

Abstract
Histone deacetylase (HDAC) proteins are transcription regulators linked to cancer. As a result, multiple small molecule HDAC inhibitors are in various phases of clinical trials as anti-cancer drugs. The majority of HDAC inhibitors non-selectively influence the activities of eleven human HDAC isoforms, which are divided into distinct classes. This tutorial review focuses on the recent progress toward the identification of class-selective and isoform-selective HDAC inhibitors. The emerging trends suggest that subtle differences in the active sites of the HDAC isoforms can be exploited to dictate selectivity.
Introduction
Histone deacetylase (HDAC) proteins play an important role in gene expression by governing the acetylation state of lysine residues located on the amino-terminal tails of histone proteins (Fig. 1). Histones comprise nucleosomes, which are the basic packaging units of chromosomes.1 By binding to genomic DNA, the accessibility of genes to transcriptional proteins is altered by histone lysine acetylation. As a result, HDAC proteins are generally associated with repression of transcription and reduced gene expression (for a review of HDAC proteins, histone acetylation, and transcription, see ref. 2).
Fig. 1.

The acetylation state of lysine amino acids are governed by the equilibrium activities of acetyltransferase enzymes and deacetylase enzymes. In the context of gene expression, the lysine residues of histone proteins are key substrates for acetylation.
HDAC proteins comprise a family of 18 members in humans and are separated into four classes based on their size, cellular localization, number of catalytic active sites, and homology to yeast HDAC proteins. Class I includes HDAC1, HDAC2, HDAC3, and HDAC8. Class II consists of six HDAC proteins that are further divided into two subclasses. Class IIa includes HDAC4, HDAC5, HDAC7, and HDAC9, which each contains a single catalytic active site. Class IIb includes HDAC6 and HDAC10, which both contain two active sites, although only HDAC6 has two catalytically competent active sites. HDAC11 is the sole member of class IV, based on phylogenetic analysis.3 Class I, II, and IV HDAC proteins operate by a metal ion-dependent mechanism, as indicated by crystallographic analysis.4 In contrast, class III HDAC proteins, referred to as sirtuins (SIRT1-7), operate by a NAD+-dependent mechanism unrelated to the other HDAC proteins (see ref. 5 for a review of the HDAC family). The metal-dependent HDAC proteins are the targets of the HDAC inhibitors discussed in this review.
Due to their fundamental role in gene expression, HDAC proteins have been associated with basic cellular events and disease states, including cell growth, differentiation, and cancer formation (see ref. 6 for a review on HDAC proteins in cancer). In particular, distinct class I and class II HDAC proteins are overexpressed in some cancers, including ovarian (HDAC1–3),7 gastric (HDAC2),8 and lung cancers (HDAC1 and 3),9 among others. In addition, a possible correlation between HDAC8 and acute myeloid leukemia (AML) has been suggested.10 With respect to the class II HDAC proteins, aberrant expression of HDAC6 was induced in some breast cancer cells.11 While individual members of class I and II HDAC proteins are linked to cancer formation, the role of each isoform in carcinogenesis is unclear. Particularly, the molecular mechanism connecting HDAC activity to cancer formation is not yet defined.
Given their association with cancer formation, class I and II HDAC proteins have emerged as attractive targets for anti-cancer therapy. Several HDAC inhibitor (HDACi) drugs are in various stages of clinical trials,12 with SAHA (suberoylanilide hydroxamic acid, Vorinostat, Fig. 2) gaining FDA approval in 2006 for the treatment of advanced cutaneous T-cell lymphoma (CTCL).13 Consistent with their clinical effects, inhibitors of HDAC proteins suppress tumor cell proliferation, induce cell differentiation, and upregulate crucial genes associated with anti-cancer effects (see ref. 14 for a review of the clinical effects of HDACi drugs). Therefore, HDACi drugs represent a promising next generation of anti-cancer therapeutics.
Fig. 2.

Pan-inhibitors TSA and SAHA.
In general, HDAC inhibitors have a standard, modular construction with structural similarities to the HDAC acetyl-lysine substrate (Fig. 1). HDAC inhibitors typically consist of a metal-binding moiety that coordinates to the catalytic metal atom within the HDAC active site and a capping group that interacts with the residues at the entrance of the active site (Fig. 2). In addition, a linker that is structurally related to the carbon chain present in the acetyl-lysine substrate appropriately positions the metal-binding moiety and capping group for interactions in the active site. Crystallographic evidence with SAHA bound in the active site of a bacterial homologue of class I HDAC proteins (HDLP) confirms that the hydroxamic acid coordinates to the zinc atom at the bottom of the active site, the linker lies in a confined hydrophobic channel, and the anilide capping group interacts with the amino acids surrounding the entrance of the active site.4
The majority of HDACi drugs in and out of clinical trials inhibit all HDAC isoforms nonspecifically (so called paninhibitors). SAHA and TSA are the canonical pan-inhibitors (Fig. 2), influencing the activity of HDAC1–9 with roughly equivalent potency.15 Selective HDAC inhibitors, which affect either a single HDAC isoform (isoform-selective HDACi) or several isoforms within a single class (class-selective HDACi), would be ideal chemical tools to elucidate the individual functions of each HDAC isoform. Specifically, selective HDAC inhibitors would aid in defining the molecular mechanism connecting HDAC activity to cancer formation. In addition, it is possible that a class-selective or isoform-selective HDAC inhibitor would provide a more effective chemotherapy compared to pan-inhibitors. Despite the utility and potential therapeutic advantage of selective HDAC inhibitors, their design has been challenging. The high sequence similarity within the active sites of the isoforms makes inhibitor design problematic. In addition, subtle differences among the active sites of each human isoform are not well characterized due to the limited crystallographic analysis; the only available crystal structures are of HDAC7 and HDAC8,16–18 although the structures of bacterial homologues related to class I and II HDAC proteins have been useful.4,19 Despite these challenges, several class-selective and isoform-selective HDAC inhibitors have been reported within the past decade.
