

Abstract
Ca2+ influx through plasma membrane wounds triggers a rapid repair response that is essential for cell survival. Earlier studies showed that repair requires the exocytosis of intracellular vesicles. Exocytosis was thought to promote resealing by “patching” the plasma membrane lesion, or by facilitating bilayer restoration through reduction in membrane tension. However, cells also rapidly repair lesions created by pore forming proteins, a form of injury that cannot be resealed solely by exocytosis. Recent studies suggest that in cells injured by pores or mechanical abrasions, exocytosis is followed by lesion removal through endocytosis. Describing the relationship between wound-induced exocytosis and endocytosis has implications for the understanding of muscular degenerative diseases that are associated with defects in plasma membrane repair.
Introduction
Ca2+ influx from the extracellular milleu into the cytosol of a damaged cell triggers rapid plasma membrane resealing, within seconds after injury 1, 2. The viability and longevity of injured cells depends on this immediate plasma membrane repair process 3, 4, as well as on longer-term responses involved in restoring cellular homeostasis 5, 6. Defects in rapid plasma membrane resealing impair the ability of cells to maintain plasma membrane integrity, a process that has significant clinical implications. In human patients and murine models, mutations in genes required for plasma membrane repair cause pathological changes in skeletal muscle, a tissue that is frequently injured in vivo 7. Forms of muscle pathology that have been associated with defective plasma membrane repair include limb-girdle muscular dystrophy type 2B and Miyoshi myopathy 8, and inflammatory myopathy in mice deficient in synaptotagmin VII 9. In this review, we discuss recent developments that are rapidly shedding light on the cellular and molecular mechanisms responsible for the rapid Ca2+-dependent plasma membrane resealing.
Ca2+-regulated exocytosis: A requirement for plasma membrane repair
Early reports of plasma membrane repair came from experiments involving in vitro manipulation of sea urchin eggs. These studies demonstrated an important concept, that extracellular Ca2+ is essential for plasma membrane resealing after injury 10, 11. Several decades later, more detailed microscopic observations detected rapid Ca2+-dependent exocytosis of intracellular vesicles at the sites of plasma membrane injury. These observations led to the hypothesis that wound resealing was mediated by intracellular membrane added to the cell surface through exocytosis 12, 13. This hypothesis provided the first mechanistic framework for the role of Ca2+ in plasma membrane resealing, since it is widely known that one of the many signaling functions of Ca2+ is to trigger the fusion of intracellular vesicles with the plasma membrane. Once considered a pathway exclusive to specialized secretory cells, Ca2+-regulated exocytosis emerged from these studies of plasma membrane repair as a cellular process that was likely to be present in most cell types. Subsequent studies confirmed that Ca2+-regulated exocytosis is indeed a ubiquitous process 14, 15. This important realization launched investigations aimed at identifying the Ca2+-responsive exocytic vesicles responsible for plasma membrane resealing.
Early insights into a ubiquitous vesicle population capable of Ca2+-regulated exocytosis were unexpectedly discovered in investigations of host cell invasion by the protozoan parasite Trypanosoma cruzi. Upon attachment to host cells, these parasites trigger signaling pathways that result in increases in intracellular free Ca2+ ([Ca2+]i) and in the recruitment of lysosomes, which provide membrane for intracellular parasitophorous vacuole formation 16. These observations led to the demonstration that conventional lysosomes can fuse with the plasma membrane in many cell types, in response to [Ca2+]i elevations above 1 μM 17. Subsequent studies using TIRF microscopy demonstrated that elevation in [Ca2+]i induces a 25 fold increase in the exocytosis rate of lysosomes, while not stimulating the secretion of a number of additional membrane proximal organelles such as ER, post-Golgi vesicles, late endosomes and early endosomes 18. Thus, instead of the conventional view that lysosomes are terminal compartments of the endocytic pathway, these studies demonstrated that conventional lysosomes behave as bona fide regulated secretory vesicles, rapidly fusing with the plasma membrane in response to Ca2+ influx 19 (see Box 1). Interfering with the exocytosis of conventional lysosomes inhibits the ability of cells to reseal plasma membrane wounds, establishing these organelles as important mediators of plasma membrane repair 2, 20, 21. The impact of the extracellular release of lysosomal enzymes is still unknown. Although it can't be ruled out that released acidic hydrolases play a role in the rapid plasma membrane resealing process, the neutral pH of the extracellular milleu is likely to prevent sustained enzymatic activity.
Box 1 — Ca2+ Regulated Exocytosis.
