diluted in 3× SDS-PAGE sample buffer and electrophoresed on 4 to 20% Tris-glycine gels. Gel bands were stained with Simply Blue Stain (Invitrogen), excised, and digested with trypsin (Promega, Madison, WI) for 8 h (9). Afterward, the samples were analyzed for phosphorylated peptides by nanoLC-ESI-MS/MS both with and without the use of immobilized metal ion affinity chromatography essentially as previously described (7, 9). The extent of phosphorylation was estimated by stable isotope-free relative and absolute quantitation essentially as described by Steen et al. (43).
Immunofluorescence studies.
U-2 OS cells stably expressing WT-, S404A-, or S404D-GR were plated onto 35-mm dishes with glass bottom inserts. The cells were treated as indicated, washed in PBS, fixed in 4% paraformaldehyde, and permeabilized by Triton X-100 (0.2% in PBS). The cells were then blocked in PBS supplemented with 1.5% normal goat serum, followed by incubation with anti-GR (1:500 dilution; BD Biosciences) or anti-pSer404-GR antibodies, followed by Alexa Fluor 488 (1:1,000; Invitrogen) or Alexa Fluor 594 antibodies (1:1,000; Invitrogen). Cells were then mounted in Vectashield containing DAPI (4′,6′-diamidino-2-phenylindole; Vecta Laboratories, Burlingame, CA). Confocal images were taken on a Zeiss LSM510-NLO meta using a C-Apochromat ×40/1.2 W Corr DIC objective lens.
cDNA microarray analysis.
U-2 OS cells stably expressing WT- or S404A-GR were incubated in growth media supplemented with 10% charcoal-dextran-stripped fetal calf serum for 48 h and then treated, in triplicate, with either vehicle (H2O) or 100 nM Dex for 6 h. The total RNA was harvested (Qiagen, Valencia, CA), and gene expression analysis was conducted by using Agilent whole-genome 4 × 44 multiplex format oligonucleotide arrays (catalog no. 014850; Agilent Technologies, Santa Clara, CA) according to the Agilent 1-color microarray-based gene expression analysis protocol. Starting with 500 ng of total RNA, Cy3-labeled cRNA was generated according to the manufacturer's protocol. For each sample, 1.65 μg of Cy3-labeled cRNAs was fragmented and hybridized for 17 h in a rotating hybridization oven. Slides were washed and then scanned with an Agilent scanner. The data were obtained using Agilent Feature Extraction software (v9.5), with the one-color defaults for all parameters. Agilent Feature Extraction software performed error modeling, adjusting for additive and multiplicative noise. In order to identify differentially expressed genes (P < 0.01), an error-weighted analysis of variance (ANOVA) with the Bonferroni correction was performed using the Rosetta Resolver system (version 7.0; Rosetta Biosoftware, Kirkland, WA). The Bonferroni test correction was used to reduce the number of false positives. Finally, the ANOVA results were subjected to Ingenuity Pathways (Ingenuity Systems, Redwood City, CA) analysis for functional analysis.
Real-time PCR analysis.
U-2 OS cells stably expressing WT- or S404A-GR were treated with 100 nM Dex or vehicle (H2O) for 6 h, and total RNA was isolated by using a Qiagen RNeasy minikit. Real-time PCR was performed by using the 7900HT sequence detection system and predesigned primer/probe sets from Applied Biosystems (Foster City, CA) according to the manufacturer's instructions. The signal obtained from each gene primer-probe set was normalized to that of the unregulated housekeeping gene β-actin primer-probe set (also available from Applied Biosystems). Each primer-probe set was analyzed with at least three different sets of RNA.
Luciferase reporter assays.
U-2 OS cells were plated into 24-well plates (35,000 per well). pGRE2-luc and pGL2-3XMHC-luc reporter constructs were used for GR transactivation and transrepression assays, respectively. Cells were treated with Dex or vehicle (20 h) 24 h after reporter transfection. The luciferase activity was measured as previously described (29). In each experiment, the firefly luciferase activity normalized to the Renilla luciferase activity was measured in duplicate and averaged. Each experiment was repeated three times.
Limited protease digestion assays.
U-2 OS cells stably expressing WT- or S404A-GR were treated with 100 nM Dex for 1 h. Then, anti-GR 57 antibodies were used to immunoprecipitate GR from cellular extracts overnight at 4°C. The immunocomplexes were then washed two times in cold PBS before incubation with 1 μM Dex for 10 min at 25°C. The GR was then incubated with 50 ng of sequence-grade trypsin (Promega) for 15 min at 25°C. A 3× SDS sample loading dye was then added, and the samples were boiled 10 min before being resolved on 4 to 20% Tris-glycine gels and probed with anti-GR antibodies (1:2,000; BD Biosciences).
Flow cytometry analysis.
Cell death was measured by propidium iodide (PI) staining. After treatment, adherent and suspended cells were collected, centrifuged, and resuspended in a PBS solution containing 10 μg/ml of PI (Invitrogen). A total of 2 × 105 control or treated cells were processed, and 1 × 104 cells were immediately analyzed by using a BD FACSort flow cytometer (Franklin Lakes, NJ) with an excitation wavelength of 488 nm and an emission wavelength of 585 nm. For GSK-3 knockdown experiments, pShuttle-GFP-shScramble and pShuttle-GFP-shGSK-3 constructs were expressed for 3 days, after which flow and Western analyses were performed in order to select for the green fluorescent protein-positive cell populations and confirm GSK-3 knockdown, respectively. Each experiment was conducted in triplicate.
Microarray data accession number.
The microarray data in this publication were deposited in NCBI's Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) (16) and are accessible through Gene Expression Omnibus series accession number GSE11205.
RESULTS
The GR is phosphorylated on Ser404.
