

Abstract
The density and distribution of single-nucleotide polymorphisms (SNPs) across the genome has important implications for linkage disequilibrium mapping and association studies, and the level of simple-sequence microsatellite polymorphisms has important implications for the use of oligonucleotide hybridization methods to genotype SNPs. To assess the density of these types of polymorphisms in P. falciparum, we sampled introns and noncoding DNA upstream and downstream of coding regions among a variety of geographically diverse parasites. Across 36,229 base pairs of noncoding sequence representing 41 genetic loci, a total of 307 polymorphisms including 248 polymorphic microsatellites and 39 SNPs were identified. We found a significant excess of microsatellite polymorphisms having a repeat unit length of one or two, compared to those with longer repeat lengths, as well as a nonrandom distribution of SNP polymorphisms. Almost half of the SNPs localized to only three of the 41 genetic loci sampled. Furthermore, we find significant differences in the frequency of polymorphisms across the two chromosomes (2 and 3) examined most extensively, with an excess of SNPs and a surplus of polymorphic microsatellites on chromosome 3 as compared to chromosome 2 (P = 0.0001). Furthermore, at some individual genetic loci we also find a nonrandom distribution of polymorphisms between coding and flanking noncoding sequences, where completely monomorphic regions may flank highly polymorphic genes. These data, combined with our previous findings of nonrandom distribution of SNPs across chromosome 2, suggest that the Plasmodium falciparum genome may be a mosaic with regard to genetic diversity, containing chromosomal regions that are highly polymorphic interspersed with regions that are much less polymorphic.
Keywords: genetic diversity, coalescent analysis, malaria
1. Introduction
Genetic variability of P. falciparum underlies its transmission success and thwarts efforts to control disease. Resistance to all antimalarial drugs arises rapidly and is mediated by mutations in key target (Peterson et al., 1990; Wang et al., 1997) or transport genes (Foote et al., 1990; Fidock et al., 2000). Antigenic variation in key proteins is the basis of immune evasion (Biggs et al., 1991) and may in part explain the lack of sterilizing immunity in humans (Bruce-Chwatt, 1963; Neva, 1977; Marsh, 1992). Thus, mutation is a key virulence determinant in the parasite. Yet, there appears to be nonrandomness in the frequency at which polymorphic mutations occur in different genes in P. falciparum. Genetic variation in antigenic (Kemp et al., 1990), drug resistance (Foote et al., 1990; Su et al., 1997; Biswas et al., 2000; Fidock et al., 2000) and pathogenesis determinants is abundant, whereas SNPs in housekeeping genes (genes involved in general cellular or metabolic processes that are not obvious targets of rapid selective change) and introns is much less abundant (Rich et al., 1998; Volkman et al., 2001) This contrast has led to the hypothesis that the P. falciparum genome may be a mosaic with regions of significant diversity juxtaposed with regions that are practically monomorphic. Possible explanations for such a mosaic pattern include chance differences in the average coalescence time of genes in different chromosomes, or that the parasite population has undergone a series of selective sweeps, resulting in a patchwork genome with different evolutionary histories. Assessing the density of polymorphisms therefore has important implications, both in terms of the population structure and in terms of the identification of genes under diversifying selection, including potential drug and vaccine targets.
There is ample evidence for recent selective sweeps within the genome associated with the emergence and subsequent spread of drug resistance. Regions of the genome that have undergone recent selective sweeps include regions of chromosome 7 surrounding the pfcrt locus (Wootton et al., 2002) and regions of chromosome 4 surrounding the dhfr (Nair et al., 2003; Roper et al., 2003) and dhps (Roper et al., 2004) loci. These selective sweeps are recent in the evolutionary history of P. falciparum, presumably because they are a consequence of recently applied drug pressure. Other selective sweeps based on biological or immune selection may also have occurred as well as older selective sweeps, but it may require a greater depth of analysis to reveal these regions using a population genetics approach.
To investigate SNP density and microsatellite polymorphisms in more detail, we carried out an in-depth analysis of sequence diversity in the noncoding flanking and intronic sequences in P. falciparum. We focused our analysis on genes from chromosomes 2 and 3, primarily to reconcile previous work that yielded conflicting estimates of the SNP frequency between these two chromosomes (Volkman et al., 2001; Mu et al., 2002; Mu et al., 2005); however, we also included sequences from other chromosomes in our analysis to provide a more global view of the genome. We identified and compared polymorphisms both on a genome-wide basis and directly between chromosomes 2 and 3. We provide evidence for dramatic differences in the frequency of polymorphisms, both among genes in each chromosome and overall between chromosomes 2 and 3. These results may imply different evolutionary histories for these chromosomes, consistent with drift events or selective sweeps.