Ca2+ triggered fusion of intracellular vesicles with the plasma membrane is a highly conserved phenomenon, based on the steep Ca2+ concentration gradient maintained between the extracellular (>1 mM) and intracellular (<100 nM) environments. In intact cells, acute rises in cytosolic free Ca2+ result from the opening of ion channels located on the plasma membrane, or on intracellular Ca2+ stores such as the endoplasmic reticulum. Extensively studied examples of Ca2+ regulated exocytosis include insulin secretion from pancreatic beta-cell granules 64, neurotransmitter release from neuronal synaptic vesicles 65, and perforin and granzyme release from the lytic granules of cytotoxic lymphocytes 66. Although such examples of specialized secretion have historically been viewed as paradigms for Ca2+ regulated exocytosis, more recently it became clear that the process is also present in non-specialized cells such as fibroblasts and epithelial cells 14, 15, 67. Investigations of regulated exocytosis in non-specialized cells identified conventional lysosomes as the major intracellular compartment capable of responding to Ca2+ by fusing with the plasma membrane 17, 18. This finding expanded significantly the concept of “secretory lysosomes”, a term initially coined to describe the modified, lysosome-like secretory granules present in hematopoietic cells 68. It is now clear that the capacity to sense Ca2+ rises and respond by “kiss-and-run” or full fusion with the plasma membrane is not exclusively a property of the modified lysosomes of hematopoietic cells. Rather, it is a process present in a large number of cell types 19. The molecular machinery mediating Ca2+ regulated exocytosis of conventional lysosomes includes the v-SNARE VAMP-7 and synaptotagmin VII on lysosomes, and the t-SNARES SNAP-23 and syntaxin-4 on the plasma membrane 69. Synaptotagmin VII is a membrane Ca2+ sensor 70 structurally related to synaptotagmin I, a synaptic vesicle protein essential for rapid neurotransmitter release in neurons 71. The identification of synaptotagmin VII on lysosomes 25 provided a molecular tool to functionally identify these organelles as the Ca2+ regulated exocytic vesicles involved in plasma membrane repair 2, 9, 21.
“Secretory lysosomes” is a term used to describe the modified lysosomal vesicles found in specialized secretory cells 22 (see Box 1). Exocytosis of these specialized lysosome-like granules also results in the release of their luminal contents into the extracellular milieu, where they perform a number of effector functions. One of the best studied examples, which actually established a paradigm for the study of secretory lysosomes, is the Ca2+ dependent degranulation and target cell killing by CD8 positive cytotoxic T lymphocytes (CTL) 22. Similar to conventional lysosomes, secretory lysosomes have an acidic lumen and contain high concentrations of acid hydrolases 23. In addition to the lytic granules of CTLs, the designation of secretory lysosomes also include melanosomes, platelet dense granules, azurophil granules of neutrophils, and the ruffled border secretory vesicles of osteoclasts 24. The role of these specialized organelles in plasma membrane repair has not been directly tested, but it is conceivable that Ca2+ responsive secretory lysosomes also participate in the resealing of injured plasma membrane. In support of this possibility, the Ca2+ sensor molecule synaptotagmin VII was shown to regulate conventional lysosome exocytosis 25 and plasma membrane repair 9, and also the exocytosis of specialized secretory lysosomes in CTLs 26 and osteoclasts 27.
To date, functional data has been obtained directly implicating the exocytosis of conventional lysosomes 2 and yolk granules (specialized secretory vesicles with lysosomal properties) 21 in plasma membrane repair. Recently, a novel Ca2+ regulated exocytic compartment termed the enlargeosome was identified in some cell types, and proposed to also participate in plasma membrane resealing 28, 29. Functional evidence for this role is still lacking, because a mechanism to specifically inhibit enlargeosome exocytosis is still not available. Involvement in plasma membrane repair was suggested by the rapid kinetics by which the enlargeosome marker desmoyokin-AHNAK appears on the plasma membrane following plasma membrane injury 28. Two isoforms are known for these giant proteins, AHNAK1 and AHNAK2, which range in size from 600 to 700 kDa 30. There is evidence that AHNAKs function as scaffold proteins important for organizing Ca2+ signaling molecules. Mouse knockout studies indicate that AHNAK1 is required for plasma membrane expression of L-type Ca2+ channels (Cav1.1), which mediate Ca2+ influx into CD4 T cells following TCR stimulation 31. In humans, mutations in AHNAK1 cause cardiac dysfunction, an observation that is also consistent with a defect in Cav1.1 Ca2+ channel function 32. Additional studies are needed to explain how desmoyokin-AHNAK (which is localized in the cytosol, nucleus, or inner leaflet of the plasma membrane, depending on cell type) can cross into the lumen of enlargeosomes, and get exposed on the plasma membrane upon exocytosis. This topology problem, which has not been given sufficient attention in the literature, is made even more significant considering the fact that AHNAK is so far the only available marker for enlargeosomes. Before attributing important functions such as exocytosis and plasma membrane repair to enlargeosomes, it will be important to find additional markers for these novel organelles, and determine whether they are ubiquitous, or present in only a few cell types.
It is still unclear if additional, ubiquitous Ca2+ regulated intracellular organelles with a role in plasma membrane repair remain to be identified. However, the number of candidate vesicles for this role is certainly not large, given that their exocytosis must be triggered by rises in cytosolic Ca2+. Ca2+-regulated exocytosis is not a property associated with most known intracellular compartments 18, except lysosomes, the still poorly characterized enlargeosomes, and a number of specialized secretory granules (see Box 1).
How does exocytosis promote plasma membrane repair?