Posttranslational alterations of numerous cellular proteins such as receptors, kinases, and transcription factors are essential for propagation of signals from the cell membrane to the nucleus to elicit a biological response (22). Based on these general phenomena, we hypothesize that posttranslational modification of the GR is important in regulating GC signaling within cells. The human GR is known to be a phospho-protein; however, previous studies have not thoroughly investigated the role of phosphorylation in GR signaling (5). Therefore, we initially utilized mass spectrometry to identify additional residues within the human GR that were phosphorylated in response to the synthetic GC, Dex. The GR isolated from control or Dex-treated U-2 OS osteosarcoma cells expressing human GRα-Flag was prepared for analysis by mass spectrometry, and a novel phosphorylation site, Ser404, was identified within the 387-to-419-amino-acid peptide fragment of GR (Fig. 1A). Furthermore, the phosphorylation of Ser404 was enhanced 2.19-fold upon Dex treatment (Fig. 1B). In order to confirm the mass spectrometry data, a phospho-specific antibody that recognizes GR when phosphorylated at Ser404 was generated. As a positive control, a phospho-specific antibody that recognizes the well-characterized GR phosphorylation site Ser211 was also generated (47). Western blot analysis utilizing these antibodies show that Ser211 in addition to Ser404 was phosphorylated in a GC-dependent manner in U-2 OS cells transiently or stably transfected with GR (Fig. 1C and D). Finally, we wanted to determine whether endogenous GR was capable of being phosphorylated at Ser404. Figure 1E shows that Ser404 was phosphorylated in cell lines of the bone (MG-63), liver (Hep-G2), cervix (HeLa), and lung (A549), and this phosphorylation event was enhanced by Dex in all of the cell lines tested. Therefore, we conclude that Ser404 of GR is phosphorylated in a GC-dependent manner in multiple cell types, suggesting that Ser404 phosphorylation may be important for GR function in a variety of cells.
FIG. 1.

The GR is phosphorylated on Ser404. (A) Samples from control or Dex (100 nM; 1 h)-treated U-2 OS cells expressing Flag-tagged human GRα were resolved by gel electrophoresis, and the band containing GR was isolated and analyzed by mass spectrometry. The tandem mass spectrometry spectrum from the peptide containing GR phosphorylated on Ser404 is shown. (B) The ion abundance of phospho-387-419AA peptide in unstimulated and Dex-treated samples. (C) U-2 OS cells transiently expressing hGRα-Flag were treated with 100 nM Dex for 0 to 4 h, immunoprecipitated, and probed with phospho-specific and total anti-GR antibodies. (D) Null U-2 OS cells or U-2 OS cells stably expressing hGRα were treated with 100 nM Dex, and cell lysates were then probed with phospho-specific and total anti-GR antibodies. (E) Cells of the bone (MG-63), liver (Hep-G2), cervix (HeLa), and lung (A549) were treated with 100 nM Dex for 1 h, and lysates were analyzed by Western blotting to determine whether endogenous GR was phosphorylated at Ser404. The levels of P-s404 (GR phosphorylated on serine 404) from three independent experiments were quantified and normalized to total GR levels (*, P < 0.05).
GSK-3β phosphorylates GR on Ser404.
Having demonstrated that the GR was phosphorylated on Ser404, we next wanted to identify the kinase responsible for this phosphorylation event. Motif scanning of the GR sequence revealed that Ser404 was contained within a possible consensus site for GSK-3 (Fig. 2A), as well as CDK5 kinase. However, since CDK5 activity is limited to the nervous system, it seems unlikely that CDK5 would phosphorylate the GR in osteosarcoma cells (45); therefore, we focused solely on GSK-3. GSK-3 kinases have been shown to have an important role in the progression of several disease states, such as cancer, diabetes, and Alzheimer's (3, 20, 33). Since enhanced GR signaling is commonly associated with some of the same pathological conditions as dysregulated GSK-3, we considered the possibility of cross talk between these two signaling pathways. Two isoforms of GSK-3 are reported in mammals: GSK-3α and GSK-3β. Therefore, we first used the well-characterized GSK-3α/β inhibitor BIO (46) to pharmacologically evaluated the role of GSK-3α/β activity in regulating GR phosphorylation. Figures 2B and C show that BIO reduced the basal and GC-induced phosphorylation of Ser404 (94.8% ± 3.6% inhibition relative to control at 5 μM BIO in Dex-treated cells), with only a small affect on Ser211 phosphorylation status (26.2% ± 2.1% inhibition relative to control at 5 μM BIO in Dex-treated cells). Furthermore, we generated a mutant of GR (S404A-GR) that is incapable of Ser404 phosphorylation, and Fig. 2B demonstrates the specificity of the phospho-Ser404 antibody solely to the Ser404 phosphorylated form of the GR. Therefore, we conclude that at least one isoform of GSK-3 is phosphorylating the GR. In order to determine which one(s), we performed immunoprecipitation assays. Figure 2D shows that both exogenous WT-GR (U2-OS cells) and endogenous GR (A549 cells) associated with GSK-3β in the presence of Dex; however, the GR failed to associate with GSK-3α. Interestingly, the phosphodeficient mutant of the GR (S404A) failed to associate with GSK-3α/β even in the presence of Dex (data not shown). Therefore, we conclude that GSK-3β is the isoform that is likely responsible for phosphorylation of GR at Ser404.
FIG. 2.

GSK-3β phosphorylates GR on Ser404. (A) Alignment of human GR sequence with the consensus site for GSK-3 kinase substrates. (B and C) U-2 OS cells stably expressing WT-GR or S404A-GR were pretreated with 0 to 5 μM concentrations of the GSK3α/β inhibitor BIO for 1 h before incubation with 100 nM Dex for 1 h. Afterward, cell extracts were analyzed by Western blot analysis to determine the amount of Ser211 and Ser404 phosphorylation of the GR. (D) U-2 OS cells stably expressing WT-GR or A549 cells that endogenously express GR were treated with 100 nM Dex for 1 h. Cell lysates were immunoprecipitated using anti-GR antibodies, and immunoblots were probed with anti-GSK-3α and anti-GSK-3β antibodies. (E) WT- and S404A-GR were immunoprecipitated from the stable U-2 OS cell lines. Immunoprecipitates were incubated with active recombinant GSK-3β, and GSK-3β-mediated phosphorylation of GR was visualized by autoradiography. (F) WT-GR-expressing U-2 OS cells transiently overexpressing WT, constitutively active (S9A), or kinase-dead (Y216F) versions of GSK-3β were lysed and assayed by Western blotting to determine pSer404-GR levels (*, P < 0.05). (G) GSK-3β+/+ or GSK-3β−/− MEFs were treated with 100 nM Dex for 1 h, and cell lysates were then analyzed by Western blotting for the presence of GR phosphorylation. Mouse Ser412 is homologous to human Ser404, and mouse Ser220 is homologous to human Ser211.