2. Materials and Methods
2.1 Identification of Polymorphisms
Genetic loci were amplified using the polymerase chain reaction, cloned and sequenced from various P. falciparum isolates (HB3, 7G8, D6, W2, Muz 12.4, 3D7, D10, Muz 37.4, Muz 51.1 and KF1776) as described (Volkman et al., 2001). These isolates represent both the K1-type and the MAD20-type alleles of msp1. Parasite DNA was genotyped using the msp2 locus (Snounou et al., 1999) to ensure independent parasite isolates were used and sequences were aligned to generate consensus sequences for each of the isolates at each of the loci to determine the polymorphisms among isolates.
All sequences were scanned for microsatellite repeats using Tandem Repeats Finder (Benson, 1999). Parameters were chosen so that repeats identified by the program encompassed as many length polymorphic microsatellites as possible, while ignoring extraneous sequences. Regions of at least 12 repetitive base pairs were identified as repeats, regardless of the number of base pairs within the repeating unit. These parameters allowed identification of greater than 98% of the polymorphic regions in the dataset. The remaining polymorphic regions that the program did not identify were entered into the dataset manually. This method allowed for an objective, consistent, and unbiased determination of microsatellite sequence boundaries. For further information about this method, as well as precise parameters values please see: http://www.oeb.harvard.edu/hartl/lab/publications/PlasmoMS/index.html
2.2 Data Analysis
The compiled dataset was analyzed by polymorphism type, polymorphisms within repetitive or nonrepetitive sequences, by chromosome, and by noncoding region (flanking or intronic sequence) to identify any significant differences regarding the frequency or distribution of polymorphisms within the dataset as described in the text. Polymorphism types included SNPs (both outside of and within microsatellites), microsatellite repeat-length polymorphisms, and other polymorphisms including small insertions and deletions. Tests of significance are noted in the text, but included both Fishers Exact Test and the Poisson Test using 2 × 2 contingency analysis.
2.3 Coalescent Analysis
The infinite-sites model was used to estimate the product (θ) of the effective population size (Ne) and the nucleotide-site mutation rate (μ) for each of Chromosomes 2 and 3. The model assumes that nucleotide sites within individual loci are tightly linked, but free recombination between loci. Under these assumptions, likelihoods for θ for each chromosome can be written as a product of likelihoods over loci. Infinite-sites likelihoods for individual loci were derived using a recursion of Griffiths and Tavare (Griffiths and Tavare, 1994; Griffiths and Tavare, 1995). Bayesian methods were used to estimateθ, so that the product likelihoods were also multiplied by a noninformative prior.
3. Results
3.1 Identification of Polymorphisms
Forty-one loci from various chromosomes were sequenced from five geographically distinct parasite genomes including HB3 (Honduras), 7G8 (Brazil), D6 (Sierra Leone), W2 (Southeast Asia), Muz 12.4 (Papua New Guinea, PNG), and 3D7 (The Netherlands). Additional sequence was derived for some loci from other PNG isolates including D10, Muz 37.4, Muz 51.1 and KF1776. Parasite DNA was genotyped using the msp2 locus to ensure independent parasite isolates were used, and products from multiple independent polymerase chain reactions were cloned and sequenced from multiple clones to ensure sequence accuracy (Volkman et al., 2001; Barry et al., 2003).
To evaluate the distribution of SNPs within microsatellite or non-microsatellite sequence, we used a tandem-repeat finding algorithm (Benson, 1999) with parameters based upon P. falciparum sequences. The computational approach allowed for an objective, consistent, and unbiased determination of microsatellite sequence boundaries. The algorithm bases the identification of microsatellites on both the number of repeats and the entire length of the repeat region, identifying repeat regions that total at least 12 or more base pairs (bp) in length, regardless of the number of repeat units in the region. Thus a region of TTTTTTTTTTTT (repeat unit length of one) or CATACATACATA (repeat unit length of four) would both be included as microsatellites, even though the number of repeat units is 12 (Ts) and 3 (CATAs) respectively. Any repeat that was found to be polymorphic in the analysis was also included, regardless of whether it met the minimum length criterion. When we used this algorithm to evaluate a comparison dataset scored manually, we found that the program correctly identified over 90% of all microsatellites and over 98% of all polymorphic microsatellites scored manually using the rule of eight repeating units (of any length) as employed in our previous study (Volkman et al 2001). Hence this approach identifies the majority of what one would otherwise call a microsatellite, with the added benefit of providing consistency in the calls from one investigator to another facilitating comparison across data sets.