Two models have been proposed to explain the role of Ca2+ triggered exocytosis in membrane repair. One model, known as the “patch hypothesis”, proposes that Ca2+ influx triggers homotypic fusion of pre-existing intracellular vesicles in the vicinity of the wound. The large vesicles postulated to arise from homotypic fusion would then somehow fuse with the plasma membrane surrounding the injured site, “patching” the wound 21. The second model proposes that the primary role of exocytosis, instead of forming a patch over the lesion, is to reduce plasma membrane tension. Reduction in membrane tension was detected experimentally in cells undergoing injury and recovery, and the process was proposed to promote repair by facilitating spontaneous bilayer resealing 33. Based on an apparent higher Ca2+ threshold for the repair of large membrane disruptions, it was suggested that the “patch” mechanism is mostly responsible for the repair of large lesions, while membrane tension reduction would preferentially reseal small wounds 21. It is important to note, however, that the experimental evidence supporting the formation of a membrane patch over large wounds consists largely of observed barriers to the diffusion of membrane impermeable dyes into injured cells. Diffusion barriers are observed immediately after large portions of the plasma membrane are removed by locally administered TX-100 34, or when Ca2+-containing water is injected into sea urchin eggs (an intervention used to demonstrate that cytosol can rapidly generate a barrier when exposed to elevated [Ca2+]i ) 35. However, the nature of these barriers (i.e. if a lipid bilayer is present) is still unclear. It is important to consider a possible role in this process for non-exocytic Ca2+ triggered pathways that can also prevent cytoplasm diffusion/loss following plasma membrane injury. One example is the wound repair pathway proposed to be mediated by tissue transglutaminase (TGase), a family of enzymes that catalyze Ca2+-dependent protein cross-linking via epsilon(gamma-glutamyl)lysine bridges 36. In mammals, crosslinking of extracellular matrix proteins by TGases was implicated in the healing and subsequent stability of tissue wounds 37-39. At the cellular level, TGase-dependent crosslinking of intracellular proteins was reported in experiments using tet-regulated expression, or antisense-mediated TGase silencing 36. TGase reaction products were detected at injured sites in the slime mold Physarum polycephalum, and RNAi-mediated knockdown of transglutaminase 2 in mammalian cells led to defective plasma membrane resealing 40. These results led to the suggestion that intracellular “clots” generated by the crosslinking activity of TGase at wound sites might be a critical early step in cell resealing. The role of Ca2+ in promoting TGase activity provides an alternative explanation for one of the key observations giving rise to the “patch hypothesis” of plasma membrane repair. Upon injection of Ca2+ containing sea water into eggs, fluorescent tracers might be blocked from diffusion into the cytosol by the Ca2+ dependent TGase protein crosslinking reaction, and not by the formation of a membranous patch generated by homotypic fusion of intracellular vesicles. Future studies are needed to investigate this intriguing possibility — that plasma membrane resealing is preceded by a clotting reaction that reduces cytosol loss through the wound. Such a reaction could significantly slow down the loss of critical intracellular molecules, allowing exocytosis and additional critical resealing steps to proceed.
Exocytosis can't do it all: Endocytosis enters the scene
Most of the studies that were discussed above, focusing on the role of Ca2+-dependent exocytosis in the repair of plasma membrane wounds, involved mechanical methods of cell injury. Interestingly, recent investigations showed that cells treated with the bacterial pore forming protein streptolysin O (SLO) also recover membrane integrity in a Ca2+ dependent fashion 41. This was an intriguing observation, because the stable nature of the lesions caused by pore-forming proteins ruled out the two previously proposed models for plasma membrane repair. Neither a patch nor a reduction in membrane tension can explain how transmembrane pores can be effectively removed from the cell surface. Importantly, we found that Ca2+ influx through SLO pores triggers a repair response very similar to that observed after mechanical wounding: Lysosomal enzymes are released into the medium, and the release of cytosolic markers such as lactate dehydrogenase (LDH) is blocked, reflecting membrane resealing. Remarkably, live imaging of cells exposed to SLO also revealed that lysosomal exocytosis is not the only membrane traffic event triggered during cell injury and repair. A rapid form of endocytosis was observed within seconds of SLO pore formation in the plasma membrane, and this occurred only under repair conditions, in the presence of extracellular Ca2+ (Figure 1) 42. Importantly, the kinetics of injury-induced endocytosis was very similar to that previously reported for the repair of mechanically induced wounds 1, 2. Subsequent studies confirmed that this rapid, Ca2+ dependent form of endocytosis also occurs in mechanically injured cells. Collectively, these results indicated that endocytosis promotes resealing by removing lesions from the plasma membrane 42. These findings provided an important new insight into how cells protect themselves, not only from mechanical injury but also from microbial toxins and other pore-forming proteins, such as those produced by the immune system 43. Although it remains to be seen if an isolated wounding event can induce prolonged endocytic responses, the observations of vigorous and sustained endocytosis in wounded cells (Figure 1) 42 may explain why plasma membrane repair is more efficient in cells that were submitted to a previous injury, either mechanical 44 or by pore forming proteins 45.
Figure 1. Rapid endocytosis is triggered by SLO in the presence of Ca2+.

HeLa cells expressing YFP-GPI on the plasma membrane were imaged in a spinning disk confocal microscope in Ca2+-containing medium. (A) Images acquired in the absence of SLO. (B) SLO (100 μg/ml) was locally added with a micropipette 89 s after initiating the imaging. Each image corresponds to 8 optical section confocal stacks acquired at 1 frame /1.4 s. The arrows point to individual endosomes that were detected inside the cells shortly after SLO addition.
