

Abstract
PURPOSE:
To develop a systematic approach for the molecular diagnosis of retinitis pigmentosa (RP) and to report new genotype-phenotype correlations for phosphodiesterase 6 (PDE6) based RP mutations.
DESIGN:
Clinical and molecular studies on a retrospective case series.
METHODS:
We screened 40 unrelated RP patients with an autosomal recessive RP microarray. Individuals with RP caused by PDE6 deficiency underwent genetic segregation and phenotype analysis.
RESULTS:
A disease-associated allele was identified in 32% of patients. Two probands (5%) had PDE6 mutations. The first proband was a compound heterozygote for known R102C and N216S alleles in PDE6A (MIM#180071). Pedigree analysis determined that the N216S variant was benign and direct sequencing discovered a novel, S303C allele. The second proband had a homozygous D600N mutation in the PDE6B gene (MIM#180072). Visual acuities of PDE6 deficient patients ranged from 20/40 to 20/200. Clinical studies showed unusual vitreomacular traction, cystoid macular edema, macular atrophy, and ring hyperfluorescence in PDE6 deficient patients. Such extensive vitreoretinal degeneration is not characteristic of photoreceptor-specific enzyme deficiencies.
CONCLUSION:
High throughput DNA microarray chips can be used in combination with clinical imaging to precisely characterize patients with RP. Identifying the precise mutation in RP may become the standard of care as gene therapy emerges.
INTRODUCTION
Death of light-sensing photoreceptors in the retina causes irreversible vision loss in individuals with retinal degenerations such as retinitis pigmentosa (RP). Various forms of RP affect approximately one in 3000 people with an estimated one and a half million RP patients worldwide 1. Genetically heterogeneous, autosomal recessive RP (arRP) is associated with mutations in at least 17 different genes <www.sph.uth.tmc.edu/Retnet/sum-dis.htm#B-diseases>. Aggregate carrier frequency for arRP in North America is estimated to be 15% 2.
Individuals with RP typically present with night blindness due to rod degeneration, but eventually they may progress to lose central vision as a result of cone death 3. About 36,000 cases of simplex and familial RP worldwide are due to defects in rod-specific cyclic guanosine monophosphate (cGMP) phosphodiesterase-6 (PDE6), estimated to account for approximately 8% of all diagnosed arRP 4, 5 Mutations in the β-subunit of cGMP-phosphodiesterase (PDE6B) (MIM#180072) can lead to either progressive retinal disease, such as RP, or stationary disease, such as congenital stationary night blindness (CSNB) 6. Heterozygous carriers of PDE6 mutations are at a risk of losing vision when excessively inhibiting PDE6 with drugs such as sildenafil, tadalafil, or vardenafil 7.
PDE6 consists of catalytic (PDE6A and PDE6B) and regulatory (PDE6G) subunits 8-13; light stimulates PDE6 activity, which eventually leads to photoreceptor activation 14. Mechanisms that initiate rod photoreceptor death are unclear; however, low levels of PDE6 activity are thought to lead to rod-cone degeneration. One hypothesis is that in PDE6 mutants an influx of calcium (Ca2+) causes rod apoptosis, due to lack of PDE activation 15. Pharmaceutical intervention to block excessive Ca2+ influx into rods has been attempted with variable success in mouse models (Pde6brd1/Pde6brd1mice) of the disease and Irish Setter dogs 16-19.
The key to gene-based treatments is efficient and accurate genotyping 20, 21. Known causes of arRP are underlied by defects in at least 17 genes, and all conventional mutation detection techniques remain labor-intensive, time-consuming, and cost inefficient. Disease-associated alleles in the 17 genes have shown substantial heterogeneity; currently >500 disease-associated variants have been identified in these genes combined with only a few alleles detected in more than 1-2 families. Comprehensive mutation analysis, therefore, involves screening of at least 120 amplicons (exons) that encompass most of the entire open reading frames of the 17 genes. To overcome these challenges and to generate a high throughput, cost-effective, and efficient screening tool, we used the arRP genotyping microarray (Asper Ophthalmics, Tartu, Estonia).
Herein, we describe the use of the arRP microarry for genetic screening of RP patients, followed by sequencing and segregation analysis. Together with a comprehensive clinical examination, the cause of arRP can be precisely determined in patients formerly left without a specific diagnosis.