Herein, we review the recent advancements in creation of class-selective and isoform-selective HDAC inhibitors. Due to the modular nature of HDAC inhibitors, this review is organized by modifications made to each region: the capping group, metal-binding moiety, and linker. In addition, the structural elements leading to selectivity are highlighted by discussing class I-selective and class II-selective inhibitors separately. The emerging trends suggest that selective inhibitors can be designed to exploit the subtle differences in the active sites of each HDAC isoform. The current challenges in the field are also highlighted to aid future inhibitor design efforts.
I. Review of class-selective and isoform-selective HDAC inhibitors
I.A Capping group modifications
The capping group region has been modified extensively towards the creation of selective HDAC inhibitors. Among the class-selective inhibitors identified, capping group moieties have been hypothesized to interact with amino acid residues near the entrance of the active site differently between the HDAC classes. Inhibitor docking studies confirm this hypothesis in the case of class II-selective inhibitors (section I.A.2). Several inhibitors with possible isoform selectivity within class I were also reported (section I.A.1), although the elements leading to selectivity in this case are less understood.
I.A.1 Class I-selective HDAC inhibitors
HDAC inhibitors with cyclic peptide moieties at the capping group region (Fig. 3) display class I selectivity. For example, natural products trapoxin A (TPX), trapoxin B, chlamydocin, and Cyl-2 displayed selectivity for HDAC1 versus HDAC6 of 640–570 000- fold with pM potency (HDAC1 IC50 = 110–820 pM).20 HC-toxin was tested against rat liver HDAC proteins (RLH), which are comprised of predominantly HDAC1–3, as well as human HDAC8 and an HDAC6-like bacterial homologue (FB188 HDAH), and demonstrated class I selectivity of greater than 1000-fold (RLH IC50 = 10 nM).21 Among the azumamides, azumamide E displayed roughly 100-fold selectivity for class I HDAC1, 2, and 3 compared to class II HDAC4, 5, 6, 7, and 9 (HDAC1–3 IC50 = 50–100 nM).22 Apicidin was tested against HDAC1–9 and showed 17–232-fold selectivity for HDAC2, 3 and 8 compared to HDAC1, 4, 6, 7, and 9 (HDAC2–3, 8 EC50 = 43–575 nM).15 Finally, romidepsin (depsipeptide, FK-228) displayed roughly 10-fold selectivity for HDAC1 and HDAC2 versus HDAC4 and roughly 300-fold selectivity versus HDAC6 (HDAC1–2 IC50 = 36–47 nM).23 Furthermore, some synthetically derived cyclic hydroxamic acid-containing peptides (CHAPs, Fig. 3) and romidepsin derivatives have also been shown to have similar class I selectivity.20,24 Interestingly, a cyclic peptide mimic (compound 1) displayed modest selectivity within class I, with a 4-fold preference for HDAC1 versus HDAC8 (HDAC1 IC50 = 57 nM), although the class II selectivity was not assessed.25 In total, the class I selectivity of inhibitors containing cyclic peptide moieties suggests that the capping group plays a significant role of discriminating the HDAC isoforms.
Fig. 3.

HDAC inhibitors with a cyclic peptide or cyclic peptide mimic capping group.
It has been hypothesized that the origin of the class I selectivity of cyclic peptide inhibitors arises from the mimicry of the peptide-derived capping group to the natural substrate associated with class I HDAC proteins.20 A comparison of the crystal structures from bacterial homologues of class I and II HDAC proteins (HDLP and FB188 HDAH, respectively) bound to HDAC inhibitors identified major differences at the entrance regions of the active sites.4,19 In particular, two loops exist in class II homologues that may prevent binding of bulky cyclic peptide capping groups.19,21 Therefore, a steric argument has been invoked to account for the observed class I specificity of cyclic peptide HDAC inhibitors.21
Consistent with the trends observed with the cyclic peptide inhibitors and the steric argument, additional class I-selective HDAC inhibitors have been created by introducing bulky hydrophobic groups at the capping group of the pan-inhibitor SAHA (Fig. 2). For example, compounds 2 and 3 contain two aromatic capping groups and display modest class I selectivity.26 Compound 2 displayed greater than 65-fold selectivity for HDAC1, 2, and 3 versus HDAC4, 5, and 7, however, it inhibited HDAC1, 2, and 3 only 10-fold greater than HDAC6 (HDAC1–3 IC50 = 0.80–4 nM). In the case of compound 3, HDAC3 and 6 (HDAC3, 6 IC50=13–14 nM) were inhibited to a greater extent than HDAC1 (4-fold), HDAC2 (12-fold), and HDAC4, 5, 7, and 8 (greater than 135-fold). The fact that compounds 2 and 3 were potent against the class II HDAC6, in addition to the class I HDAC1, 2, and 3, suggests some structural similarity among these isoforms that is not discriminated by SAHA analogues (see section I.A.2 for more discussion).