The discovery of injury-induced endocytosis provides a new framework for understanding plasma membrane repair pathways in mammalian cells. For several years Ca2+ triggered exocytosis was regarded as the major mechanism for the repair of membrane injury. However, in light of these new findings, it is no longer clear whether new membrane addition by exocytosis directly mediates plasma membrane resealing, or whether it acts indirectly, by triggering a subsequent compensatory endocytic response that represents the true resealing event. In support of the latter possibility, acute cholesterol depletion effectively blocks both endocytosis and plasma membrane resealing in response to mechanically induced injury. Conversely, disassembly of the actin cytoskeleton markedly facilitates both injury induced endocytosis and plasma membrane resealing 42. These results provide a mechanistic explanation for prior observations of increased repair efficiency after disruption of the cortical cytoskeleton 44, 46. The recent discovery of injury induced endocytosis also suggests an intriguing new interpretation for the large vesicles observed close to the wound in several earlier studies of plasma membrane repair 13, 47, 48 (Figure 2). The endocytic vesicles (identified through their content of endocytic tracers - Figure 2 E-F) found in cells wounded by pore forming toxins or scraping (Figure 2 C-D) are remarkably similar to the large and heterogeneous vesicles observed in earlier studies involving mechanical cell wounding (Figure 2 A-B).
Figure 2. Endocytic tracers reveal that the large intracellular vesicles associated with injury sites correspond to rapidly formed endosomes.

(A-B) Endothelial cells were injured by passing through a 30-gauge needle in the presence of horse radish peroxidase (HRP), fixed and prepared for transmission EM observation. An accumulation of HRP-positive vesicles was observed at sites near a plasma membrane disruption (arrowheads). Reproduced with permission from Ref. 13. (C) Transmission EM of NRK cells fixed 4 min after scraping in the presence of Ca2+. (D) Transmission EM of NRK cells fixed 4 min after exposure to 200 ng/ml SLO in the presence of Ca2+ and the endocytic tracer BSA-gold. The arrows point to large endosomes containing BSA-gold. Bars = 5 μm. (E) Close-up of a BSA-gold containing endosome in a NRK cell wounded by scraping (a procedure that generated numerous plasma membrane lesions, due to disruption of focal contacts). (F) Close-up of a BSA-gold containing endosome in a NRK cell wounded by SLO. Arrows point to individual gold particles. Bars = 200 nm. Reproduced with permission from Ref. 42.
Interestingly, a rapid form of compensatory endocytosis was also reported following enlargeosome exocytosis triggered by Ca2+ ionophores in PC12−27 cells 29. In their assays, the authors observed internalization of desmoyokin-AHNAK, an enlargeosome marker, within a short period after its delivery to the plasma membrane. Although this study did not propose that the compensatory endocytosis of enlargeosomes was responsible for plasma membrane repair, it is intriguing that the Ca2+-dependent desmoyokin-AHNAK positive endosomes 29 are very similar in size and morphology to injury-induced endosomes (Figure 2) 42, and are also distinct from classical dynamin-dependent endosomes 29, 42. Future studies involving direct inhibition of lysosome and enlargeosome exocytosis should clarify whether exocytosis of one or both of these organelle types is required for induction of endocytosis and plasma membrane repair. Simultaneous imaging of lysosomal exocytosis and endocytosis by total internal reflection (TIRF) microscopy should also provide very useful information to define the spatial and temporal relationship of both processes.
Novel mechanistic insights on membrane repair: Implications for understanding human disease
Ca2+ influx into the cytosol of injured cells is thought to trigger exocytosis by binding and activating cytosolic or membrane resident Ca2+ sensors. In mammals, synaptotagmin VII (Syt VII) 2 and dysferlin 8 have been identified as strong candidates for membrane Ca2+ sensors mediating plasma membrane repair. Loss of function mutations in either of these genes gives rise to pathology 8, 9, 49, and interestingly, the disease characteristics reflect at least in part the tissue distribution of these proteins. The ubiquitously expressed SytVII regulates the exocytosis of lysosomes and some other Ca2+-regulated secretory vesicles, and mutations in Syt VII cause an autoimmune inflammatory disease in the skin and skeletal muscle of mice 9. Dysferlin is most abundantly expressed in muscle, and it is the gene carrying the mutations responsible for Miyoshi myopathy and Limb-girdle muscular dystrophy 2B, which are adult onset forms of distal muscular dystrophy in humans 49. Dysferlin is largely localized on the plasma membrane of muscle fibers, although under certain conditions it has also been detected on intracellular vesicles 50. Unlike Syt VII, which has been directly demonstrated to confer Ca2+ sensitivity to membrane fusion reactions 51, a role for dysferlin in linking Ca2+ transients to membrane traffic has not yet been directly demonstrated. However, such a role is inferred from the presence of several Ca2+-binding C2 domains in the cytosolic domain of dysferlin. The two C2 domains present on the cytoplasmic region of synaptotagmins are responsible for promoting Ca2+-dependent interactions with membrane phospholipids and SNARE molecules, events that precede membrane fusion 52. Interestingly, mutations in either Syt VII or dysferlin disrupt the function of skeletal muscle. Thus, both Syt VII and dysferlin control membrane traffic events that are required for maintaining integrity of the sarcolemma, in face of the frequent injuries suffered by this contractile tissue in vivo.