METHODS
Imaging and Psychophysical Testing
Patients were enrolled with the approval of the Institutional Review Board (protocol #AAAB6560) at Columbia University. The tenets of the Declaration of Helsinki were followed. Forty unrelated individuals with autosomal recessive RP were phenotyped. If applicable, additional family members were examined. Fundus photographs, optical coherence tomography (OCT, Zeiss Stratus and Zeiss Cirrus), and fundus autofluorescence (AF) images were reviewed. AF was obtained by illuminating the fundus with argon laser light (488 nm) and viewing the resultant fluorescence through a band pass filter with a short wavelength cut off at 495nm 22. Microperimetry (MP1, Nidek Technologies, Milan, Italy) static perimetry program with monitoring of fixation of a 3-degree red cross was used to monitor macular sensitivity. For each test point location light sensitivity threshold values were recorded.
Electrophysiological Testing
Pupils were dilated before full-field electroretinogram testing using tropicamide (1%) and phenylephrine hydrochloride (2.5%). Full-field ERGs were performed using extended testing protocols incorporating the International Society for Clinical Electrophysiology of Vision (ISCEV) standard 22. The protocol incorporated the rod-specific and standard bright flash ERGs, both recorded after a minimum of 20 minutes dark adaptation. Following 10 minutes light adaptation, the photopic 30 Hz flicker cone and transient photopic cone ERGs were recorded. A stimulus 0.6 log units greater than the ISCEV standard flash was also used to better demonstrate the a-wave, as suggested in the recent revision of the ISCEV standard for ERG.
Mutation Analyses
DNA extracted from blood samples of 40 unrelated simplex and arRP patients was screened on the arRP genotyping microarray. Microarrays were designed and manufactured with the arrayed primer extension (APEX) technology.23-24, 28 Briefly, 5′-modified sequence-specific oligonucleotides, designed with their 3′ end immediately adjacent to the variable site, are arrayed on a glass slide. Fragmented patient DNA are amplified using polymerase chain reaction and annealed to oligonucleotides on the slide, followed by sequence-specific extension of the 3′ ends of primers with dye-labeled nucleotide analogs (ddNTPs) by DNA polymerase. The APEX reaction is, in essence, a sequencing reaction on a solid support. Every known disease-associated sequence change (501) described in all 17 known arRP genes was included on the chip via sequence-specific oligonucleotides.
For screening purposes, a total of 120 amplicons (exons) from 17 known arRP genes (CERKL, CNGA1, CNGB1, MERTK, LRAT, PDE6A, PDE6B, PNR, RDH12, RGR, RLBP1, SAG, TULP1, CRB1, RPE65, USH2A, USH3A) were PCR-amplified as described previously 23-25. Primer sequences are available upon request. In the amplification mixture, 20% of the dTTP was substituted by dUTP 23, 24, 26. UTP is added to fragment the PCR products with uracil n-glycosylase. The amplification products were concentrated and purified by Generall PCR kit (Biosystems, Seoul, Korea). Fragmentation of amplification products was achieved by adding thermolabile uracil n-glycosylase (Epicentre Biotechnologies, Madison, WI) and the following heat treatment 26.
One-sixth of every amplification product was utilized in the primer extension reaction on the arRP array (http://www.asperophthalmics.com/ARRPgenetest.htm). Each APEX reaction consisted of a fragmented and denatured PCR product, 4 units of ThermoSequenase DNA Polymerase (Amersham Biosciences, Piscataway, NJ), 1x reaction buffer and 1.4 μM final concentration of each fluorescently labeled ddNTP: Texas Red-ddATP, fluorescein-ddGTP (Amersham Biosciences, Piscataway, NJ), Cy3-ddCTP, Cy5-ddUTP (NEN). The reaction mixture was applied to a microarray slide for 15 minutes at 58° C. The reaction was stopped by washing the slide at 95° C in Milli-Q water 27. The slides were imaged with the Genorama™ QuattroImager (Asper Ophthalmics, Tartu, Estonia) and the sequence variants were identified by Genorama Genotyping Software 23, 24, 27. Further details about APEX arrays and analysis can be found elsewhere 24, 28.