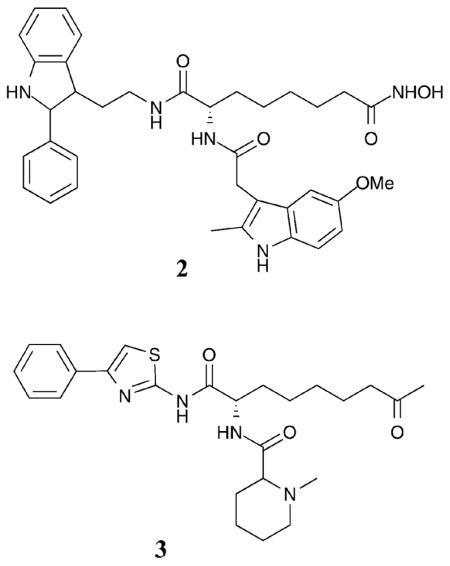
While the cyclic peptides and SAHA analogues point towards bulky capping groups to discriminate among the HDAC classes, smaller aromatic capping groups in conjunction with aromatic linker regions have also been associated with selectivity. For example, an HDACi bearing an extended aromatic capping group attached via an aromatic amide linker (compound 4) demonstrated modest selectivity for HDAC1 and HDAC4 versus HDAC6 (3-fold and 8-fold, respectively, with HDAC4 IC50 = 98 nM).27 Capping group modifications were investigated in an iterative fashion, leading to the discovery of compound 5, which displayed 10–250-fold selectivity for HDAC1, 2, and 3 compared to HDAC6 and 8 (HDAC1 IC50 = 36 nM). Interestingly, compound 5 also displayed isoform selectivity within class I, with a 10-fold and 20-fold selectivity for HDAC1 versus HDAC2 and HDAC3, respectively.28 Finally, several 6-amino nicotinamides bearing aromatic capping groups were also tested against HDAC1–3, 6, and 8, with compound 6 displaying roughly 20-fold selectivity for class I HDAC1, 2, and 3 versusHDAC6 and 8 (HDAC1 IC50=73 nM). Again, in this case, within class I, compound 6 demonstrated 4-fold and 13-fold selectivity for HDAC1 over HDAC2 and HDAC3, respectively.29 Although the capping groups were extensively investigated in the reports for compounds 4–6, they also contain aromatic linkers that may play a role in the observed selectivity (see section I.C). Furthermore, it is notable that compounds 5 and 6 contain benzamide metal-binding moieties, which may play a role in the observed selectivity (see section I.B).
In total, the trends observed with capping group derivatives suggest that compounds containing smaller capping groups have generally less class I selectivity (10–100-fold class I preference) compared to most cyclic peptide derivatives (Fig. 3) or compounds 2 and 3 (100–10 000-fold preference for class I). Therefore, the size of the capping group is likely important for governing class selectivity. In terms of isoform selectivity and discrimination between isoforms within class I, the selectivities are modest (4–20-fold preference for HDAC1 with compounds 1, 5, and 6), making trends less obvious.
I.A.2 Class II-selective HDAC inhibitors
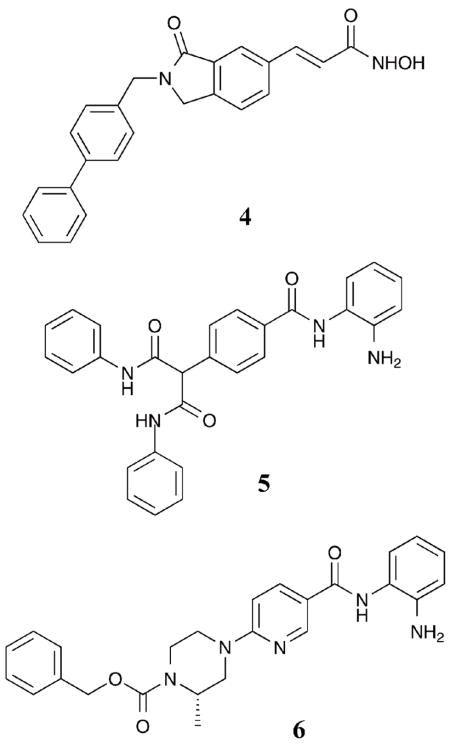
The class I-selective inhibitors described in section I.A.1 were identified via screening of acetyl-histone derived protein or peptide substrates against individual HDAC isoforms. In contrast, many class II-selective inhibitors were discovered by screening against various acetylated substrates. For example, the HDACi tubacin was originally identified in an in vivo screen monitoring the acetylation state of α-tubulin.30 Because α-tubulin is a substrate for HDAC6, tubacin is considered a selective HDAC6 inhibitor. In fact, tubacin inhibits HDAC6 by 4-fold over HDAC1,31 although inhibition of other class I HDAC proteins has not been reported.21 While HDAC6 uniquely contains two functional catalytic domains, tubacin influences the activity of only one of the active sites to inhibit deacetylation of both histone and α-tubulin substrate proteins in vitro.32 Tubacin is structurally related to SAHA but contains a very large capping group that putatively mimics the natural substrate, acetylated α-tubulin, for HDAC6. Interestingly, tubacin has structural similarities to SAHA analogs with class I selectivity (see section I.A.1), suggesting that the chemical nature of the capping group is critical for subtle discrimination between HDAC6 and the class I HDAC isoforms.
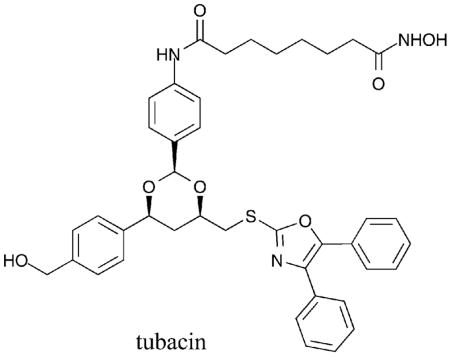
Towards development of assays for screening selective HDAC inhibitors, the deacetylation of fluorescent acetyl-lysine substrate derivatives was studied with several HDAC isoforms to identify the determinants of substrate selectivity.33 For example, substrate 7 was deacetylated preferentially by HDAC6 versus HDAC1 and HDAC3. Using the selective substrates, small molecules were screened for HDAC1 versus HDAC6 selectivity.34 Unfortunately, the isoform selectivity predicted by the substrate screening was not always validated in secondary assays with purified proteins,35 suggesting that caution should be used when screening with selective substrates. However, several inhibitors with biaryl and halogenated aryl capping groups were identified.35 Of the best hits, compound 9 demonstrated 14-fold selectivity for HDAC6 versus HDAC1 (HDAC6 IC50 = 1.69 μM). Inhibitor docking studies using homology models of HDAC1 and HDAC6 revealed favorable interactions between non-polar residues at the entrance of the HDAC6 active site and the capping group. Specifically, compound 9 is accommodated optimally by the wider entrance to the active site of HDAC6 where distinct cavities offer selective binding modes.35 With structural similarities to SAHA, as with tubacin, the observations with compound 9 are consistent with the capping group playing a role in class II, and possible HDAC6, selectivity.