The recent identification of injury-induced, Ca2+ dependent endocytosis as an event important for plasma membrane repair offers new opportunities for understanding molecular defects linked to degenerative muscle disease. It has been known for some time that mutations in caveolin-3, a muscle specific protein associated with the endocytic structures known as caveolae, cause Limb-girdle muscular dystrophy 1C and other forms of myopathy 53, 54. In addition to being a major structural component of caveolae at the sarcolemma , caveolin-3 is also required for normal T-tubule development and for numerous signaling pathways 55-57. Thus, it is not clear which specific functional defects associated with the lack of caveolin-3 lead to muscular dystrophy. However, recent observations revealed that caveolin-3 is important for retaining dysferlin at the plasma membrane. In the absence of caveolin-3 dysferlin is rapidly endocytosed through a clathrin-independent pathway, and this process is counteracted by caveolin-3 expression 58. These findings highlight an intriguing link between dysferlin function, endocytosis and the maintenance of muscle fiber integrity. It is important to note, however, that the endocytic vesicles generated in injured cells do not have a morphology that is consistent with caveolae (Figure 2), and behave differently from most characterized endocytic pathways (which are dynamin and/or actin-dependent - see Box 2). The endosomes generated in response to plasma membrane injury are dynamin-independent, and are actually stimulated after disruption of the actin cytoskeleton 42. Thus, the available lines of evidence suggest that caveolin-3 and dysferlin may function in concert in the regulation of a rapid, non-conventional form of endocytosis that may be also present in muscle cells. It is noteworthy that the C2 domain-containing Ca2+ sensors synaptotagmins Syt I and Syt VII also have a dual role in membrane traffic, simultaneously coordinating Ca2+ triggered exocytosis of intracellular vesicles and recruiting protein complexes to promote their own endocytosis from the plasma membrane 59, 60. In future studies, it will be important to characterize the domains in the cytosolic region of dysferlin responsible for its interactions with caveolin-3 61 and for its rapid endocytosis when caveolin-3 is absent. Collectively, these observations make it enticing to speculate that some forms of human muscular dystrophy may result from aberrant injury-induced endocytosis pathways.
Box 2 — Endocytosis.
Endocytosis is the term used to describe the multiple pathways by which cells internalize macromolecules and particles into vesicles derived from the plasma membrane. It is becoming increasingly clear that the cellular mechanisms of endocytosis are highly diverse and tightly regulated, in order to fulfill essential physiological roles 72. Phagocytosis is the actin-dependent, typically receptor-mediated particle uptake process that gives rise to the largest endosomes known, phagosomes. Phagocytic uptake is a function essential for the clearance of pathogens and of apoptotic cells, and for antigen presentation and the initiation of immune responses. Pinocytosis, on the other hand, involves the uptake of fluid and soluble macromolecules, and can give rise to intracellular vesicles of different sizes, ranging from >1 μm (macropinocytosis, which is also actin-dependent) to smaller vesicles of 90−120 nm. The pathways generating these smaller types of endosomes fall into three categories: clathrin-mediated endocytosis, caveolae-mediated endocytosis and clathrin and caveolae-independent endocytosis. Interestingly, several of these otherwise mechanistically diverse endocytic pathways are regulated by the large GTPase dynamin. The function of dynamin in endocytosis is best understood in the context of clathrin-mediated endocytosis, where it self-assembles into a “collar” thought to facilitate the pinching off of vesicles from the plasma membrane. However, dynamin is also required for at least two types of non-clathrin-dependent endocytosis, caveolae-mediated and RhoA-regulated endocytosis 73.
Clathrin and caveolae-independent mechanisms of endocytosis are less well understood, but some of these pathways are also dynamin-independent and regulated by small GTPases, such as CDC42 or Arf6. Although cholesterol extraction inhibits most forms of endocytosis, a cholesterol-sensitive form of CDC42 activation may regulate recruitment of the actin-polymerization machinery that is required for generation of the GPI anchored protein-enriched endosomal compartment (GEEC) 73. Interestingly, the large endosomes (300−500 nm) recently identified in cells wounded in the presence of extracellular Ca2+ fall into yet another unique category: they are independent of dynamin, sensitive to cholesterol extraction, but are not inhibited (and are actually enhanced) by drugs that disrupt the actin cytoskeleton 42. Further characterization of the form of endocytosis involved in plasma membrane repair is likely to add to our current knowledge of the multiple mechanisms developed by eukaryotic cells to maintain plasma membrane and cellular homeostasis.
Concluding Remarks
The role of Ca2+-dependent exocytosis of intracellular vesicles in mediating plasma membrane repair has been studied extensively. The current belief is that exocytosis provides extra membrane to seal the wound, and a decrease in membrane tension that is required for bilayer resealing. An important role for these mechanisms in the repair of plasma membrane injury cannot be ruled out, but a study of how cells restore membrane integrity after attack by pore-forming proteins has led to surprising findings. Ca2+ influx through wounds caused by transmembrane pores or mechanical abrasion triggers the rapid formation of large endocytic vesicles, which remove lesions from the plasma membrane 42. These data led us to hypothesize that this novel form of endocytosis is a compensatory reaction to Ca2+ triggered lysosomal exocytosis, and that it may represent a general mechanism by which cells can repair many forms of membrane damage (see model and outstanding questions in Figure 3) .