Array-identified variants were confirmed by direct sequencing with the Taq Dyedeoxy Terminator Cycle Sequencing kit (Applied Biosystems, Framingham, MA), according to the manufacturer's instructions. Sequencing reactions were resolved on an ABI 377 automated sequencer.
RESULTS
Genetic Screening
Screening of 40 unrelated patients diagnosed with autosomal recessive or sporadic RP on the arRP array resulted in identification of at least 1 (out of 2, as per definition of a recessive disease) possible disease-associated mutation in 32.5% (13/40) individuals. The likely disease-associated variants were identified in the following genes: PDE6A (R102C and N216S), PDE6B (D600N), CRB1 (T745M and P836T), USH2A (E478D, L555V, W4149R, P1978S, G713R, E767fs, and C759F) and RPE65 (A132T).
A proband with array-identified R102C and N216S variants in the PDEA gene was a member of a large family from which eight people were available for examination (Figure 1). Of these, two additional siblings (cases II-4, II-5 and II-8) were clinically diagnosed with RP. Both parents and five other siblings were unaffected. Segregation analysis showed that all three affected family members and one unaffected sibling carried a 304 C>T transition in exon 1 causing the R102C mutation. However, the N216S variant was found in all family members, including the carrier of R102C. Therefore, it was defined as a benign missense polymorphism. The PDE6A gene was completely sequenced in the proband, which resulted in confirmation of the R102C and N216S variants but, more importantly, in finding a novel mutation 908C>G (S303C, Figure 2). The S303C variant segregated with the disease in the family; only affected family members possessed both R102C and S303C mutations (Figure 1).
Figure 1.

Figure 2.

In the second RP proband of no relation to the previous family, genetic analysis showed homozygous 1798 G>A mutation in exon 14 of PDE6B gene. This mutation introduces a D600N amino acid change.
Case II-4. (Figure 3)
Figure 3.
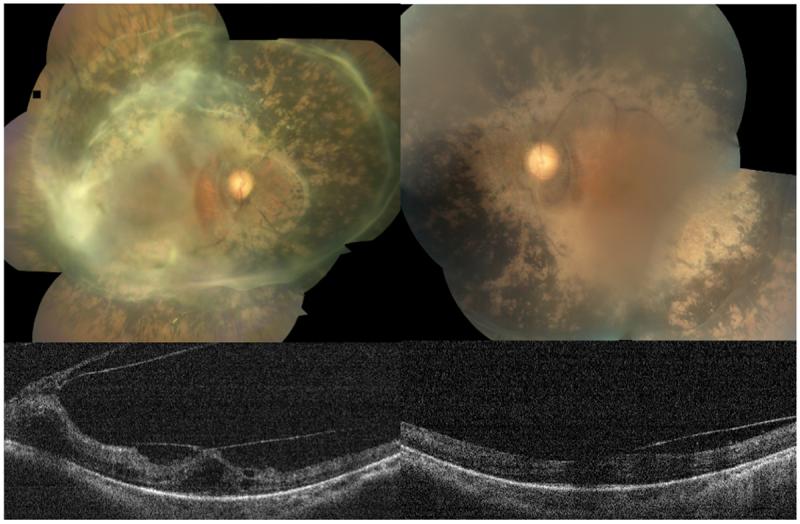
The proband was a 41-year old man who presented with 20/100 vision in each eye. Cataract extraction in the right eye had improved his vision from 20/200 to 20/100. He had a posterior intraocular lens in his right eye and there was a visually significant posterior subcapsular cataract in his left eye. Dilated funduscopic exam showed extensive non-nummular intraretinal pigmentation from the arcades to the mid-peripheral retina. There were extensive epiretinal membranes with thickened hyaloid in both eyes, which extended to cover circumferentially the entire arcades in both maculae. He had bilateral vitreomacular traction and cystoid edema confirmed by optical coherence tomography (OCT). Central macular thicknesses were 304 microns in the right and 472 microns in the left. We felt that the patient was unlikely to benefit from surgical intervention to remove the epiretinal membranes at the present time.
Case II-5. (Figure 4)
Figure 4.