3.2 Excess Polymorphisms within Microsatellites with Repeat Unit Length of One or Two
We found a nonrandom distribution of microsatellite polymorphisms among microsatellite sequences with different repeat lengths, and a nonrandom distribution of polymorphic microsatellites between chromosomes 2 and 3. In particular, we observed that microsatellite repeats with unit lengths of one or two were significantly more polymorphic than microsatellite sequences with repeat unit lengths of three or more (P = 5.2E-31, Fisher Exact Test). Furthermore, there was an excess of these short-repeat microsatellites on chromosome 3 relative to chromosome 2 (P = 0.026, Fisher Exact Test), resulting in a greater abundance of microsatelllite polymorphisms on chromosome 3 relative to chromosome 2 (Figure 1). In our previous study (Volkman et al., 2001) we found a significant enrichment of SNPs within microsatellite regions, presumably because of mechanisms such as replication slippage and mismatch repair. However, in this larger dataset analyzed with automated annotation of microsatellites we did not observe an excess of SNPs within microsatellite repeats.
Figure 1.
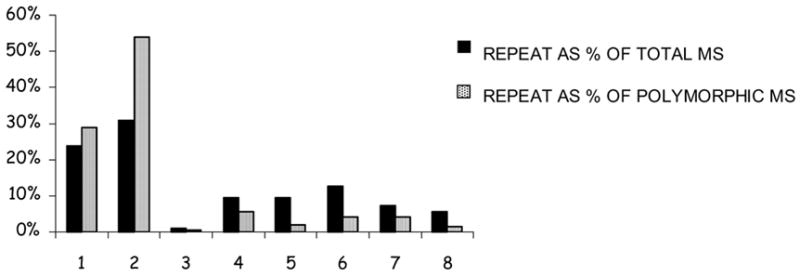
Microsatellites with a Repeat Unit Length of 1 or 2 are Highly Polymorphic. Microsatellite (MS) repeats of unit length one or two were significantly more polymorphic than microsatellite repeats of unit length three or more (P = 5.2E-31) and there was an excess of these short-repeat microsatellites on chromosome 3 relative to chromosome 2 (P = 0.026).
3.3 SNP Distribution is Nonrandom
The SNPs were also nonrandomly distributed across the sequences in the dataset, with an excess of SNPs in only a few genes. About one-third of the 41 genetic loci (16/41, 39%) contained at least one SNP within the noncoding sequence analyzed, but only three of these loci (plm6, pfmdr1, and hsp86) contained almost half the total SNPs (18/39, 46%) across a relatively small amount of sequence (6,393 bp; P < 0.01, Poisson Test of individual loci). The noncoding regions of these three genes contain 42% of the SNPs in only 19% of the sequence (P = 0.00001, Fisher Exact Test).