Figure 3. Model for the rapid membrane traffic events triggered by Ca2+ influx in injured cells.
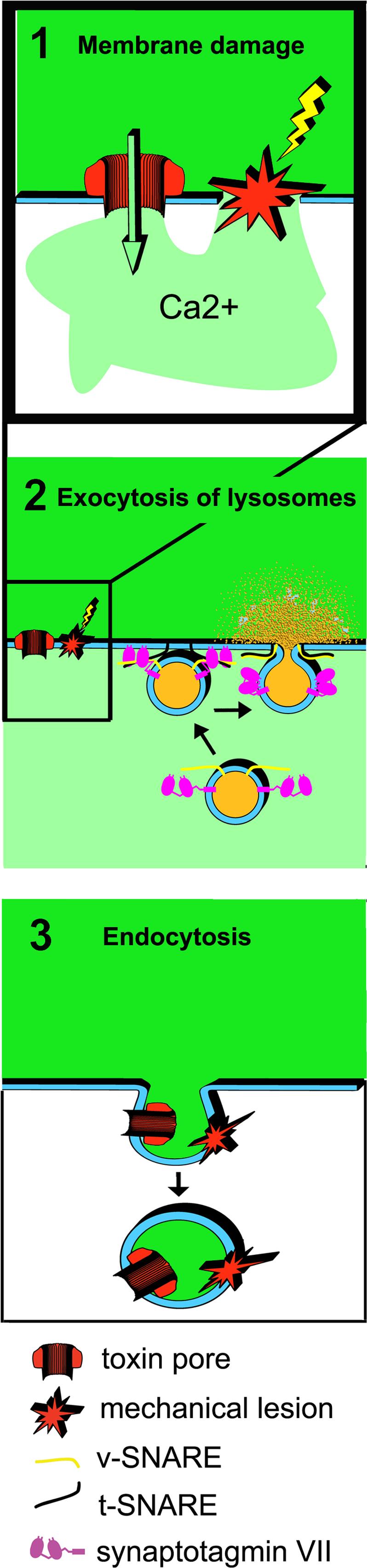
1. Transmembrane pores formed by toxin monomers or mechanical abrasions (microinjection, scratching, laser wounding) wound the plasma membrane. 2. Ca2+ influx triggers lysosomal exocytosis, mediated by v-SNARES and the Ca2+ sensor synaptotagmin VII on lysosomes and t-SNARES on the plasma membrane. Exocytosis results in the release of lysosomal contents extracellularly. 3. Exocytosis is rapidly followed by the formation of large endosomes, which can internalize the lesions and promote plasma membrane repair. Questions that remain to be answered at each of these steps are shown in the right panels.
These recent results have opened up new avenues to be pursued, towards the goal of understanding plasma membrane repair dynamics and the diseases resulting from failures in this mechanism. There are several questions that need to be addressed in light of these recent discoveries. First, what is the role of Ca2+ in injury-induced endocytosis? Is it critical because Ca2+-dependent exocytosis triggers a compensatory endocytic response, or because there are Ca2+ sensors specifically involved in regulating endocytosis? In order to answer these questions, it will be necessary to develop methods to independently block exocytosis and endocytosis, and assess the contribution of each pathway on plasma membrane repair. Second, what are the molecular machineries involved in regulating injury-induced exocytosis and endocytosis? This goal will require the development of genome-wide loss-of-function screens of plasma membrane repair. Third, it will be important to determine whether additional Ca2+-responsive molecules proposed to participate in plasma membrane repair, such as calpain 62 and annexin A1 63, regulate exocytosis or endocytosis, or additional steps that still remain to be characterized. A deeper mechanistic knowledge of the membrane traffic pathways involved in rapid plasma membrane repair is likely to facilitate the identification of new genes associated with human disease, and to advance the search for therapeutic strategies.
Acknowledgements
Work in the author's laboratory was supported by grants from the National Institutes of Health (R37AI34867 and R01GM0646625 to N.W.A and F32AI069785 to C.T.).