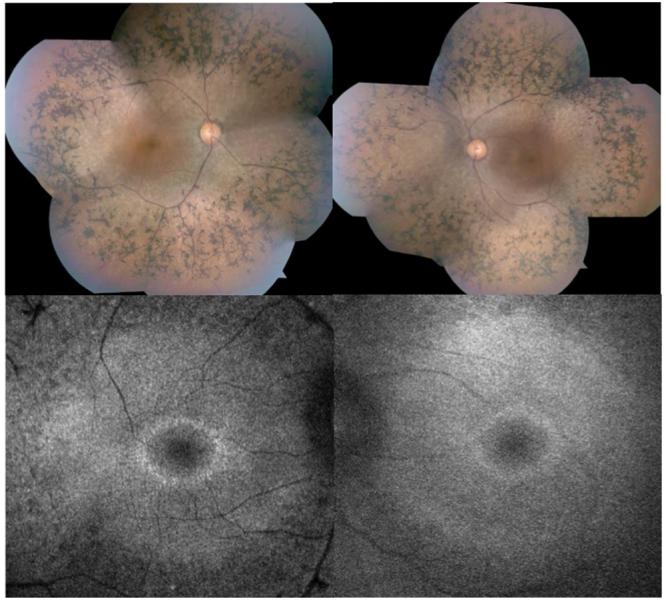
A 38-year old man presented with visual acuity of 20/60 in each eye. Anterior segment examination was remarkable for trace posterior subcapsular cataracts. Ophthalmoscopy showed extensive intraretinal pigment migrations extending from the mid-periphery equatorial region to the arcades in both eyes with extensive arterial attenuation. There was some central sparing of the RPE in both maculae, and the eyes were without edema (confirmed with OCT). Scanning laser ophthalmoscopy autofluorescence (AF) examination showed central high density AF rings.
Case II-8. (Figure 5)
Figure 5.

A 27-year old man presented with 20/50 vision in each eye. Anterior segment examination was within normal limits. There was extensive non-nummular intraretinal pigment migration from the equatorial region to the arcades in both eyes with extensive arteriolar attenuation. There was mild bilateral cystoid macular edema, which was confirmed by OCT. Scanning laser ophthalmoscopy AF showed rings of high-density signal in both maculae with pigment displacement and a petalloid pattern within the larger ring. MP1 sensitivities were decreased in the area of hyperfluorescence. (Figure 5) The patient was treated with 1 gram daily of oral acetazolamide for three months with improvement of visual acuity to 20/25- as well as functional improvement as measured by MP1.
Case of PDE6B Mutation. (Figure 6)
Figure 6.
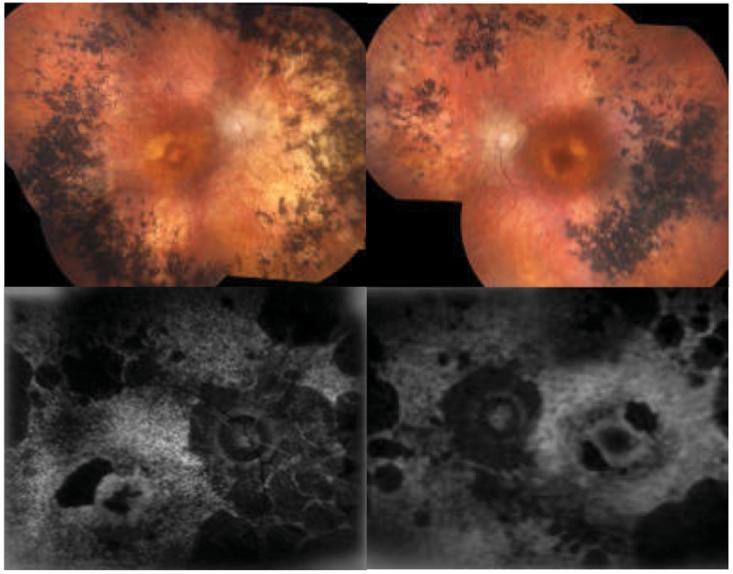
An 81-year old woman presented with 20/200 vision in her right eye and 20/30 vision in her left eye. There was no known family history of visual impairment. Her past medical history was significant for poorly differentiated gastric adenocarcinoma with metastasis to her esophagus. She was pseudophakic in both eyes with posterior intraocular lenses. Fundus photos showed atrophic RPE in both maculae with pale optic nerves and intraretinal pigment migration in the mid-periphery of both eyes. AF showed severe loss of RPE function in both eyes, except a central foveal island in the left.