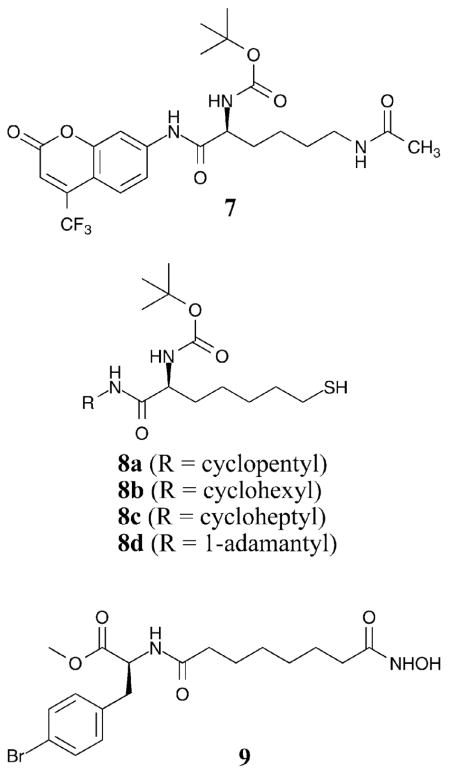
While selective substrates were used to screen for selective HDAC inhibitors, the HDAC6-selective substrate 7 was also exploited towards development of a selective inhibitor. In particular, several thiolate analogs of 7 were synthesized with a variety of different cyclic capping groups (compounds 8a–c) to create selective HDAC6 inhibitors.36 Testing with HDAC1, 4, and 6 revealed a 32–42-fold selective inhibition of HDAC6 over HDAC1 and HDAC4, respectively, with the thiolate analogs (HDAC6 IC50 = 23–29 nM). Further exploration of the bulky alkyl capping groups lead to the identification of analog 8d, which demonstrated 46–51-fold isoform selectivity of HDAC6 over HDAC1 and HDAC4, respectively (HDAC6 IC50 = 82 nM).37 These results suggest that an understanding of the substrate selectivity of various HDAC isoforms can be exploited to create selective HDAC inhibitors.
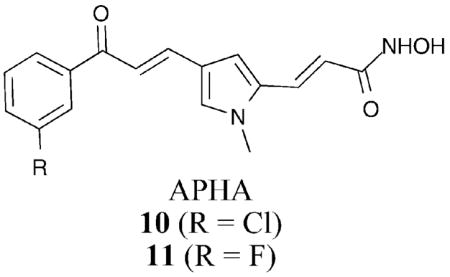
While the capping group has influence on class selectivity, the linker and capping group were simultaneously varied in a series of aroyl pyrrolyl hydroxamide (APHA) compounds. In this series, compounds were screened against maize HD1-B and HD1-A, which represent mammalian class I and class IIa homologues, respectively. The initial hit compound 10 demonstrated 78-fold preference for maize class IIa HD1-A compared to class I HD1-B.38 Further modification of the capping group region led to compound 11 which displayed a 176-fold selectivity for HD1-A over HD1-B.31 Importantly, non-halogenated or differently substituted APHA derivatives showed no HD1-B/HD1-A selectivity, underscoring the importance of the position of the halogen in the capping group. Interestingly, the APHA compounds and compound 9 contain halogenated phenyl capping groups and prefer the class II proteins, although with opposing class IIa and IIb selectivity, respectively, suggesting that halogens impart class preference. Because APHA compounds contain an aromatic pyrrole in the linker chain, the elevated class selectivity of compounds 10 and 11 relative to tubacin and compounds 8d and 9 may reflect the influence of the linker in addition to the capping group (see section I.C).
In total, the capping group has a significant impact on HDAC inhibitor selectivity for both class I and class II. Unfortunately, the specific functional groups in the capping group required for discriminating class I or class II isoforms are not clear from the available examples. It is notable, however, that compared to the most robust class I-selective inhibitors, such as trapoxin A, B, chlamydocin, and Cyl-2 (640–570 000-fold preference for HDAC1 versus HDAC6), the class II-selective HDAC inhibitors are generally less discriminating (4–176-fold preferences), possibly reflecting the fewer studies reported with class II-selective inhibitors.
I.B Metal-binding moiety modifications
Capping group modifications likely exploit structural differences at the entrance of the active site of each isoform to afford class selectivity or isoform selectivity (section I.A). In the case of modification near the metal-binding moiety, the high sequence similarity near the catalytic metal among the HDAC isoforms makes a similar approach problematic. However, selective inhibitor design has centered in some cases on the presence of distinct binding pockets adjacent to the catalytic metal.
I.B.1 Class I-selective HDAC inhibitors
Many class I-selective HDAC inhibitors contain benzamides as metal-binding groups (Fig. 4). Most notably, MS-275 displayed class I selectivity with at least 135-fold preference for HDAC1 and 3 compared to HDAC6 and 8 (HDAC1 IC50 = 180 nM).39 Interestingly, MS-275 also showed selectivity within class I, showing preference for HDAC1 compared to HDAC3 by either 4-fold or 27-fold.39,40 Similar to MS-275, CI-994 and MGCD0103 showed both class and isoform selectivity when tested against HDAC1, 3, 6, and 8. With CI-994, at least a 133- fold preference for HDAC1 and 3 compared to HDAC6 and 8 was reported, while also displaying a roughly 2-fold preference for HDAC1 versus HDAC3 (HDAC1 IC50 = 410 nM).39 In the case of MGCD0103, at least a 40-fold preference for HDAC1 and 3 compared to HDAC6 and 8 was observed, while an 8-fold selectivity for HDAC1 versus HDAC3 was documented (HDAC1 IC50 = 82 nM).39 It is noteworthy that while MS-275, CI-994, and MGCD0103 contain benzamide metal-binding groups, they also contain aromatic linkers that may contribute to selectivity (section I.C.1). However, a trend of the data suggests that the benzamide moiety contributes to class I selectivity. In fact, a direct comparison of SAHA analogs (Fig. 2) containing either a benzamide or hydroxamic acid metal binding moiety showed that the benzamide derivative discriminated against the class II HDAC6 isoform, suggesting class I selectivity.41 While the benzamide may take advantage of sequence differences in the region adjacent to the catalytic metal, some have suggested an alternative allosteric binding mode for MS-275.42
Fig. 4.