3.4 Chromosome 3 Is Significantly More Polymorphic Than Chromosome 2 Overall
We also observed dramatic differences in the frequency of polymorphisms among individual chromosomes. Comparing chromosomes 2 and 3, for which we had acquired the most sequence data, there were 42 polymorphic microsatellites, 5 SNPs, and 1 other polymorphism across 7,848 bp on chromosome 2, whereas there were 129 polymorphic microsatellites, 18 SNPs, and 9 other polymorphisms across 13,737 bp on chromosome 3 (P = 0.0001, Fisher Exact Test). Collectively, these data argue that the frequency and distribution of the different types of polymorphisms identified within the falciparum genome are markedly different between chromosomes 2 and 3. Furthermore, the difference in the frequency of SNPs between chromosomes 2 and 3 (5 SNPs/7,848 bp versus 18 SNPs/13,737 bp) is greater than a factor of two and nominally statistically significant in a one-tailed test (P ≈ 0.05)
3.5 Comparison of Chromosomes 2 and 3 Using Coalescent Methods
The unexpected observation that chromosome 3 had almost double the relative number of SNPs as chromosome 2 led us to ask if the evolutionary history of these two chromosomes may in fact be distinct. To address this issue we used a coalescent analysis of the ancestry of the current alleles, which traces the sequences at each locus backwards in time to their common ancestor assuming selective neutrality. For this analysis we made two changes to our dataset for chromosomes 2 and 3. First, we used only sequence data for which there was complete coverage of all isolates; and, second, we considered a group of four clustered SNPs that appeared in a single isolate in one locus as one independent event. This resulted in a total of 5 SNPs across 7,789 bp on chromosome 2 and 14 SNPs across 12, 595 bp of sequence on chromosome 3. All 19 SNPs have two variable nucleotides. Of the 19 SNPs, 10 are transversions and 9 are transitions. Of the 10 transversion SNPs, 7 are AT (that is, have As and Ts only) while 3 are CA or CG. Summing over loci the 23 alignments have 130 sequences with 30 distinct haplotypes. Curiously, the two chromosomes have almost identical AT content, namely 86.5% for chromosome 2 and 86.3% for chromosome 3. We treated each of the 23 loci as a non-recombining block of the genome with free recombination between loci. We also assumed that there was at most one mutation at any one site since the coalescent at that locus. This is the classical “infinite sites” assumption and is consistent with our data. We assumed constant population size. We also examined models with exponential growth, but these yielded excessively large confidence intervals for the estimates, and they are therefore not reported.
We used the coalescent analysis to estimate theta (θ), which is equal to the product of the haploid effective population size Ne and the mutation rate (μ) per nucleotide site (Table 1). Theta also corresponds to the mean number of mutations or the mutation rate per site per Ne generations. To estimate θ we calculated the likelihood of the set of haplotypes at each locus as a fixed function of θ by iterating a recurrence equation due to Griffiths and Tavare (Griffiths and Tavare, 1994; Griffiths and Tavare, 1995). The full likelihood was the product of within-locus likelihoods across loci, with the product also multiplied by a Bayesian γ(a, b) prior distribution with a = b = 0.001. The likelihood for each chromosome was integrated numerically to find credible intervals for θ. Markov Chain Monte Carlo based on the full likelihood was also used to estimate θ with nearly identical results. Point-wise estimates of θ were found based on the median value of the posterior distribution. Markov Chain Monte Carlo was also used to find the posterior distribution of θ3/θ2, where θ2 and θ3 are the values of θ for those chromosomes (Table 1).
Table 1.
Estimates of θ for Chromosomes 2 & 3.
| Comparison | Value | Lower 95% CI | Median | Upper 95% CI |
|---|---|---|---|---|
| Ch 2 | θ | 0.88 E-4 | 2.79 E-4 | 6.80 E-4 |
| Ch 3 | θ | 2.92 E-4 | 5.65 E-4 | 10.22 E-4 |
| Ch 3: Ch 2 | θ3/θ2 | 0.656 | 2.033 | 7.486 |
| Bayes | P = 0.1230 | |||
| Permutation | P = 0.1050 |
Two tests of the hypothesis that chromosome 2 has a smaller value of θ than that of chromosome 3 were carried out: (i) a Bayesian test whose P value is the posterior probability that θ3 ≤ θ2 and (ii) a permutation test, in which the ratio of the maximum likelihood estimates (MLEs) of θ3 and θ2 for the observed data is compared with that of 10,000 artificial datasets in which the 23 loci were randomly permuted among the two chromosomes in such a way to preserve the number of loci on each chromosome. The P value for the permutation test is the proportion of artificial datasets with a larger or equal value of the ratio of the MLEs of θ3 andθ2. The results for the two tests were P = 0.1230 (i) and 0.1050 (ii) (Table 1), which are both one-sided P values and non significant. Nevertheless, the observed ratio of the θ values between chromosomes 2 and 3 is relatively large (more than a factor of two), and it is also noteworthy that the minimum 95% confidence interval for the estimate of θ for chromosome 3 is greater than the median estimate of θ for chromosome 2 (Table 1).