Electrophysiological Features of PDE6 Deficiency
In all four PDE6-deficient RP patients tested, ERG showed severe generalized rod-cone dysfunction. Scotopic rod specific ERG, maximal ERG, photopic 30 Hz flicker ERG and transient photopic ERG were all severely abnormal. There was relative preservation of the photopic 30 Hz flicker ERG (II-8) compared to rod-specific ERG responses. This signified the rod system was more severely affected than the cone system, which is consistent with PDE6A and PDE6B mutations (Figure 7).
Figure 7.

Neither R102C or S303C mutations caused any abnormal physiological or funduscopic features in the four heterozygous carriers analyzed (Figure 7).
DISCUSSION
Genetic heterogeneity in retinitis pigmentosa (RP) makes it challenging to advise patients regarding their visual prognoses. Genotyping and comprehensive phenotyping with ERG, AF, OCT, and MP1 mapping are helpful to specify prognosis and for gene-based treatment trials 3, 14, 29. Therefore, we applied a high throughput genotyping array platform, electrophysiological testing, imaging and psychophysical studies to genotype and phenotype patients with PDE6-related RP.
In one patient, AF imaging revealed abnormal foci of high density that corresponded with retinal sensitivity map obtained with the MP1 (Figure 5). High density AF lesions suggest localized accumulation of lipofuscin and disruption of RPE metabolism that may eventually be associated with photoreceptor death and RPE atrophy. These annuli of high density have been shown to constrict with time 30. The increase in autofluorescence may represent an intermediate stage of increased photoreceptor-RPE metabolic load before apoptosis initiates.
Although PDE6A is a photoreceptor-specific gene, two of our PDE6A deficient patients exhibited extensive vitreoretinal degenerations (Figure 3 and 5) as well as tractional vitreous rings (Figure 3). One mechanism to explain this may be that loss of the photoreceptors elicits new glial barriers causing Muller cells to migrate. In a ten year follow-up on a PDE6B deficient patient, there were structural changes in the inner retinal (retinal remodeling) 31, which may also account for the changes in the vitreoretinal interphase. Mice lacking PDE6 also display extensive retinal remodeling following rod death 32-34.
Phenotype-genotype correlation studies are essential as genetic testing is more readily available and gene-based therapies are emerging. After mutations in the USH2A gene, PDE6 defects are the second most common dentifiable cause of arRP 4, 5. Two probands with PDE6 defects were identified from a pool of 40 unrelated RP patients. Three affected individuals (cases 1 to 3) from the family of the first proband carried compound heterozygous mutations in PDE6A. Both R102 and S303 are conserved residues in PDE6A in different species (Figure 2). N216S was a benign polymorphism found in all non-affected and affected family members. Case 4 had a homozygous mutation in PDE6B. D600 is located in a highly conserved zinc (Zn2+) and magnesium (Mg2+) binding site of PDE6B based on modeling of the PDE4 crystal structure 35.
Loss of rod PDE6 catalytic activity and photoreceptor dysfunction can be detected by electroretinography (Figure 7). Electrophysiological testing helps to diagnose and predict patient's course of disease. A mouse model (Pde6brd1/Pde6brd1) with extinguished PDE6 function has been used to study RP for the past 80 years 36. Mice lacking PDE6 activities exhibit a dramatic elevation of retinal cGMP with subsequent rapid rod degeneration within 3 weeks after birth. Abnormally high outer segment cGMP 37-39, and Ca2+ 15 levels underlie degeneration of rods seen in Pde6brd1/Pde6brd1. Since high cGMP levels opens cGMP-gated Na+/Ca2+ channels (CNG), it is believed that high Ca2+ concentrations accompany high cGMP levels in Pde6brd1/Pde6brd1mutants between P10 and P14.
High throughput array-based DNA screening and functional clinical evaluation, such as electrophysiology and AF, help verify and prognosticate PDE6-related retinal degenerations. This information is important for genetic counseling of families with retinal degenerations. Early diagnosis enables better utilization of special educational services for children who will eventually have learning difficulties.
Gene specific therapeutics for early onset retinal dystrophy patients are already undergoing clinical trials, and to recruit patients for any genetic trial, their specific gene-defects must be known 28. Furthermore, in theory patients with PDE6 defects may specifically benefit from Ca2+channel blockers 17, or other PDE6 gene-based therapies 24, 40-43.