HDAC inhibitors displaying benzamide metal binding moieties.
According to crystallographic analysis of bacterial homologues of class I (HDLP) and class II (FB188 HDAH) HDAC proteins, as well as human HDAC8, a 14 Å internal cavity exists deep within the HDAC active site near the catalytic metal atom.4,17–19 The internal channel is hypothesized to function as an exit channel for release of the acetate byproduct after acetyl-lysine deacetylation.43 Critical for selective inhibitor design, sequence differences in the internal cavity exist between HDLP and FB188 HDAH, which can be exploited to create a selective HDACi.43,44 Specifically, docking of MS-275 in the active site of an HDAC1 homology model suggested that substituents could be placed on the benzamide and fit favorably into the 14 Å internal cavity.45 Based on this analysis, compound 12 was synthesized and displayed a 5-fold selectivity for HDAC1 over HDAC2 (HDAC1 IC50 = 20 nM). Because no selectivity between HDAC1 and HDAC2 was observed with MS-275,45 the presence of the thienyl substituent on the benzamide imparts selectivity. Interestingly, truncation of the capping group and linker regions of compound 12 (compounds 13–15) did not significantly influence the potency of the compounds (HDAC1 IC50 = 40–50 nM). In addition, compounds 13–15 demonstrated roughly 100-fold selectivity for HDAC1 and 2 versus HDAC3 and 8, although the preference for HDAC1 versus HDAC2 was reduced to between 1.6- and 4-fold.45
Recently, another set of HDAC inhibitors were designed to target the 14 Å internal cavity for the specific inhibition of HDAC1 and 2 versus HDAC3–8.46 Compound 12 was synthesized in this study, as well as compounds 16–19, all bearing biaryl substituents extending from the metal-binding benzamide moiety. In this study, compound 12 demonstrated greater than 145-fold selectivity for HDAC1 and 2 versus HDAC3 and 8 with greater than 260-fold selectivity versus HDAC4–7 (HDAC1 IC50 = 6 nM). Compound 12 also showed a 32-fold selectivity for HDAC1 over HDAC2, similar to the previous work.46 Compounds 16–17 demonstrated greater than 86-fold selectivity for HDAC1 and 2 versus HDAC3 and greater than 275-fold versus HDAC4–8, while showing a 7–9-fold selectivity for HDAC1 over HDAC2 (HDAC1 IC50 = 10–13 nM). In addition, compounds 18–19 demonstrated greater than 475- fold selectivity for HDAC1 and 2 versus HDAC3–8 with roughly a 10-fold selectivity for HDAC1 over HDAC2 (HDAC1 IC50 = 7–10 nM).46
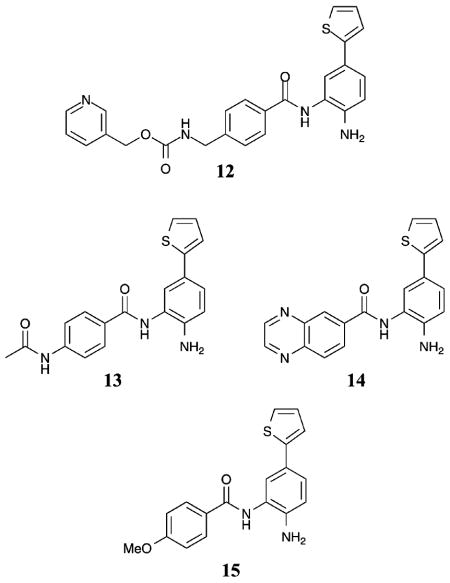
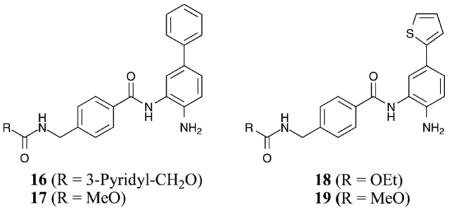
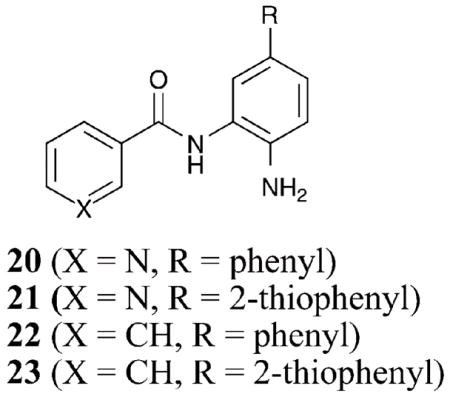
With the selective inhibition observed with compounds 12–19, a small-molecule library of amide derivatives was synthesized to probe the influence of the benzamide substituents on selectivity.47 Compounds 20–23 demonstrated 12–30- fold selectivity for HDAC1 and 2 versus HDAC3 with a 6–19- fold selectivity for HDAC1 over HDAC2 (HDAC1 IC50 = 48–65 nM). Analyses of homology models of HDAC1 and HDAC3 based on the crystal structure of an HDAC8 mutant verified that the 14 Å internal cavity is hydrophobic, suggesting that nonpolar substituents on the benzamide are preferred over polar groups.47 Significantly, the homology models indicate that a Ser113 present in the cavity of HDAC1 is replaced by Tyr96 in HDAC3, perhaps explaining the observed selectivity for HDAC1 over HDAC3 by the substituted benzamides.47 Although targeting the 14 Å internal cavity to impart inhibitor isoform selectivity is an exciting new possibility, additional experiments are needed to verify the binding mode of the substituted benzamides in the active site channel and 14 Å internal cavity. However, a significant conclusion of these studies is that the metal binding moiety can be substituted to create selective HDAC inhibitors.