To further investigate the role of GSK-3β in Ser404 phosphorylation, we performed in vitro kinase assays. Figure 2E shows that WT-GR was phosphorylated in the presence of active, recombinant GSK-3β. Interestingly, only minimal GR phosphorylation was observed when GSK-3β was incubated with a phospho-deficient mutant of GR (S404A-GR), suggesting that Ser404 is the major site of GSK-3β phosphorylation of the GR. Next, we overexpressed WT, constitutively active (S9A), or kinase-dead (Y216F) versions of GSK-3β in U-2 OS cells expressing WT-GR and determined the relative amount of Ser404 phosphorylation. Our results show that increased expression of catalytically active GSK-3β enhanced GC-mediated GR phosphorylation. Conversely, overexpression of the kinase-dead version of GSK-3β had a dominant-negative effect and blocked Ser404 phosphorylation (Fig. 2F). Finally, WT and GSK-3β-null MEFs were assayed for Ser412 (mouse pSer412 is the homologous serine to human Ser404) phosphorylation of the GR. We found that MEFs lacking GSK-3β expression did not exhibit Dex-induced phosphorylation of the GR at Ser412 (Fig. 2G). Together, our results show that Dex promoted a novel complex formation between the GR and GSK-3β and that GSK-3β activity is required for Ser404 phosphorylation of the GR.
Effects of Ser404 phosphorylation on GR cellular localization.
Since we found that the GR was phosphorylated at Ser404 in an agonist-dependent manner, we next investigated whether the phosphorylation status of Ser404 affected GR signaling. GR is a nuclear receptor and functions primarily to enhance and/or repress gene transcription (19); therefore, we first studied the effects of Ser404 phosphorylation on nuclear localization of the GR. In U-2 OS cells expressing WT-GR, GR was predominantly cytoplasmic, and stimulation with Dex for 1 h drove its nuclear accumulation (Fig. 3A). In addition, U-2 OS cells expressing the unphosphorylatable GR (S404A) or a Ser404 to aspartic acid mutant of GR (S404D-GR), a mutation that mimics both the size and the negative charge of a phosphorylated serine residue, were assayed for GR localization. We found that cells expressing S404A-GR or S404D-GR had complete nuclear localization of the GR after 1 h of Dex stimulation (Fig. 3A). These results suggest that Ser404 phosphorylation does not affect the ability of the receptor to translocate from the cytoplasm to the nucleus upon hormone stimulation. Since Dex stimulation enhanced Ser404 phosphorylation and GSK-3β is primarily a nuclear kinase, we hypothesized that phosphorylation of the GR occurs in the nucleus. Therefore, we determined the cellular location of pSer404-GR. Immunofluorescent staining of U-2 OS cells expressing WT-GR demonstrated that the GR was cytoplasmic and contained a basal amount of pSer404 phosphorylation. Upon Dex treatment, GR translocated to the nucleus and Ser404 phosphorylation of GR increased. Interestingly, pSer404-GR was mainly localized to the perinuclear region (Fig. 3B), suggesting that once phosphorylated in the nucleus, the GR maybe exported from the nucleus. To address this question, we inhibited Ser404-GR phosphorylation with the GSK-3α/β inhibitor BIO and found enhanced nuclear localization of GR and a prevention of the accumulation of pSer404 in the perinuclear space, further supporting the hypothesis of receptor phosphorylation leading to nuclear export. To confirm these results via an alternative approach, WT-GR-expressing U-2 OS cells were stimulated with 100 nM Dex for 1 h and separated into cytoplasmic and nuclear fractions. Figure 3C shows that GR was primarily in the cytoplasm in the absence of hormone and, upon Dex treatment, GR accumulated in the nucleus. Hormone treatment also increased GR phosphorylation on Ser211 and Ser404. However, while pSer211-GR was both cytoplasmic and nuclear (47), pSer404-GR was largely cytoplasmic (Fig. 3C). Together, these results suggest that Ser404-GR phosphorylation does not affect the import of the GR into the nucleus but may affect its export into the cytoplasm.
FIG. 3.

Effects of Ser404 phosphorylation on GR cellular localization. (A) WT-, S404A-, or S404D-GR-expressing U-2 OS cells were treated with 100 nM Dex for 1 h. Cells were then fixed and stained with antibodies directed against human GR. (B) WT- and S404A-GR-expressing U-2 OS cells were treated with Dex (100 nM) and/or the GSK-3α/β inhibitor BIO (5 μM) for 1 h. Cells were then fixed and stained with the nuclear dye DAPI (blue), antibodies directed against human GR (red), and antibodies directed against phospho-S404-GR (green). Arrows indicate phosphorylated GR. All images were captured on a Zeiss LSM 410 laser-scanning microscope. (C) WT-GR-expressing U-2 OS cells were treated with Dex (100 nM) for 1 h before whole-cell lysates were subjected to nuclear and/or cytoplasmic fractionation.
Ser404 phosphorylation enhances GR protein downregulation.
Our results above raise the possibility that Ser404 phosphorylation may affect GR export from the nucleus. Once GR exits the nucleus, it can be targeted by the proteasome for degradation, which limits the cellular response to GCs (27). The GR protein has been shown to have a half-life of 15 to 20 h, and this half-life decreases to 7 to 9 h upon agonist stimulation (30). Previous work by our lab demonstrated that the phosphorylation status of mouse GR affected receptor half-life (48). Therefore, we wanted to determine whether Ser404 phosphorylation of the human GR also alters the stability of the protein in a ligand-dependent or -independent manner. In accordance with previous reports, we found that WT-GR had a half-life of 21.9 h, and this half-life was decreased to 9.8 h upon exposure to Dex (Fig. 4A). Interestingly, the half-life of unphosphorylatable GR (S404A) had an increased half-life of 32.6 h that was decreased to 13.1 h when exposed to Dex (Fig. 4B). In contrast, the phospho-mimic mutant of GR (S404D) had a decreased half-life of 18.1 h, which was further decreased to only 2.5 h upon Dex exposure (Fig. 4C). These data suggest that the phosphorylation of Ser404 has an important role in GR protein stability. Furthermore, our results raise the possibility that cells with aberrant GSK-3β activity resulting in enhanced Ser404 phosphorylation may preferentially downregulate GR protein, leading to states of GC resistance.
FIG. 4.

Ser404 phosphorylation enhances GR protein downregulation. The expression of WT-GR (A), S404A-GR (B), or S404D-GR (C) was turned off by doxycycline (10 ng/ml) in U-2 OS cells stably expressing GR. The level of GR expression was determined in the absence or presence of Dex (100 nM) over a time course of 0 to 50 h. After treatments, the total cell lysate was quantitated, and 30 μg of protein/lane was analyzed by Western blotting. The protein half-life was then determined by averaging the GR protein levels from three independent experiments.