In conclusion, a combination of comprehensive phenotyping and precise genotyping improves patient counseling and contributes to the future of gene-based therapy in arRP, one of most heterogeneous and complex disorders in human genetics.
ACKNOWLEDGEMENTS
Funding / Support; SHT: Burroughs-Wellcome Program in Biomedical Sciences Fellow, Charles Culpeper Scholarship, Foundation Fighting Blindness, Hirschl Trust, Schneeweiss Stem Cell Fund, Joel Hoffmann Foundation, Jonas Family Fund, Crowley Research Fund, Jahnigen/Hartford/American Geriatrics Society, Eye Surgery Fund, Bernard Becker-Association of University Professors in Ophthalmology-Research to Prevent Blindness (RPB), Jules Stein-RPB Omics Laboratory and NIH grant K08-EY004081. RA: Schneeweiss Stargardt Fund and NHI grant R01EY013435-07.
Charlie Martin at Jules Stein Eye Institute for photographs.
Biography
 Since 1992, Stephen H. Tsang, MD, PhD, Bernard-Becker-AUPO-RPB scholar and Assistant Professor in Retina Division at New York-Presbyterian Hospital has been studying cGMP-phosphodiesterase (PDE6), a key component of the phototransduction cascade. Dr. Tsang's team provided evidence on how light induced post-translational modification of PDE6 regulates signaling in the living retina. Dr. Tsang generated lines of transgenic mice that provide models for testing novel treatment approaches for patients with RP.
Since 1992, Stephen H. Tsang, MD, PhD, Bernard-Becker-AUPO-RPB scholar and Assistant Professor in Retina Division at New York-Presbyterian Hospital has been studying cGMP-phosphodiesterase (PDE6), a key component of the phototransduction cascade. Dr. Tsang's team provided evidence on how light induced post-translational modification of PDE6 regulates signaling in the living retina. Dr. Tsang generated lines of transgenic mice that provide models for testing novel treatment approaches for patients with RP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE
None: I.T., C.L.C., J.Z., E.H., R.I., J.T.,
Financial Disclosures; E.H. is a scientist at Asper Ophthalmics. R.A. and J.Z. are scientific partners with Asper Ophthalmics. None: S.H.T., I.T., C.L.C., R.I., J.T.
Statement about conformity with Author Information: Institutional Review Board Protocol #AAAB6560 approved at Columbia University
REFERENCES
- 1.Bird AC. Retinal photoreceptor dystrophies: the LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 1995;119:543–562. doi: 10.1016/s0002-9394(14)70212-0. [DOI] [PubMed] [Google Scholar]
- 2.Rivolta C, Sharon D, DeAngelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002;11:1219–1227. doi: 10.1093/hmg/11.10.1219. [DOI] [PubMed] [Google Scholar]
- 3.Tsui I, Song B, Lin C-S, Tsang S. A Practical Approach to Retinal Dystrophies. Retinal Physician. 2007;4:18–26. doi: 10.1007/978-3-319-95046-4_51. http://www.retinalphysician.com/article.aspx?article=100310. [DOI] [PubMed]
- 4.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 5.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsang SH, Woodruff ML, Jun L, et al. Transgenic mice carrying the H258N mutation in the gene encoding the beta-subunit of phosphodiesterase-6 (PDE6B) provide a model for human congenital stationary night blindness. Hum Mutat. 2007;28:243–54. doi: 10.1002/humu.20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stockman A, Sharpe LT, Tufail A, Kell PD, Ripamonti C, Jeffery G. The effect of sildenafil citrate (Viagra) on visual sensitivity. J Vis. 2007;7:4. doi: 10.1167/7.8.4. http://journalofvision.org//7/8/4/ [DOI] [PubMed]