I.B.2 Class II-selective inhibitors
To probe the impact of the metal binding moiety on the selectivity of the cyclic peptide HDAC inhibitors (see section I.A.1), several derivatives of chlamydocin were generated with a variety of metal-binding substituents.48 While all discriminated against HDAC6, similar to chlamydocin, several of the compounds tested showed a slight preference for class IIa HDAC4 versus class I HDAC1. For example, compound 24 demonstrated a 4-fold and 15-fold selectivity for HDAC4 over HDAC1 and HDAC6, respectively (HDAC4 IC50=60 nM).48 Although the capping group is important for class selectivity (section I.A), the data suggests that the metal-binding moiety also plays a role in selectivity within class II. Consistent with the interplay between the capping group and metal binding moiety in dictating selectivity, changing the metal-binding moiety of a cyclic peptide HDACi resulted in loss of class selectivity.20,49
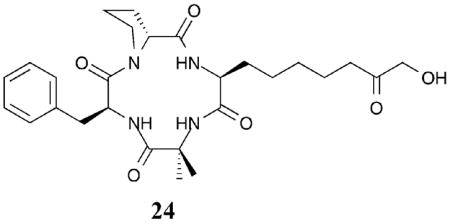
In total, the metal binding moiety is an important attribute in imparting selectivity in HDACi design. Interestingly, positioning groups to interact favorably with the 14 Å internal cavity can be exploited to create potent and selective HDAC inhibitors.
I.C Linker region modifications
The residues in the hydrophobic active site channel are highly conserved among the HDAC isoforms. Similar to modifications to the metal-binding moiety, selective HDAC inhibitors designed to exploit structural differences in the linker region have been few. As a result, only class I-selective HDAC inhibitors have been reported.
I.C.1 Class I-selective HDAC inhibitors
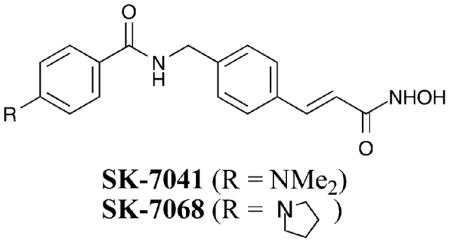
The pan-HDACi TSA (Fig. 2) was altered to create compounds SK-7041 and SK-7068. In these cases, an amide bond and phenyl ring are present in the linker, unlike in TSA.50 SK-7041 and SK-7068 showed preferential inhibition of HDAC1 and 2 over HDAC3, 4, 5, and 6, although a quantitative analysis was not performed. Because TSA is a pan-inhibitor and maintains an identical metal binding moiety and capping group compared to SK-7041, the data show that modifications in the linker region govern selectivity. Particularly, the aromatic group and/or polar amide bond are selectivity dictating elements in the linker region, perhaps by interacting differentially with residues in the hydrophobic active site channel or altering the spatial relationship between the metal binding moiety and capping group.
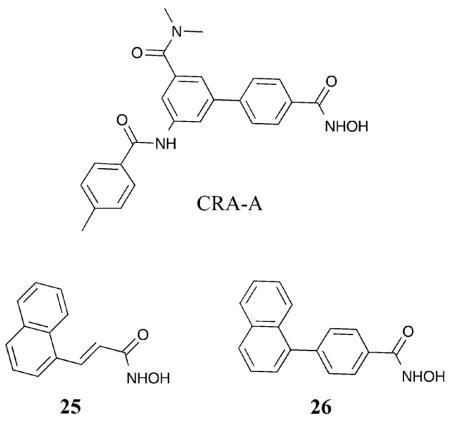
Analysis of the crystal structure of HDAC8 bound with an aryl linker containing HDACi (CRA-A) revealed a large subpocket formed in the side of the hydrophobic active site channel that was not apparent when HDAC8 was bound to the non-aryl linker containing SAHA (Fig. 2).17 Specifically, when CRA-A was bound to HDAC8, analysis showed Phe152 located in the hydrophobic active site channel shifted significantly from its normal position to create the large sub-pocket. Based on the crystal structure analysis, HDAC inhibitors were designed to specifically target this sub-pocket and create HDAC8-selective inhibitors with structural similarities to CRA-A in the aromatic linker region.10 Compound 25 displayed potent inhibition of HDAC8 with a greater than 140- and 115-fold isoform selectivity over HDAC1 and HDAC6, respectively (HDAC8 IC50 = 700 nM). Compound 26 displayed greater than 330-fold and 180-fold isoform selectivity for HDAC8 over HDAC1 and HDAC6, respectively (HDAC8 IC50 = 300 nM). Although only three HDAC isoforms were tested, the presence of an aromatic linker adjacent to the metal-binding moiety may influence HDAC8 selectivity.
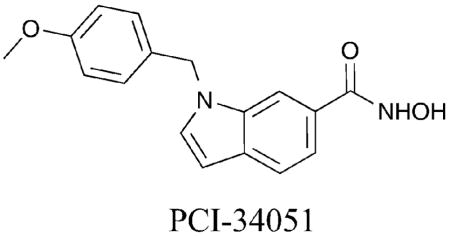
Consistent with the design strategies of compounds 25 and 26, PCI-34051 was rationally designed with an indole present in the linking domain.51 PCI-34051 demonstrated greater than 290-fold selectivity for HDAC8 over HDAC1–3, 6, and 10 (HDAC8 IC50 = 10 nM). Compounds 25, 26, and PCI-34051 all contain aromatic linkers with extended bulky aromatic groups. Although consistent with the possibility of targeting the unique sub-pocket formed in HDAC8, further crystallographic analysis is needed to confirm the mode of binding.