References
- 1.Steinhardt RA, et al. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- 2.Reddy A, et al. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 3.McNeil PL, Steinhardt RA. Loss, restoration and maintenance of plasma membrane integrity. J. Cell Biol. 1997;137:1–4. doi: 10.1083/jcb.137.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrews NW. Membrane resealing: synaptotagmin VII keeps running the show. Sci STKE. 20052005:pe19. doi: 10.1126/stke.2822005pe19. [DOI] [PubMed] [Google Scholar]
- 5.Gurcel L, et al. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126:1135–1145. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 6.Huffman DL, et al. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc Natl Acad Sci U S A. 2004;101:10995–11000. doi: 10.1073/pnas.0404073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- 8.Bansal D, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabarti S, et al. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol. 2003;162:543–549. doi: 10.1083/jcb.200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambers R, Chambers E. Explorations into the nature of the living cell. Harvard University Press; 1961. [Google Scholar]
- 11.Heilbrunn L. The dynamics of living protoplasm. Academic Press; 1956. [Google Scholar]
- 12.Bi GQ, et al. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131:1747–1758. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. 1995;131:1737–1745. doi: 10.1083/jcb.131.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coorsen JR, et al. Ca2+ triggers massive exocytosis in Chinese hamster ovary cells. EMBO J. 1996;15:3787–3791. [PMC free article] [PubMed] [Google Scholar]
- 15.Chavez RA, et al. A biosynthetic regulated secretory pathway in constitutive secretory cells. J Cell Biol. 1996;133:1177–1191. doi: 10.1083/jcb.133.6.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tardieux I, et al. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez A, et al. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaiswal JK, et al. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol. 2002;159:625–635. doi: 10.1083/jcb.200208154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews NW. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000;10:316–321. doi: 10.1016/s0962-8924(00)01794-3. [DOI] [PubMed] [Google Scholar]
- 20.McNeil PL. Repairing a torn cell surface: make way, lysosomes to the rescue. J Cell Sci. 2002;115:873–879. doi: 10.1242/jcs.115.5.873. [DOI] [PubMed] [Google Scholar]
- 21.McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- 22.Blott EJ, Griffiths GM. Secretory lysosomes. Nat Rev Mol Cell Biol. 2002;3:122–131. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 23.Holt OJ, et al. Regulating secretory lysosomes. J Biochem. 2006;140:7–12. doi: 10.1093/jb/mvj126. [DOI] [PubMed] [Google Scholar]
- 24.Dell'Angelica EC, et al. Lysosome-related organelles. Faseb J. 2000;14:1265–1278. doi: 10.1096/fj.14.10.1265. [DOI] [PubMed] [Google Scholar]
- 25.Martinez I, et al. Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol. 2000;148:1141–1149. doi: 10.1083/jcb.148.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fowler KT, et al. Expression and function of synaptotagmin VII in CTLs. J Immunol. 2007;178:1498–1504. doi: 10.4049/jimmunol.178.3.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, et al. Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Dev Cell. 2008;14:914–925. doi: 10.1016/j.devcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borgonovo B, et al. Regulated exocytosis: a novel, widely expressed system. Nature Cell Biology. 2002;4:955–962. doi: 10.1038/ncb888. [DOI] [PubMed] [Google Scholar]
- 29.Cocucci E, et al. Enlargeosome, an exocytic vesicle resistant to nonionic detergents, undergoes endocytosis via a nonacidic route. Mol Biol Cell. 2004;15:5356–5368. doi: 10.1091/mbc.E04-07-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komuro A, et al. The AHNAKs are a class of giant propeller-like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc Natl Acad Sci U S A. 2004;101:4053–4058. doi: 10.1073/pnas.0308619101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matza D, et al. A scaffold protein, AHNAK1, is required for calcium signaling during T cell activation. Immunity. 2008;28:64–74. doi: 10.1016/j.immuni.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haase H, et al. Ahnak is critical for cardiac Ca(V)1.2 calcium channel function and its beta-adrenergic regulation. Faseb J. 2005;19:1969–1977. doi: 10.1096/fj.05-3997com. [DOI] [PubMed] [Google Scholar]
- 33.Togo T, et al. A decrease in membrane tension precedes successful cell-membrane repair. Mol Biol Cell. 2000;11:4339–4346. doi: 10.1091/mbc.11.12.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNeil PL, et al. Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci. 2000;113:1891–1902. doi: 10.1242/jcs.113.11.1891. [DOI] [PubMed] [Google Scholar]
- 35.Terasaki M, et al. Large plasma membrane disruptions are rapidly resealed by Ca2+ -dependent vesicle-vesicle fusion events. J Cell Biol. 1997;139:63–74. doi: 10.1083/jcb.139.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicholas B, et al. Cross-linking of cellular proteins by tissue transglutaminase during necrotic cell death: a mechanism for maintaining tissue integrity. Biochem J. 2003;371:413–422. doi: 10.1042/BJ20021949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haroon ZA, et al. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. Faseb J. 1999;13:1787–1795. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 38.Nardacci R, et al. Transglutaminase type II plays a protective role in hepatic injury. Am J Pathol. 2003;162:1293–1303. doi: 10.1016/S0002-9440(10)63925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gross SR, et al. Importance of tissue transglutaminase in repair of extracellular matrices and cell death of dermal fibroblasts after exposure to a solarium ultraviolet A source. J Invest Dermatol. 2003;121:412–423. doi: 10.1046/j.1523-1747.2003.12353.x. [DOI] [PubMed] [Google Scholar]