Ser404 phosphorylation alters the ability of GR to regulate gene transcription.
Our results thus far identified Ser404 as a potentially important residue in regulating GR signaling. Therefore, we wanted to determine whether there was physiological significance of Ser404 phosphorylation. To accomplish this goal, we first examined the ability of WT-GR and phosphorylation-deficient GR (S404A) to regulate gene transcription in U-2 OS cells. For these experiments, WT- and S404A-GR-expressing U-2 OS cells were treated with vehicle or 100 nM Dex for 6 h, and the total cellular RNA was collected for whole human genomic microarray analysis. It is important to note that the level of GR expression after Dex treatment was comparable in both WT- and S404A-GR samples (Fig. 5A). We found that the WT receptor regulated 7,772 genes, while the phosphorylation-deficient mutant S404A regulated 9,617 genes. Of these genes, 4,993 were common to both WT- and S404A-GR, 2,779 were unique to WT-GR, and 4,624 were unique to S404A-GR (Fig. 5A). To help elucidate the mechanism of enhanced gene regulation by S404A-GR, we separated the Dex-induced genes from the Dex-repressed genes and found that S404A-GR had an increased number of genes repressed compared to the WT receptor (Fig. 5B). The receptor phosphorylation-specific regulation of endogenous genes was also confirmed by real-time PCR (Fig. 5C). Some genes such as FOXO4 (induced) and Bcl 3 (repressed) were commonly regulated by WT- and S404A-GR. However, S404A-GR regulated additional genes such as the genes encoding Kip2 (induced) and interleukin-1β (repressed). Finally, in order to understand the repertoire of gene regulation by WT and phospho-deficient GR, data were loaded into Ingenuity Pathways analysis software and compared for biological pathway regulation. Genes involved in cell growth and proliferation, hematological system development and function, and immune response were highly regulated in the WT-GR-expressing cells (Fig. 6A). Interestingly, the regulation of these same genes was significantly different in cells expressing S404A-GR. Of greater interest, we found that S404A-GR-expressing cells had the ability to regulate genes involved in lipid metabolism, small molecule biochemistry, and cancer, whereas the WT-GR-expressing cells did not significantly regulate this same pathway (Fig. 6B). Real-time PCR analysis was also performed to verify differentially regulated genes involved in these two pathways and is shown in the right panels of Fig. 5C. Indeed, SLC16A5, STAT4, GPR126, and interleukin-4 were differentially regulated by Dex in WT-GR and S404A-GR-expressing cells. These data indicate that the lack of Ser404 phosphorylation not only enhances GR-mediated gene regulation but also significantly alters the repertoire of GR-regulated genes. These results also suggest that the GSK-3β activity in cells is an important determinant in the GR transcriptional response to GC hormones.
FIG. 5.

Ser404 phosphorylation alters the ability of GR to regulate gene transcription. (A) WT- and S404A-GR-expressing U-2 OS cells were treated with Dex (100 nM; 6 h) before the total RNA was isolated and subjected to whole-human-genome microarray analysis. Identification of differentially expressed genes (P < 0.01) was performed by an error-weighted ANOVA using the Bonferroni correction. (B) Dex-regulated genes were separated into those induced or those repressed. (C) Microarray results of representative Dex-regulated genes were confirmed by real-time PCR and include three independent RNA samples per group (*, P < 0.05).
FIG. 6.

GSK-3β-mediated GR phosphorylation redirects gene transcription. ANOVA results from microarray analysis were loaded into Ingenuity Pathways analysis software to compare the biological pathway regulation in WT- and S404A-GR microarray samples. The most statistically significant biological pathways regulated by WT-GR (A) and S404A-GR (B) are shown. The shade of red (induction) or green (repression), lighter to darker, signifies the least to the greatest degree of gene induction or repression, respectively.
Ser404 phosphorylation decreases GR function.
Due to the ability of Ser404 phosphorylation to globally affect GR-mediated gene transcription, we next sought to determine the mechanism for this altered response. First, we examined the ability of the GR to regulate different promoter elements. To accomplish this goal, we transiently transfected GR-expressing U-2 OS cells with various reporter constructs and analyzed Dex-induced luciferase activity. As expected, WT-GR-expressing U-2 OS cells treated with 0 to 100 nM Dex for 18 h induced luciferase expression driven by the synthetic GC response element (GRE) promoter with a 50% effective concentration of 1 nM (Fig. 7A). Interestingly, Dex-stimulated S404A-GR-expressing cells induced GRE-driven luciferase with a similar 50% effective concentration but with a significantly increased efficacy over WT-GR (Fig. 7A). In addition, S404D-GR-expressing U-2 OS cells had a slightly decreased response to Dex relative to WT-GR-expressing cells (Fig. 7A). In addition to the ability of GR to induce the expression of GRE-containing promoters, GR also mediates the transrepression of several genes. The classical promoters transrepressed by the GR are promoters containing NF-κB binding elements (42). Therefore, to examine GR-mediated transrepression, a construct with luciferase driven by the MHC promoter (containing NF-κB binding elements) was utilized. As predicted, WT-GR-expressing cells treated with Dex repressed luciferase expression to 34.4% ± 2.1% relative to the control, with an 50% inhibitory concentration of >500 pM. Interestingly, S404A-GR-expressing cells had a stronger Dex-induced transrepressive effect, decreasing the luciferase reporter activity to 5.4% ± 1.1% of the control, untreated levels with a decreased 50% inhibitory concentration of <50 pM (Fig. 7B). In contrast, S404D-GR-expressing U-2 OS cells treated with Dex had a diminished ability to transrepress the activity of the NF-κB reporter (57.7% ± 3.1% relative to the control). To further evaluate the impact of Ser404 GR phosphorylation on NF-κB-mediated gene expression, we analyzed the effect of Dex on the expression of well-known NF-κB-regulated prosurvival proteins (c-IAP, survivin, and Bcl-xl). Figure 7C demonstrates that Dex had an enhanced ability to repress NF-κB-regulated prosurvival genes when Ser404 phosphorylation was blocked with the GSK-3α/β inhibitor BIO. Next, WT- and S404D-GR-expressing U-2 OS cells were compared for their ability to repress prosurvival proteins in response to Dex. Figure 7D shows that in WT-GR-expressing cells, Dex was able to repress the expression of Bcl-xl, survivin, and c-IAP. In contrast, S404D-GR-expressing cells failed to repress these prosurvival genes in response to Dex stimulation.