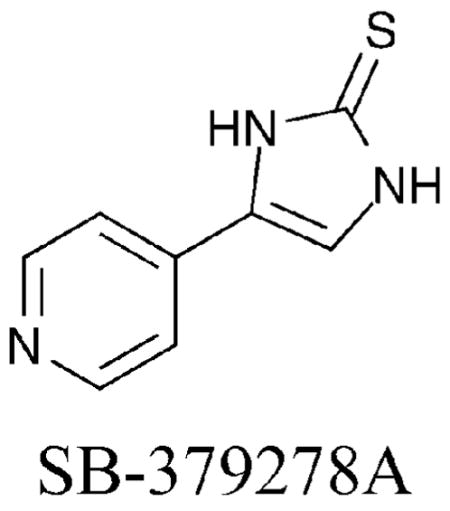
Taking advantage of the fact that HDAC8 is the only isoform expressed in active form in bacteria, an HDAC8 enzyme-based high-throughput screen was performed.40 SB-379278A was identified and has a related structure to compounds 25, 26 and PCI-34051 by containing aromatic functionality within the linker region, although a different metal-binding moiety is present. Similar to these HDAC8-selective compounds, SB-379278A displayed roughly a 60-fold preference for HDAC8 inhibition compared to HDAC1 and 3, suggesting HDAC8 isoform selectivity (HDAC8 IC50 = 500 nM). The results with 25, 26, PCI-34051, and SB-379278A taken together evidence the role of an aromatic linker in dictating HDAC8 selectivity.
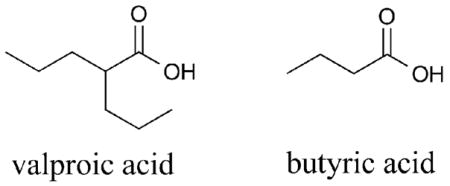
Valproic acid (VPA) is a moderately potent μM HDACi that contains an unusual branched aliphatic linker region. Although VPA has been suggested to be a class I-selective inhibitor,52 more recent data has shown VPA to discriminate only against the class IIb HDAC6 and HDAC10 isoforms.53 Similarly, butyrate also inhibits the activity of HDAC1, but not HDAC6 or HDAC10, in this case with μM to mM potency.54 The data suggest that compounds containing an aliphatic linker, but lacking a distinct capping group, are not tolerated in the hydrophobic active site region of the class IIb HDAC isoforms. It is also notable that VPA and butyrate display carboxylic acid metal binding moieties, which may have an influence on selectivity.
The linker region is a relatively unexplored area of HDACi design. However, several compounds bearing aromatic linker regions display selective HDAC inhibition. While SB-379278A, PCI-34051 and compounds 25 and 26 show a preference for HDAC8, the benzamide-containing HDAC inhibitors (Fig. 4) also contain aromatic groups within the linker region and discriminate against HDAC8 (section I.B.1). The combined data suggest that the metal-binding moiety and aromatic linker together contribute to selectivity among the class I HDAC isoforms. In addition, APHA derivatives contain an aromatic linker region and a halogenated capping group and display class II selectivity (section I.A.2), while VPA and butyrate contain an aliphatic linker but lack a capping group and discriminate against the class IIb isoforms, HDAC6 and HDAC10. In these cases, the combination of capping group and aromatic linker may contribute to class selectivity. With the multiple modifications and the variable selectivities observed in the tested compounds, the role of the aromatic linker alone in the observed class selectivity or isoform selectivity remains unclear.
Analyses of the crystal structures of the bacterial class I and II homologues (HDLP and FB188 HDAH, respectively) and human HDAC8 bound to an HDACi reveal a residue orientation in the hydrophobic active site channel that may confer selectivity.4,17–19 Specifically, two Phe residues are in parallel alignment and could allow for favorable π–π-interactions with an appropriately placed aromatic substituent in the linker region of an HDACi. Therefore, aromatic groups in the linker region may make favorable or unfavorable interactions with residues in the hydrophobic channel, possibly resulting in discrimination among the isoforms. With the linker region likely playing a role in selectivity, further analysis is warranted to systematically study the influence of bulky aromatic or aliphatic linkers on selectivity.
I.D Miscellaneous HDAC inhibitors
Some HDAC inhibitors do not fit the standard modular structural mode described in Fig. 2. The selective HDAC inhibitors that are of this “miscellaneous” class and are not easily categorized in the fashion outlined in this review are discussed here.
I.D.1 Class I-selective HDAC inhibitors
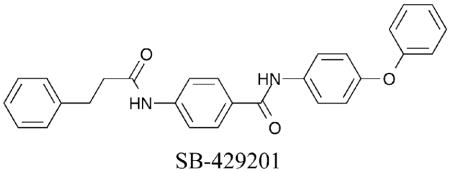
The high-throughput screen that identified SB-379278A (section I.C.1) also yielded a potent HDAC1-selective compound, SB-429201.40 SB-429201 displayed at least a 20-fold preference for HDAC1 inhibition over HDAC3 and HDAC8, suggesting selectivity among class I isoforms (HDAC1 IC50 = 1.5 μM). The authors make no comment on the possible mode of binding.