- 40.Kawai Y, et al. Transglutaminase 2 activity promotes membrane resealing after mechanical damage in the lung cancer cell line A549. Cell Biol Int. 2008 doi: 10.1016/j.cellbi.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 41.Walev I, et al. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci U S A. 2001;98:3185–3190. doi: 10.1073/pnas.051429498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Idone V, et al. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol. 2008;180:905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan BP. Complement membrane attack on nucleated cells: resistance, recovery and non-lethal effects. Biochem J. 1989;264:1–14. doi: 10.1042/bj2640001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Togo T, et al. The mechanism of facilitated cell membrane resealing. J Cell Sci. 1999;112:719–731. doi: 10.1242/jcs.112.5.719. [DOI] [PubMed] [Google Scholar]
- 45.Reiter Y, et al. Complement membrane attack complex, perforin, and bacterial exotoxins induce in K562 cells calcium-dependent cross-protection from lysis. J Immunol. 1995;155:2203–2210. [PubMed] [Google Scholar]
- 46.Miyake K, et al. An actin barrier to resealing. J Cell Sci. 2001;114:3487–3494. doi: 10.1242/jcs.114.19.3487. [DOI] [PubMed] [Google Scholar]
- 47.Krause TL, et al. Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci. 1994;14:6638–6651. doi: 10.1523/JNEUROSCI.14-11-06638.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casademont J, et al. Vacuolation of muscle fibers near sarcolemmal breaks represents T-tubule dilatation secondary to enhanced sodium pump activity. J Neuropathol Exp Neurol. 1988;47:618–628. doi: 10.1097/00005072-198811000-00005. [DOI] [PubMed] [Google Scholar]
- 49.Liu J, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 50.Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Bhalla A, et al. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell. 2005;16:4755–4764. doi: 10.1091/mbc.E05-04-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapman ER. Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat Rev Mol Cell Biol. 2002;3:498–508. doi: 10.1038/nrm855. [DOI] [PubMed] [Google Scholar]
- 53.Woodman SE, et al. Caveolinopathies: mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology. 2004;62:538–543. doi: 10.1212/wnl.62.4.538. [DOI] [PubMed] [Google Scholar]
- 54.Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. 2006;7:762–773. doi: 10.1038/nrm2024. [DOI] [PubMed] [Google Scholar]
- 55.Parton RG, et al. Caveolin-3 associates with developing T-tubules during muscle differentiation. J Cell Biol. 1997;136:137–154. doi: 10.1083/jcb.136.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galbiati F, et al. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276:21425–21433. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 57.Carozzi AJ, et al. Inhibition of lipid raft-dependent signaling by a dystrophy-associated mutant of caveolin-3. J Biol Chem. 2002;277:17944–17949. doi: 10.1074/jbc.M110879200. [DOI] [PubMed] [Google Scholar]
- 58.Hernandez-Deviez DJ, et al. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J Biol Chem. 2008;283:6476–6488. doi: 10.1074/jbc.M708776200. [DOI] [PubMed] [Google Scholar]
- 59.Jarousse N, et al. Endocytosis of synaptotagmin 1 is mediated by a novel, tryptophan-containing motif. Traffic. 2003;4:468–478. doi: 10.1034/j.1600-0854.2003.00101.x. [DOI] [PubMed] [Google Scholar]
- 60.Dasgupta S, Kelly RB. Internalization signals in synaptotagmin VII utilizing two independent pathways are masked by intramolecular inhibitions. J Cell Sci. 2003;116:1327–1337. doi: 10.1242/jcs.00290. [DOI] [PubMed] [Google Scholar]
- 61.Matsuda C, et al. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet. 2001;10:1761–1766. doi: 10.1093/hmg/10.17.1761. [DOI] [PubMed] [Google Scholar]
- 62.Mellgren RL, et al. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem. 2007;282:2567–2575. doi: 10.1074/jbc.M604560200. [DOI] [PubMed] [Google Scholar]
- 63.McNeil AK, et al. Requirement for annexin A1 in plasma membrane repair. J Biol Chem. 2006;281:35202–35207. doi: 10.1074/jbc.M606406200. [DOI] [PubMed] [Google Scholar]
- 64.Easom RA. Beta-granule transport and exocytosis. Semin Cell Dev Biol. 2000;11:253–266. doi: 10.1006/scdb.2000.0174. [DOI] [PubMed] [Google Scholar]
- 65.Sollner TH. Regulated exocytosis and SNARE function (Review). Mol Membr Biol. 2003;20:209–220. doi: 10.1080/0968768031000104953. [DOI] [PubMed] [Google Scholar]
- 66.Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- 67.Ninomiya Y, et al. Ca2+-dependent exocytotic pathways in Chinese hamster ovary fibroblasts revealed by a caged-Ca2+ compound. J Biol Chem. 1996;271:17751–17754. doi: 10.1074/jbc.271.30.17751. [DOI] [PubMed] [Google Scholar]
- 68.Griffiths GM. Secretory lysosomes - a special mechanism of regulated secretion in haemopoietic cells. Trends Cell Biol. 1996;6:329–332. doi: 10.1016/0962-8924(96)20031-5. [DOI] [PubMed] [Google Scholar]
- 69.Rao SK, et al. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J Biol Chem. 2004;279:20471–20479. doi: 10.1074/jbc.M400798200. [DOI] [PubMed] [Google Scholar]
- 70.Bhalla A, et al. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol. 2006 doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- 71.Koh TW, Bellen HJ. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 2003;26:413–422. doi: 10.1016/S0166-2236(03)00195-4. [DOI] [PubMed] [Google Scholar]
- 72.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 73.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nature Reviews Molecular Cell Biology. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]