FIG. 7.

Ser404 phosphorylation decreases GR function. (A and B) GR-null U-2 OS cells or those expressing WT-, S404A-, or S404D-GR were transiently transfected with pRL-Renilla and the GRE-luc (A) or MHC-luc NF-κB (B) luciferase reporters. Cells were then treated with Dex (0 to 100 nM) as indicated. After 20 h, the cells were lysed and analyzed for luciferase activity, which was normalized to the Renilla activity. Plotted are the normalized percentages of GRE transactivation or NF-κB transrepression by Dex (*, P < 0.05). (C) WT-GR-expressing U-2 OS cells were treated with the GSK-3α/β inhibitor BIO (5 μM) and Dex (100 nM) for 20 h. Cells were then lysed, analyzed by Western blotting, and probed with antibodies directed to the NF-κB regulated prosurvival genes Bcl-xL, c-IAP, and survivin. The protein band intensities were quantitated from three independent experiments and normalized to actin. (D) WT-and S404D-GR-expressing U-2 OS cells were treated with Dex for 20 h (100 nM), lysed, analyzed by Western blotting, and probed with antibodies directed to Bcl-xL, c-IAP, and survivin. (E) U-2 OS cells stably expressing WT- or S404A-GR were treated with 100 nM Dex for 1 h, and the cell lysates were immunoprecipitated and then probed with anti-p65/RelA, anti-CBP/p300, anti-GRIP1, or anti-GR antibodies. (F) WT- or S404A-GR were incubated with or without 1 μM Dex before digestion with 50 ng of trypsin/ml and analyzed by PAGE. See Materials and Methods for details.
Our results demonstrated that Ser404 phosphorylation affected GR-mediated NF-κB repression, so we next sought to determine whether phosphorylation alters the ability of the GR to form a complex with NF-κB (p65/RelA). Several recent reports suggest it is mainly the interaction with p65/RelA that finally leads to GR-mediated NF-kB repression (35). Figure 7E shows that upon Dex treatment, GR formed a complex with p65/RelA. Interestingly, when Ser404 phosphorylation was blocked via mutation, GR had a stronger association with p65 after Dex stimulation. Based on this result, we wanted to determine whether other cofactors were also differentially recruited by GR phosphorylation. We found that in the presence of Dex, WT and phospho-deficient GRs (S404A) were able to interact similarly with GR interacting protein 1 (GRIP1); however, the phospho-mutant GR (S404A) had a weaker association with the transcriptional coactivator CBP/p300. Therefore, our results indicate that the presence of Ser404 phosphorylation on the GR affects cofactor recruitment and partially explain the altered gene expression patterns observed. Finally, due to the altered cofactor recruitment to the GR, we speculated that Ser404 phosphorylation might induce a conformational change within the GR that would result in different cofactor associations with unphosphorylated and phosphorylated receptors. Therefore, limited trypsin digestion analysis was carried out on WT- and S404A-GR to test this hypothesis. When digested with trypsin, WT-GR is cleaved at several positions, producing a number of fragments, including the production of a characteristic 16-kDa fragment. In addition, previous reports show that a conformational change within GR upon Dex binding prevents the formation of this fragment (15). In accordance with previous results, we, too, found that digestion of WT-GR with trypsin produced a 16-kDa fragment, whereas Dex treatment prevented it (Fig. 7F). However, we found this 16-kDa fragment was far less apparent in the S404A receptor than the WT receptor both in the absence and in the presence of Dex (Fig. 7F). Therefore, the altered amount of 16-kDa fragment of GR suggests that upon Dex binding, phosphorylated and unphosphorylated GRs adopt different conformations. Together, our results indicate that Ser404 phosphorylation of the GR affects the ability of GR to repress NF-κB, and cells lacking Ser404 phosphorylation may have an altered ability to respond to hormone due to different cofactor recruitment to the GR.
Phosphorylation of Ser404 of GR protects against Dex-dependent death in U-2 OS cells.
Together, Dex and GR promote osteoblast apoptosis primarily by their ability to induce proapoptotic gene expression and repress prosurvival gene expression (6, 30). Due to Ser404 phosphorylation status affecting the ability of the GR to repress prosurvival genes, we hypothesized that receptor phosphorylation may affect GC-dependent cell death in U-2 OS cells. We evaluated this possibility by determining the viability of GR-expressing U-2 OS cells in the presence of Dex. Dex treatment of WT-GR-expressing U-2 OS cells resulted in 45.9% ± 6.2% cell death after 48 h, while S404A-GR-expressing cells showed increased cell death of 75.8% ± 4% (Fig. 8A) after Dex treatment. Interestingly, cells expressing the phospho-mimic, S404D, were highly resistant to GC-mediated cell death (8.2% ± 0.9%; Fig. 8A). We also show that addition of the GSK-3α/β inhibitor BIO enhanced cell death in Dex-treated WT-GR-expressing cells but not in Dex-treated S404A-GR-expressing cells (Fig. 8B). To further support that the results seen in Fig. 8B were not inhibitor specific, WT-GR-expressing cells were treated with several different GSK-3α/β inhibitors (lithium chloride, SB415286, and AR-A014418), and each exhibited the similar result of enhanced GC-dependent cell death (data not shown). Finally, to evaluate the requirement of GSK-3β in mediating Ser404 phosphorylation, we used short hairpin RNA constructs to knockdown the expression of GSK-3α/β. Figure 8D shows that after 3 days in culture, there was a >80% knockdown of GSK-3β protein and a >80% decrease in Dex-mediated Ser404 GR phosphorylation. Interestingly, the lack of GSK-3β expression significantly enhanced GC-dependent cell death in WT-GR-expressing U-2 OS cells but not in phosphorylation-deficient S404A-GR-expressing cells (Fig. 8C). The knockdown was repeated with an additional short hairpin RNA construct targeted to a similar region of GSK-3α/β and produced similar results (data not shown). Our results indicate that cells incapable of GR phosphorylation at Ser404 are more responsive to Dex-induced cell death than are cells that are capable of GR Ser404 phosphorylation. These data also suggest that cells with high levels of Ser404 phosphorylation due to increased GSK-3β activity may be resistant to GC-dependent apoptosis.
FIG. 8.