- 8.Tsang SH, Burns ME, Calvert PD, et al. Role of the Target Enzyme in Deactivation of Photoreceptor G Protein in Vivo. Science. 1998;282:117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- 9.Tsang SH, Gouras P, Yamashita CK, et al. Retinal degeneration in mice lacking the γ subunit of rod cGMP phosphodiesterase. Science. 1996;272:1026–1029. doi: 10.1126/science.272.5264.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsang SH, Woodruff ML, Chen CK, et al. GAP-independent termination of photoreceptor light response by excess gamma subunit of the cGMP-phosphodiesterase. J Neurosci. 2006;26:4472–4480. doi: 10.1523/JNEUROSCI.4775-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsang SH, Woodruff ML, Janisch KM, Cilluffo MC, Farber DB, Fain GL. Removal of phosphorylation sites of {gamma} subunit of phosphodiesterase 6 alters rod light response. J Physiol. 2006;579:303–312. doi: 10.1113/jphysiol.2006.121772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsang SH, Yamashita CK, Doi K, et al. In vivo studies of the gamma subunit of retinal cGMP-phophodiesterase with a substitution of tyrosine-84. Biochem J. 2001;353:467–474. doi: 10.1042/0264-6021:3530467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsang SH, Yamashita CK, Lee WH, et al. The positive role of the carboxyl terminus of the gamma subunit of retinal cGMP-phosphodiesterase in maintaining phosphodiesterase activity in vivo. Vision Res. 2002;42:439–445. doi: 10.1016/s0042-6989(01)00213-9. [DOI] [PubMed] [Google Scholar]
- 14.Tsang SH, Gouras P. Photoreceptors and photoreceptor dysfunctions. In: Adelman G, Smith B, editors. Encyclopedia of Neuroscience. Elsevier Science; Amsterdam: 2004. pp. 1633–1644. [Google Scholar]
- 15.Lisman J, Fain G. Support for the equivalent light hypothesis for RP. Nat Med. 1995;1:1254–1255. doi: 10.1038/nm1295-1254. [DOI] [PubMed] [Google Scholar]
- 16.Bush RA, Kononen L, Machida S, Sieving PA. The effect of calcium channel blocker diltiazem on photoreceptor degeneration in the rhodopsin Pro213His rat. Invest Ophthalmol Vis Sci. 2000;41:2697–2701. [PubMed] [Google Scholar]
- 17.Vallazza-Deschamps G, Cia D, Gong J, et al. Excessive activation of cyclic nucleotide-gated channels contributes to neuronal degeneration of photoreceptors. Eur J Neurosci. 2005;22:1013–1022. doi: 10.1111/j.1460-9568.2005.04306.x. [DOI] [PubMed] [Google Scholar]
- 18.Pawlyk BS, Li T, Scimeca MS, Sandberg MA, Berson EL. Absence of photoreceptor rescue with D-cis-diltiazem in the rd mouse. Invest Ophthalmol Vis Sci. 2002;43:1912–1915. [PubMed] [Google Scholar]
- 19.Pearce-Kelling SE, Aleman TS, Nickle A, et al. Calcium channel blocker D-cis-diltiazem does not slow retinal degeneration in the PDE6B mutant rcd1 canine model of retinitis pigmentosa. Mol Vis. 2001;7:42–47. [PubMed] [Google Scholar]
- 20.Stone EM. Genetic testing for inherited eye disease. Arch Ophthalmol. 2007;125:205–212. doi: 10.1001/archopht.125.2.205. [DOI] [PubMed] [Google Scholar]
- 21.Stone EM. Leber Congenital Amaurosis-A Model for Efficient Genetic Testing of Heterogeneous Disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007;144:791–811. doi: 10.1016/j.ajo.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 22.Tsang SH, Vaclavik V, Bird AC, Robson AG, Holder GE. Novel phenotypic and genotypic findings in X-linked retinoschisis. Arch Ophthalmol. 2007;125:259–267. doi: 10.1001/archopht.125.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaakson K, Zernant J, Kulm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 24.Zernant J, Kulm M, Dharmaraj S, et al. Genotyping microarray (disease chip) for Leber congenital amaurosis: detection of modifier alleles. Invest Ophthalmol Vis Sci. 2005;46:3052–3059. doi: 10.1167/iovs.05-0111. [DOI] [PubMed] [Google Scholar]
- 25.Klevering BJ, Yzer S, Rohrschneider K, et al. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone-rod dystrophy and retinitis pigmentosa. Eur J Hum Genet. 2004;12:1024–1032. doi: 10.1038/sj.ejhg.5201258. [DOI] [PubMed] [Google Scholar]
- 26.Kurg A, Tonisson N, Georgiou I, Shumaker J, Tollett J, Metspalu A. Arrayed primer extension: solid-phase four-color DNA resequencing and mutation detection technology. Genet Test. 2000;4:1–7. doi: 10.1089/109065700316408. [DOI] [PubMed] [Google Scholar]