3.6 SNPs are Nonrandomly Distributed Between Coding and Noncoding Regions of a Gene
To address the question of nonrandom distribution of SNPs within individual genetic loci, we analyzed whether flanking regions of highly polymorphic genes were similarly highly polymorphic. For this analysis we compared the msp1 (Holder and Freeman, 1984; Tanabe et al., 1987; Miller et al., 1993) gene on chromosome 9 and the pfmdr1 (Foote et al., 1989; Wilson et al., 1989) gene on chromosome 5, and either analyzed 1000 bp of flanking sequence or until the next predicted gene. Both of these genes are likely to be under selective pressure, either immune or drug pressure, respectively, and are known to be highly polymorphic within their coding regions. When we examined the flanking regions for pfmdr1 we found additional polymorphisms in the flanking sequences, many of which were localized to the 5′ region that is presumably associated with gene regulation. Across 2,775 bp upstream of pfmdr1 we find 4 SNPs, and across 768 bp downstream of pfmdr1 we find 2 SNPs (Figure 2). Furthermore, it has been shown that one of these SNPs is present in both laboratory and field isolates, and several of these mutations may be functionally relevant due to the proximity to the start of transcription (Myrick et al., 2003).
Figure 2.
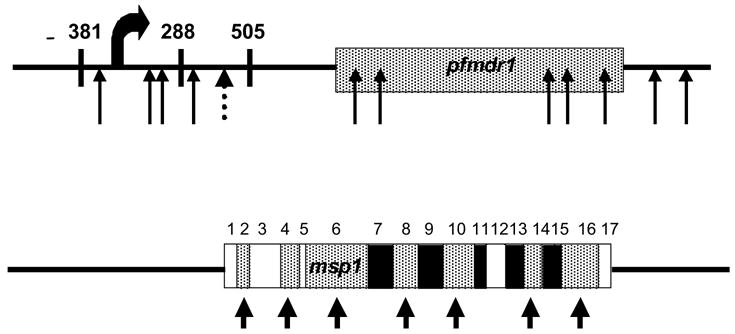
SNPs are Nonrandomly Distributed Between Coding and Noncoding Regions of a Gene. The pfmdr1 gene (PFE1150w; coding region from position 957,885 – 962,144; shown from position 955,110 – 962,912) contains SNPs both within the coding region (arrows corresponding to Tyr86, Phe184, Cys1034, Asp1042, Tyr1246) and in the 5′ (2775 base pairs (bp)) and 3′ (768 bp) flanking regions. One of these SNPs (dotted arrow) is present in both field and laboratory isolates. Several of these mutations may be functionally relevant due to the proximity to the start of transcription (curved arrow). The msp1 gene (PFI1475w; coding region from position 1,201,802 – 1,206,964; shown from position 1,200,803 – 1,208,021) contains several SNPs within the coding region (represented by arrows) within the highly polymorphic blocks (2, 4, 6, 8, 10, 14, 16) defined by Tanabe and colleagues (Miller et al., 1983) shown as conserved (open), semiconserved (hatched), and variable (solid) regions. Most of this variation is dimorphic and grouped into the allelic groups of K1-type and MAD20-type alleles. Exception to this dimorphism is present in Block 2 with additional degenerate tripeptide repeats that represent the RO33-type allele. Both the 5′ (999 bp) and the 3′ (1057 bp) flanking regions are monomorphic among eight or more genetically diverse parasites and lack SNPs in the dataset analyzed.
In contrast, flanking sequences for msp1 were monomorphic, with no SNPs identified in the 1000 bp either upstream or downstream of the msp1 coding region (Figure 2). This result is surprising, because the coding sequence of msp1 is one of the most polymorphic regions in eukaryotes (Hughes, 1999). There appears to be extensive recombination between sites within conserved regions even between otherwise highly divergent msp1 alleles, as linkage disequilibrium declines rapidly and is essentially nonexistent for sites in the coding region separated by ≥ 800 bp (Conway et al., 1999). Recombination within the coding region would also allow the flanking regions of divergent msp1 alleles to be swapped, and then random genetic drift within each class of alleles could fix the same or a similar sequence.
4. Discussion
Analysis of noncoding regions from multiple chromosomes across the P. falciparum genome reveals a nonrandom distribution of polymorphisms, including excess SNPs within few genetic loci, the surplus of polymorphic microsatellites on chromosome 3, and the juxtaposition of monomorphic flanking sequences and highly polymorphic coding sequences, that describe a mosaic organization of the P. falciparum genome. These variable patterns of polymorphism are observed when comparing individual chromosomes, and we demonstrate that, with respect to SNPs and microsatellites taken together, there is an overall significant difference between the frequency of polymorphisms across chromosomes 2 and 3. The approximate twofold difference in the frequency of SNPs in chromosomes 2 and 3 largely accounts for the discrepancy in the estimated age of the most recent common ancestor of genes in these chromosomes reported previously (Volkman et al. 2001; Mu et al. 2002), as one estimate was based on genes in chromosome 2 and the other on genes in chromosome 3.