Phosphorylation of Ser404 of GR protects against Dex-dependent death in U-2 OS cells. (A) U-2 OS cells expressing WT-, S404A-, or S404D-GR were treated with Dex (100 nM) for 48 h. (B) Parental, WT-, or S404A-GR-expressing U-2 OS cells were treated with the GSK-3α/β inhibitor BIO (5 μM) and/or Dex (100 nM) for 24 h. (C and D) WT- or S404A-GR-expressing U-2 OS cells were transfected with control or shGSK-3 knockdown plasmids for 3 days. Cells were subsequently treated with Dex (100 nM) for 30 h. (A, B, and C) After the indicated treatments, the cells were resuspended in 10 mg of PI/ml. Dead cells were analyzed for PI incorporation on a flow cytometer. Western blot analysis was performed in parallel with PI staining to confirm GSK-3β protein knockdown and the level of Ser404 phosphorylation of GR (*, P < 0.05).
DISCUSSION
GCs are currently used to treat a wide variety of pathological conditions from an isolated skin rash to the chronic condition of rheumatoid arthritis. Therefore, the sensitivity of tissues to GCs plays a key role in their ability to treat pathological conditions. Recent work has shown that several factors contribute to the hormonal actions of GR signaling, and alterations in these factors lead to GC resistance or hypersensitivity. The major contributing factors in GR signaling are ligand availability, receptor isoform expression, promoter association, and GR stability (25). In addition, several recent studies suggest that covalent additions such as acetylation, ubiquitylation, and phosphorylation affect steroid receptor stability and activity (17). Therefore, the presence or absence of posttranslational modifications within different tissues would provide a potential mechanism for cell- or gene-specific regulation of GR function.
The work presented here describes a novel convergence point between GSK-3β and the GR pathway. Specifically, we found that upon hormone stimulation, GSK-3β can directly phosphorylate GR on Ser404 (Fig. 1 and 2). Furthermore, we found that this phosphorylation event alters the ability of GR to function as a transcription factor (Fig. 5, 6, and 7) and ultimately effects how U-2 OS osteosarcoma cells respond to GCs (Fig. 9). Therefore, the phosphorylation status of Ser404 has an important role in dictating how the GR will respond to GC stimulation. One could also speculate that cells with aberrant GSK-3β activity would have an altered response to GCs.
FIG. 9.
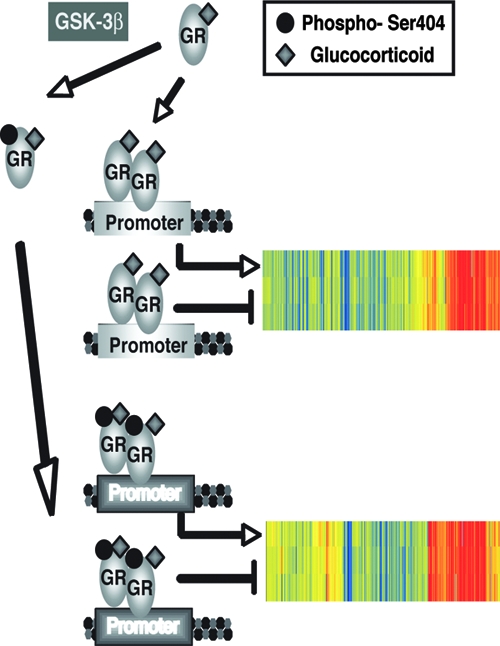
The binding of hormone to the GR allows for DNA binding and the subsequent alteration of gene transcription. The phosphorylation of the GR on Ser404 by GSK-3β significantly alters the GR-mediated transcriptional response. Shown is a cluster analysis of the microarray data to include the GC-regulated genes involved in the cell death pathway of WT GR (805 genes) and phospho-deficient GR (991 genes).
GSK-3α/β kinase was originally identified as a regulator of glycogen synthesis (49) and has since been shown to regulate, both positively and negatively, a broad range of substrates by phosphorylation (3). The majority of GSK-3α/β substrates are formed via prior phosphorylation by an additional kinase at position P+4 (pS/TXXXpS/T), also referred to as the priming site. However, several substrates, such as β-catenin, tau, and axin, do not require a priming site prior to GSK-3α/β phosphorylation (18). We did not discover a priming site for GSK-3β within the GR; however, further experiments will determine whether such a site exists and the requirement of a priming event.
GSK-3α/β is active in resting cells and regulates glucose metabolism by phosphorylating and inactivating glycogen synthase. An insulin stimulus to normal cells inhibits GSK-3α/β kinase activity and thus relieves the inhibitory control of glucose metabolism. However, cells that have become resistant to insulin fail to regulate glucose and display elevated GSK-3α/β activity (20). Due to the ability of chronic GC treatment to induce insulin resistance, it would be interesting to explore the role of GSK-3β-mediated GR phosphorylation in pathological conditions such as metabolic syndrome and obesity. Our lab is currently exploring such possibilities.
Recently, several groups have demonstrated a role for GSK-3β kinase in NF-κB activation and cell survival in a variety of cell types (13, 21, 36, 37). Their results show that loss of GSK-3β function in cells leads to excessive tumor necrosis factor alpha toxicity, resulting in enhanced cellular death. These observations are consistent with our present findings that decreased GSK-3β activity leads to enhanced cellular death in osteoblasts. Similarly, we observed that GSK-3β activity can affect NF-κB-mediated gene regulation. Our work here opens the possibility that GSK-3β activity may control NF-κB transcriptional responses by Ser404 phosphorylation of the GR and altered binding to p65 (Fig. 5 and 7). Therefore, it would be of interest to explore the possibility that aberrant NF-κB activation, as in cancer and autoimmunity, may be partially due to an alteration in the phosphorylation status of the GR.
Activation of the GR by hormone resulted in a transcription alteration of nearly 20% of the human genome (Fig. 5). Therefore, pathways that modify the GR response will have a global impact on the function and properties of the cell. Here, we illustrate that a single posttranslational modification to the GR can significantly redirect the transcriptional output of the cell in response to hormone. As shown in Fig. 6A, hormone-activated WT and phospho-deficient GRs are capable of activating the same pathways, though with significantly different gene regulation patterns. More surprisingly, the phospho-deficient GR mutant can selectively regulate additional pathways in excess of the WT receptor (Fig. 6B). Therefore, we show a global importance of GSK-3β-mediated phosphorylation of the GR to significantly redirect the transcriptional output of cells and thus alter their response to hormone. Finally, our results are not without precedence. While the present study was in review, Chen et al. published a study showing that GR phosphorylation at Ser211 and Ser226 alters the expression profile of several GC-responsive genes (8).