- 27.Tonisson N, Zernant J, Kurg A, et al. Evaluating the arrayed primer extension resequencing assay of TP53 tumor suppressor gene. Proc Natl Acad Sci USA. 2002;99:5503–5508. doi: 10.1073/pnas.082100599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koenekoop RK, Lopez I, den Hollander AI, Allikmets R, Cremers FP. Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions. Clin Experiment Ophthalmol. 2007;35:473–485. doi: 10.1111/j.1442-9071.2007.01534.x. [DOI] [PubMed] [Google Scholar]
- 29.Tsang SH, Gouras P. Molecular Physiology and Pathology of the Retina. In: Tasman W, Jaeger EA, editors. Duane's Clinical Ophthalmology. Lippincott-Raven; Philadelphia: 1996. Chapter 2. [Google Scholar]
- 30.Robson AG, Saihan Z, Jenkins SA, et al. Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Br J Ophthalmol. 2006;90:472–479. doi: 10.1136/bjo.2005.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobson SG, Sumaroka A, Aleman TS, Cideciyan AV, Danciger M, Farber DB. Evidence for retinal remodelling in retinitis pigmentosa caused by PDE6B mutation. Br J Ophthalmol. 2007;91:699–701. doi: 10.1136/bjo.2006.104463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. J Comp Neurol. 2007;500:222–238. doi: 10.1002/cne.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003;22:607–655. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- 34.Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and functional abnormalities in the inner retina of the rd/rd mouse. J Neurosci. 2002;22:5492–5504. doi: 10.1523/JNEUROSCI.22-13-05492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W, Colman RW. Conserved amino acids in metal-binding motifs of PDE3A are involved in substrate and inhibitor binding. Blood. 2000;95:3380–3386. [PubMed] [Google Scholar]
- 36.Farber DB, Flannery JG, Bowes-Rickman C. The rd mouse story: Seventy years of research on an animal model of inherited retinal degeneration. Prog Retin Eye Res. 1994;13:31–64. [Google Scholar]
- 37.Farber DB, Lolley RN. Cyclic Guanosine monophosphate: Elevations in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–451. doi: 10.1126/science.186.4162.449. [DOI] [PubMed] [Google Scholar]
- 38.Farber DB, Lolley RN. Enzyme basis for cyclic GMP accumulation in degenerative photoreceptor cells of mouse retina. J Cyclic Nucleotide Res. 1976;2:139–148. [PubMed] [Google Scholar]
- 39.Lolley RN, Farber DB, Rayborn ME, Hollyfield JG. Cyclic GMP accumulation causes degeneration of photoreceptor cells: Simulation of an inherited disease. Science. 1977;196:664–666. doi: 10.1126/science.193183. [DOI] [PubMed] [Google Scholar]
- 40.Kumar-Singh R, Farber DB. Encapsidated adenovirus mini-chromosome-mediated delivery of genes to the retina: application to the rescue of photoreceptor degeneration. Hum Mol Genet. 1998;7:1893–1900. doi: 10.1093/hmg/7.12.1893. [DOI] [PubMed] [Google Scholar]
- 41.Bennett J, Tanabe T, Sun D, et al. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat Med. 1996;2:649–654. doi: 10.1038/nm0696-649. [DOI] [PubMed] [Google Scholar]
- 42.Jomary C, Vincent KA, Grist J, Neal MJ, Jones SE. Rescue of photoreceptor function by AAV-mediated gene transfer in a mouse model of inherited retinal degeneration. Gene Ther. 1997;4:683–690. doi: 10.1038/sj.gt.3300440. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi M, Miyoshi H, Verma IM, Gage FH. Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J Virol. 1999;73:7812–7816. doi: 10.1128/jvi.73.9.7812-7816.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayanagi K, Ikuno Y, Tano Y. Different fundus autofluorescence patterns of retinoschisis and macular hole retinal detachment in high myopia. Am J Ophthalmol. 2007;144:299–301. doi: 10.1016/j.ajo.2007.03.049. [DOI] [PubMed] [Google Scholar]