Other studies also support the mosaic nature of polymorphisms across the P. falciparum genome. For example, in an analysis of chromosome 3 sequences, many of the 31 SNPs in noncoding regions reported were concentrated in few of the genetic loci (Mu et al., 2002; Mu et al., 2005). Analysis of SNPs within nine aspartyl protease genes revealed that one gene (plasmepsin 10) contains a significantly higher density of SNPs than the others in both coding and noncoding regions, while another (plasmepsin 6) contained a disproportionate number of SNPs in introns compared to coding sequences (Barry et al. 2006). More detailed information on the mosaic nature of the genome would emerge from complete genomic sequencing of additional isolates of P. falciparum. The question of whether noncoding polymorphisms are selectively neutral or nearly neutral also remains undetermined, and other methods to address this important issue must be taken. One such approach might involve comparison of the level of polymorphism in P. falciparum with the level of divergence of orthologous regions in the chimpanzee parasite P. reichenowi. This approach will be feasible when the genome sequence for P. reichenowi becomes available.
The mechanisms that maintain polymorphism within the P. falciparum genome are largely unknown, yet it is important to understand the population structure in order to identify rapidly evolving genes and to develop intervention strategies for newly emerging drug resistance. There is evidence of recent selective sweeps across specific regions of the genome, presumably as a consequence of recent drug pressure (Wootton et al., 2002; Nair et al., 2003; Roper, 2003; Roper, 2004). Older selective sweeps as a consequence of natural selection may have occurred across the genome, but identification of these regions might require a genome-wide haplotype map and analysis of linkage disequilibrium.
Our data suggest that microsatellites with a repeat length of one or two may be less stable genetically than microsatellites with longer repeats. There may be other sources of increased polymorphism such as gene conversion, which has been shown to account for most of the genetic polymorphism between two closely related paralogous genes on chromosome 11 (Nielsen et al., 2003). Another factor that may contribute to the variable diversity across the genome in the extant population is recombination. A high recombination rate, estimated at 1cM per 15–30 kb (Su et al., 1999), may shuffle ancestral sequences to make a mosaic of different sequence patterns, as apparently accounts for the high polymorphism of the msp1 coding sequence, where the K1 and MAD20 types of alleles can differ by over 60% and yet be flanked by nearly monomorphic 5′ and 3′ sequences. At all events, individual genetic loci across the genome there may have markedly different levels of polymorphism.
Acknowledgments
This work was supported by a grant from the National Institutes of Health. SAS was partially supported by a grant from the National Science Foundation. AEB was supported by a Howard Florey Fellowship from The Royal Society, UK and National Health and Medical Research Council, Australia 2001–2003. We are also grateful for support from the Ellison Medical Foundation. The authors acknowledge the efforts of the Malaria Genome Project consortium for reference data and curators of PlasmoDB for online access to the annotated genome data.