Finally, chronic GC treatments often lead to tissue- and cell-type-specific resistance. Mechanisms resulting in GC resistance are rarely attributed to mutation of the GR but often attributed to the convergence of additional activated cell signaling components with the GR pathway. Such pathways include mitogen-activated kinases, protein kinase A, CDKs, and receptor tyrosine kinase (2, 26, 32, 39, 44). We show here the convergence of another pathway, the GSK-3β signaling pathway, which also exerts a form of cellular resistance to GC administration. Specifically, we find that when Ser404 of GR is mutated to aspartic acid (S404D), a cellular mutation to mimic GSK-3 phosphorylation of GR, osteoblasts are protected against apoptosis (Fig. 8). These results suggest that Ser404 phosphorylation of GR is a possible mechanism to explain the ability of some cells to become resistant to GC-induced apoptosis. Taken together, our results demonstrate the important and novel role of GSK-3β-mediated phosphorylation of Ser404 of the GR in regulating GC signaling.
Acknowledgments
We thank Katina Johnson for assistance with the mass spectrometry analysis. We thank Laura Wharey and the rest of the NIEHS Microarray Core for help with the microarray data and analysis. We also thank Jeff Tucker for guidance with the confocal imagery. We are also grateful to Carl D. Bortner and Maria Sifre for technical assistance with the flow cytometric analysis. We thank the members of the Cidlowski lab, Katherine L. Gross, Robert H. Oakley, and Alyson B. Scoltock for critical reading of the manuscript. Finally, we thank Paul Housley for help in characterizing the phospho-specific antibodies to the GR.
This research was supported by the Intramural Research Program of the NIH National Institute of Environmental Health Sciences (Z01E5090057-12).
Footnotes
Published ahead of print on 6 October 2008.
REFERENCES
- 1.Ali, A., K. P. Hoeflich, and J. R. Woodgett. 2001. Glycogen synthase kinase-3: properties, functions, and regulation. Chem. Rev. 1012527-2540. [DOI] [PubMed] [Google Scholar]
- 2.Bachmann, P. S., R. Gorman, R. A. Papa, J. E. Bardell, J. Ford, U. R. Kees, G. M. Marshall, and R. B. Lock. 2007. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res. 674482-4490. [DOI] [PubMed] [Google Scholar]
- 3.Billadeau, D. D. 2007. Primers on molecular pathways: the glycogen synthase kinase-3β. Pancreatology 7398-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blind, R. D., and M. J. Garabedian. 2008. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J. Steroid Biochem. Mol. Biol. 109150-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodwell, J. E., E. Orti, J. M. Coull, D. J. Pappin, L. I. Smith, and F. Swift. 1991. Identification of phosphorylated sites in the mouse glucocorticoid receptor. J. Biol. Chem. 2667549-7555. [PubMed] [Google Scholar]
- 6.Canalis, E. 2005. Mechanisms of glucocorticoid action in bone. Curr. Osteoporos. Rep. 398-102. [DOI] [PubMed] [Google Scholar]
- 7.Cao, H., L. J. Deterding, J. D. Venable, E. A. Kennington, J. R. Yates III, K. B. Tomer, and P. J. Blackshear. 2006. Identification of the anti-inflammatory protein tristetraprolin as a hyperphosphorylated protein by mass spectrometry and site-directed mutagenesis. Biochem. J. 394285-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, W., T. Dang, R. D. Blind, Z. Wang, C. N. Cavasotto, A. B. Hittelman, I. Rogatsky, S. K. Logan, and M. J. Garabedian. 2008. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol. Endocrinol. 221754-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi, J. H., J. Williams, J. Cho, J. R. Falck, and S. B. Shears. 2007. Purification, sequencing, and molecular identification of a mammalian PP-InsP5 kinase that is activated when cells are exposed to hyperosmotic stress. J. Biol. Chem. 28230763-30775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chrousos, G. P., and T. Kino. 2007. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress 10213-219. [DOI] [PubMed] [Google Scholar]
- 11.Cidlowski, J. A., D. L. Bellingham, F. E. Powell-Oliver, D. B. Lubahn, and M. Sar. 1990. Novel antipeptide antibodies to the human glucocorticoid receptor: recognition of multiple receptor forms in vitro and distinct localization of cytoplasmic and nuclear receptors. Mol. Endocrinol. 41427-1437. [DOI] [PubMed] [Google Scholar]
- 12.DeFranco, D. B., M. Qi, K. C. Borror, M. J. Garabedian, and D. L. Brautigan. 1991. Protein phosphatase types 1 and/or 2A regulate nucleocytoplasmic shuttling of glucocorticoid receptors. Mol. Endocrinol. 51215-1228. [DOI] [PubMed] [Google Scholar]
- 13.Demarchi, F., C. Bertoli, P. Sandy, and C. Schneider. 2003. Glycogen synthase kinase-3β regulates NF-κB1/p105 stability. J. Biol. Chem. 27839583-39590. [DOI] [PubMed] [Google Scholar]
- 14.Dephoure, N., C. Zhou, J. Villen, S. A. Beausoleil, C. E. Bakalarski, S. J. Elledge, and S. P. Gygi. 2008. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 10510762-10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong, D. D., C. M. Jewell, R. J. Bienstock, and J. A. Cidlowski. 2006. Functional analysis of the LXXLL motifs of the human glucocorticoid receptor: association with altered ligand affinity. J. Steroid Biochem. Mol. Biol. 101106-117. [DOI] [PubMed] [Google Scholar]
- 16.Edgar, R., M. Domrachev, and A. E. Lash. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30207-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faus, H., and B. Haendler. 2006. Post-translational modifications of steroid receptors. Biomed Pharmacother. 60520-528. [DOI] [PubMed] [Google Scholar]
- 18.Harwood, A. J. 2001. Regulation of GSK-3: a cellular multiprocessor. Cell 105821-824. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi, R., H. Wada, K. Ito, and I. M. Adcock. 2004. Effects of glucocorticoids on gene transcription. Eur. J. Pharmacol. 50051-62. [DOI] [PubMed] [Google Scholar]
- 20.Henriksen, E. J., and B. B. Dokken. 2006. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 71435-1441. [DOI] [PubMed] [Google Scholar]
- 21.Hoeflich, K. P., J. Luo, E. A. Rubie, M. S. Tsao, O. Jin, and J. R. Woodgett. 2000. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 40686-90. [DOI] [PubMed] [Google Scholar]
- 22.Hunter, T. 2007. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28730-738. [DOI] [PubMed] [Google Scholar]
- 23.Ismaili, N., and M. J. Garabedian. 2004. Modulation of glucocorticoid receptor function via phosphorylation. Ann. N. Y. Acad. Sci. 102486-101. [DOI] [PubMed] [Google Scholar]
- 24.Kassel, O., and P. Herrlich. 2007. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol. Cell Endocrinol. 27513-29. [DOI] [PubMed] [Google Scholar]
- 25.Kino, T. 2007. Tissue glucocorticoid sensitivity: beyond stochastic regulation on the diverse actions of glucocorticoids. Horm. Metab. Res. 39420-424. [DOI] [PubMed] [Google Scholar]
- 26.Kino, T., T. Ichijo, N. D. Amin, S. Kesavapany, Y. Wang, N. Kim, S. Rao, A. Player, Y. L. Zheng, M. J. Garabedian, E. Kawasaki, H. C. Pant, and G. P. Chrousos. 2007. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol. Endocrinol. 211552-1568. [DOI] [PubMed] [Google Scholar]
- 27.Kinyamu, H. K., J. Chen, and T. K. Archer. 2005. Linking the ubiquitin-proteasome pathway to chromatin remodeling/modification by nuclear receptors. J. Mol. Endocrinol. 34281-297. [DOI] [PubMed] [Google Scholar]
- 28.Krstic, M. D., I. Rogatsky, K. R. Yamamoto, and M. J. Garabedian. 1997. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol. Cell. Biol. 173947-3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu, N. Z., and J. A. Cidlowski. 2005. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell 18331-342. [DOI] [PubMed] [Google Scholar]
- 30.Lu, N. Z., J. B. Collins, S. F. Grissom, and J. A. Cidlowski. 2007. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol. Cell. Biol. 277143-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manoukian, A. S., and J. R. Woodgett. 2002. Role of glycogen synthase kinase-3 in cancer: regulation by Wnts and other signaling pathways. Adv. Cancer Res. 84203-229. [DOI] [PubMed] [Google Scholar]
- 32.Miller, A. L., A. S. Garza, B. H. Johnson, and E. B. Thompson. 2007. Pathway interactions between MAPKs, mTOR, PKA, and the glucocorticoid receptor in lymphoid cells. Cancer Cell Int. 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muyllaert, D., A. Kremer, T. Jaworski, P. Borghgraef, H. Devijver, S. Croes, I. Dewachter, and F. Van Leuven. 2008. Glycogen synthase kinase-3β, or a link between amyloid and tau pathology? Genes Brain Behav. 7(Suppl. 1)57-66. [DOI] [PubMed] [Google Scholar]
- 34.Necela, B. M., and J. A. Cidlowski. 2004. Mechanisms of glucocorticoid receptor action in non-inflammatory and inflammatory cells. Proc. Am. Thorac. Soc. 1239-246. [DOI] [PubMed] [Google Scholar]
- 35.Neumann, M., and M. Naumann. 2007. Beyond IκBs: alternative regulation of NF-κB activity. FASEB J. 212642-2654. [DOI] [PubMed] [Google Scholar]
- 36.Ougolkov, A. V., N. D. Bone, M. E. Fernandez-Zapico, N. E. Kay, and D. D. Billadeau. 2007. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor κB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 110735-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ougolkov, A. V., M. E. Fernandez-Zapico, D. N. Savoy, R. A. Urrutia, and D. D. Billadeau. 2005. Glycogen synthase kinase-3β participates in nuclear factor κB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 652076-2081. [DOI] [PubMed] [Google Scholar]
- 38.Rhen, T., and J. A. Cidlowski. 2005. Anti-inflammatory action of glucocorticoids-new mechanisms for old drugs. N. Engl. J. Med. 3531711-1723. [DOI] [PubMed] [Google Scholar]
- 39.Rogatsky, I., S. K. Logan, and M. J. Garabedian. 1998. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 952050-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogatsky, I., C. L. Waase, and M. J. Garabedian. 1998. Phosphorylation and inhibition of rat glucocorticoid receptor transcriptional activation by glycogen synthase kinase-3 (GSK-3). Species-specific differences between human and rat glucocorticoid receptor signaling as revealed through GSK-3 phosphorylation. J. Biol. Chem. 27314315-14321. [DOI] [PubMed] [Google Scholar]
- 41.Scoltock, A. B., G. Heimlich, and J. A. Cidlowski. 2007. Glucocorticoids inhibit the apoptotic actions of UV-C but not Fas ligand in hepatoma cells: direct evidence for a critical role of Bcl-xL. Cell Death Differ. 14840-850. [DOI] [PubMed] [Google Scholar]
- 42.Smoak, K. A., and J. A. Cidlowski. 2004. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech. Ageing Dev. 125697-706. [DOI] [PubMed] [Google Scholar]
- 43.Steen, H., J. A. Jebanathirajah, M. Springer, and M. W. Kirschner. 2005. Stable isotope-free relative and absolute quantitation of protein phosphorylation stoichiometry by MS. Proc. Natl. Acad. Sci. USA 1023948-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szatmary, Z., M. J. Garabedian, and J. Vilcek. 2004. Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J. Biol. Chem. 27943708-43715. [DOI] [PubMed] [Google Scholar]
- 45.Tang, D., and J. H. Wang. 1996. Cyclin-dependent kinase 5 (Cdk5) and neuron-specific Cdk5 activators. Prog. Cell Cycle Res. 2205-216. [DOI] [PubMed] [Google Scholar]
- 46.Tseng, A. S., F. B. Engel, and M. T. Keating. 2006. The GSK-3 inhibitor BIO promotes proliferation in mammalian cardiomyocytes. Chem. Biol. 13957-963. [DOI] [PubMed] [Google Scholar]
- 47.Wang, Z., J. Frederick, and M. J. Garabedian. 2002. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J. Biol. Chem. 27726573-26580. [DOI] [PubMed] [Google Scholar]
- 48.Webster, J. C., C. M. Jewell, J. E. Bodwell, A. Munck, M. Sar, and J. A. Cidlowski. 1997. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J. Biol. Chem. 2729287-9293. [DOI] [PubMed] [Google Scholar]
- 49.Woodgett, J. R. 1990. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 92431-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]