Abbreviations
- SNPs
single nucleotide polymorphisms
- MLEs
maximum likelihood estimates
- Ne
effective population size
- μ
nucleotide-site mutation rate
- bp
base pair(s)
- θ
theta
- kb
kilobase(s)
- cM
centimorgan
- CI
confidence interval
- MS
microsatellite
- Tyr
tyrosine
- Phe
phenylalanine
- Cys
cysteine
- Asp
asparagines
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barry AE, Leliwa A, Choi M, Nielsen KM, Hartl DL, Day KP. DNA sequence artifacts and the estimation of time to the most recent common ancestor (TMRCA) of Plasmodium falciparum. Mol Biochem Parasitol. 2003;130:143–147. doi: 10.1016/s0166-6851(03)00164-6. [DOI] [PubMed] [Google Scholar]
- Barry AE, et al. Variable SNP density in aspartyl-protease genes of the malaria parasite Plasmodium falciparum. Gene. 2006;376:163–173. doi: 10.1016/j.gene.2006.02.029. [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs BA, Gooze L, Wycherley K, Wollish W, Southwell B, Leech JH, Brown GV. Antigenic variation in Plasmodium falciparum. Proc Natl Acad Sci U S A. 1991;88:9171–9174. doi: 10.1073/pnas.88.20.9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S, Escalante A, Chaiyaroj S, Angkasekwinai P, Lal AA. Prevalence of point mutations in the dihydrofolate reductase and dihydropteroate synthetase genes of Plasmodium falciparum isolates from India and Thailand: a molecular epidemiologic study. Trop Med Int Health. 2000;5:737–743. doi: 10.1046/j.1365-3156.2000.00632.x. [DOI] [PubMed] [Google Scholar]
- Bruce-Chwatt LJ. A Longitudinal Survey of Natural Malaria Infection in a Group of West African Adults. West Afr Med J. 1963;12:199–217. [PubMed] [Google Scholar]
- Conway DJ, Roper C, Oduola AM, Arnot DE, Kremsner PG, Grobusch MP, Curtis CF, Greenwood BM. High recombination rate in natural populations of Plasmodium falciparum. Proc Natl Acad Sci U S A. 1999;96:4506–4511. doi: 10.1073/pnas.96.8.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, bir Singh Sidhu A, Naude B, Deitsch KW, Su X, Wootton JC, Roepe PD, Wellems TE. Mutations in the P. falciparum Digestive Vacuole Transmembrane Protein PfCRT and Evidence for Their Role in Chloroquine Resistance. Mol Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote SJ, Kyle DE, Martin RK, Oduola AM, Forsyth K, Kemp DJ, Cowman AF. Several alleles of the multidrug-resistance gene are closely linked to chloroquine resistance in Plasmodium falciparum. Nature. 1990;345:255–258. doi: 10.1038/345255a0. [DOI] [PubMed] [Google Scholar]
- Foote SJ, Thompson JK, Cowman AF, Kemp DJ. Amplification of the multidrug resistance gene in some chloroquine-resistant isolates of P. falciparum. Cell. 1989;57:921–930. doi: 10.1016/0092-8674(89)90330-9. [DOI] [PubMed] [Google Scholar]
- Griffiths RC, Tavare S. Ancestral inference in population genetics. Statistical Science. 1994;9:307–319. [Google Scholar]
- Griffiths RC, Tavare S. Unrooted genealogical tree probabilities in the infinitely-many-sites model. Math Biosci. 1995;127:77–98. doi: 10.1016/0025-5564(94)00044-z. [DOI] [PubMed] [Google Scholar]
- Holder AA, Freeman RR. Protective antigens of rodent and human bloodstage malaria. Philos Trans R Soc Lond B Biol Sci. 1984;307:171–177. doi: 10.1098/rstb.1984.0117. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Adaptive Evolution of Genes and Genomes. Oxford University Press; New York: 1999. [Google Scholar]
- Kemp DJ, Cowman AF, Walliker D. Genetic diversity in Plasmodium falciparum. Adv Parasitol. 1990;29:75–149. doi: 10.1016/s0065-308x(08)60105-0. [DOI] [PubMed] [Google Scholar]
- Marsh K. Malaria--a neglected disease? Parasitology. 1992;104(Suppl):S53–69. doi: 10.1017/s0031182000075247. [DOI] [PubMed] [Google Scholar]
- Miller LH, Roberts T, Shahabuddin M, McCutchan TF. Analysis of sequence diversity in the Plasmodium falciparum merozoite surface protein-1 (MSP-1) Molecular and Biochemical Parasitology. 1993;59:1–14. doi: 10.1016/0166-6851(93)90002-f. [DOI] [PubMed] [Google Scholar]
- Mu J, Awadalla P, Duan J, McGee KM, Joy DA, McVean GA, Su XZ. Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 2005;3:1734–1741. doi: 10.1371/journal.pbio.0030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Duan J, Makova KD, Joy DA, Huynh CQ, Branch OH, Li WH, Su XZ. Chromosome-wide SNPs reveal an ancient origin for Plasmodium falciparum. Nature. 2002;418:323–326. doi: 10.1038/nature00836. [DOI] [PubMed] [Google Scholar]
- Myrick A, Munasinghe A, Patankar S, Wirth DF. Mapping of the Plasmodium falciparum multidrug resistance gene 5′-upstream region, and evidence of induction of transcript levels by antimalarial drugs in chloroquine sensitive parasites. Mol Microbiol. 2003;49:671–83. doi: 10.1046/j.1365-2958.2003.03597.x. [DOI] [PubMed] [Google Scholar]
- Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann JP, Smithuis FM, Hien TT, White NJ, Nosten F, Anderson TJ. A selective sweep driven by pyrimethamine treatment in southeast asian malaria parasites. Mol Biol Evol. 2003;20:1526–1536. doi: 10.1093/molbev/msg162. [DOI] [PubMed] [Google Scholar]
- Neva FA. Looking back for a view of the future: observations on immunity to induced malaria. Am J Trop Med Hyg. 1977;26:211–215. doi: 10.4269/ajtmh.1977.26.211. [DOI] [PubMed] [Google Scholar]
- Nielsen KM, Kasper J, Choi M, Bedford T, Kristiansen K, Wirth DF, Volkman SK, Lozovsky ER, Hartl DL. Gene conversion as a source of nucleotide diversity in Plasmodium falciparum. Mol Biol Evol. 2003;20:726–734. doi: 10.1093/molbev/msg076. [DOI] [PubMed] [Google Scholar]
- Peterson DS, Milhous WK, Wellems TE. Molecular basis of differential resistance to cycloguanil and pyrimethamine in Plasmodium falciparum malaria. Proc Natl Acad Sci U S A. 1990;87:3018–3022. doi: 10.1073/pnas.87.8.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich SM, Licht MC, Hudson RR, Ayala FJ. Malaria’s Eve: evidence of a recent population bottleneck throughout the world populations of Plasmodium falciparum. Proc Natl Acad Sci U S A. 1998;95:4425–4430. doi: 10.1073/pnas.95.8.4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper C, Pearce R, Bredenkamp B, Gumede J, Drakeley C, Mosha F, Chandramohan D, Sharp B. Antifolate antimalarial resistance in southeast Africa: a population-based analysis. Lancet. 2003;361:1174–1181. doi: 10.1016/S0140-6736(03)12951-0. [DOI] [PubMed] [Google Scholar]
- Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- Snounou G, Zhu X, Siripoon N, Jarra W, Thaithong S, Brown KN, Viriyakosol S. Biased distribution of msp1 and msp2 allelic variants in Plasmodium falciparum populations in Thailand. Trans R Soc Trop Med Hyg. 1999;93:369–374. doi: 10.1016/s0035-9203(99)90120-7. [DOI] [PubMed] [Google Scholar]
- Su X, Ferdig MT, Huang Y, Huynh CQ, Liu A, You J, Wootton JC, Wellems TE. A genetic map and recombination parameters of the human malaria parasite Plasmodium falciparum. Science. 1999;286:1351–1353. doi: 10.1126/science.286.5443.1351. [DOI] [PubMed] [Google Scholar]
- Su X, Kirkman LA, Fujioka H, Wellems TE. Complex polymorphisms in an approximately 330 kDa protein are linked to chloroquine-resistant P. falciparum in Southeast Asia and Africa. Cell. 1997;91:593–603. doi: 10.1016/s0092-8674(00)80447-x. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Mackay M, Goman M, Scaife JG. Allelic dimorphism in a surface antigen gene of the malaria parasite Plasmodium falciparum. J Mol Biol. 1987;195:273–287. doi: 10.1016/0022-2836(87)90649-8. [DOI] [PubMed] [Google Scholar]
- Volkman SK, Barry AE, Lyons EJ, Nielsen KM, Thomas SM, Choi M, Thakore SS, Day KP, Wirth DF, Hartl DL. Recent origin of Plasmodium falciparum from a single progenitor. Science. 2001;293:482–484. doi: 10.1126/science.1059878. [DOI] [PubMed] [Google Scholar]
- Wang P, Read M, Sims PF, Hyde JE. Sulfadoxine resistance in the human malaria parasite Plasmodium falciparum is determined by mutations in dihydropteroate synthetase and an additional factor associated with folate utilization. Mol Microbiol. 1997;23:979–986. doi: 10.1046/j.1365-2958.1997.2821646.x. [DOI] [PubMed] [Google Scholar]
- Wilson CM, Serrano AE, Wasley A, Bogenschutz MP, Shankar AH, Wirth DF. Amplification of a gene related to mammalian mdr genes in drug-resistant Plasmodium falciparum. Science. 1989;244:1184–6. doi: 10.1126/science.2658061. [DOI] [PubMed] [Google Scholar]
- Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, Su XZ. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–3. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]