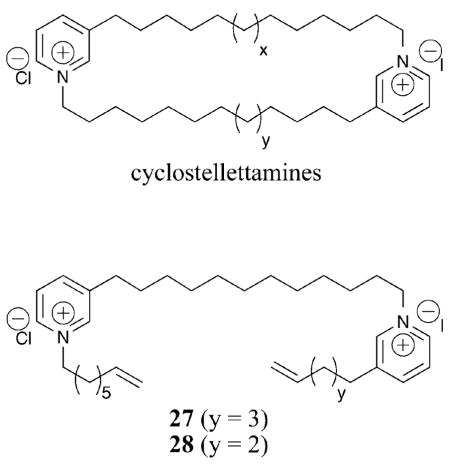
The cyclostellettamines are natural products that inhibit HDAC proteins at μM concentrations, although they do not maintain the modular structure of most HDAC inhibitors. Based on the cyclostellettamines, acyclic derivatives, represented by compounds 27 and 28, were synthesized and tested against HDAC1, 2, 3, and 4.55 In the case of compound 27, at least a 370-fold preference for HDAC1 compared to HDAC4 was observed and at least a 10-fold selectivity for HDAC1 versus HDAC2 and 3 was shown, suggesting it is a class I-selective and possibly HDAC1-selective inhibitor (HDAC1 IC50 = 530 nM). Due to the dissimilar structure of compound 27 compared to known HDACi, it is difficult to ascertain the origin of the observed isoform selectivity. The authors propose binding to the entrance of the acetate exit cavity or active site channel, allosteric binding to distal regions that influence catalysis, or disruption of HDAC–histone interactions.55 Similar to compounds 12, 16–19 (section I.B.1), we propose an alternative binding mode in which one pyridinium moiety sits near the catalytic metal with its olefin group situated in the internal cavity and the second pyridinium representing the capping group. Based on the isoform selectivity observed with these atypical HDAC inhibitors, further analysis of the mode of inhibition is warranted.
I.D.2 Class II-selective HDAC inhibitors
Among the acyclic cyclostellettamine derivatives, while compound 27 displayed class I selectivity, compound 28 displayed modest class II selectivity. In the case of compound 28, at least a 3-fold preference for HDAC4 compared to HDAC1, 2, and 3 was observed (HDAC4 IC50 = 540 nM).55 Furthermore, compound 28 was unable to alter α-tubulin acetylation levels, indicating possible discrimination against HDAC6. It is notable that compounds 27 and 28 have opposing selectivity profiles, yet differ by only a single methylene in their structures. The comparison of compounds 27 and 28 suggest that subtle differences in inhibitor structure can significantly influence selectivity.
II. Conclusions
Efforts to create selective HDACi have seen increased attention over the past several years, leading to several class-selective and some isoform-selective inhibitors. To summarize the compounds discussed in this review, Table 1 lists each compound with the observed selectivity. Of the class I-selective inhibitors, HDAC1, 2, and 3 are targeted by multiple inhibitors, with discrimination against HDAC8. In the cases where isoform selectivity is observed, HDAC1 and HDAC8 are targeted exclusively. Fewer class II-selective HDAC inhibitors have been identified, with HDAC4 and HDAC6 targeted exclusively. Therefore, while progress has been made to create selective inhibitors, only a few of the isoforms are targeted in the available compounds, suggesting that additional attention is needed to direct inhibitors to all isoforms.
Table 1.
Selective HDAC inhibitors, subdivided by class and isoform selectivity
| Inhibitor | HDAC Isoform | Selectivity |
|---|---|---|
| Class I-selective inhibitors | ||
| Trapoxin A and derivatives | HDAC1 | 640–570 000-fold vs. HDAC6 |
| Apicidin | HDAC2–3 | 5–83-fold vs. HDAC1, 4–9 |
| Romidepsin | HDAC1–2 | 10-fold vs. HDAC4 |
| Azumamide E | HDAC1–3 | 37–500-fold vs. HDAC4–9 |
| 2 | HDAC1–3 | 10-fold vs. HDAC6 |
| 65-fold vs. HDAC4,5,7 | ||
| 3 | HDAC3,6 | 4–135-fold vs. |
| HDAC1,2,4,5,7,8 | ||
| 4 | HDAC1,4 | 3–8-fold vs. HDAC6 |
| MS-275 | HDAC1,3 | 135-fold vs. HDAC6,8 |
| CI-994 | HDAC1,3 | 133-fold vs. HDAC6,8 |
| MGCD0103 | HDAC1,3 | 40-fold vs. HDAC6,8 |
| 13–15 | HDAC1,2 | 100-fold vs. HDAC3,8 |
| SK-7041 | HDAC1,2 | HDAC3–6 |
| SK-7068 | HDAC1,2 | HDAC3–6 |
| VPA | HDAC1–5,7 | >20-fold vs. HDAC6, 10 |
| Isoform-selective inhibitors within class I | ||
| 1 | HDAC1 | 4-fold vs. HDAC8 |
| 5 | HDAC1 | 10-fold vs. HDAC2 |
| 20-fold vs. HDAC3 | ||
| 6 | HDAC1 | 4-fold vs. HDAC2 |
| 13-fold vs. HDAC3 | ||
| 12 | HDAC1 | 5–32-fold vs. HDAC2 |
| >145-fold vs. HDAC3,8 | ||
| 16–23 | HDAC1 | 6–19-fold vs. HDAC2 |
| SB-429201 | HDAC1 | 20-fold vs. HDAC3,8 |
| 27 | HDAC1 | 370-fold vs. HDAC4 |
| 10-fold vs. HDAC2,3 | ||
| 25 | HDAC8 | 140-fold vs. HDAC1 |
| 115-fold vs. HDAC6 | ||
| 26 | HDAC8 | 330-fold vs. HDAC1 |
| 180-fold vs. HDAC6 | ||
| PCI-34051 | HDAC8 | >290-fold vs. |
| HDAC1–3,6,10 | ||
| SB-379278A | HDAC8 | 60-fold vs. HDAC1,3 |
| Class II-selective inhibitors | ||
| 10 | HD1-A | 78-fold vs. HD1-B |
| 11 | HD1-A | 176-fold vs. HD1-B |
| Tubacin | HDAC6 | 4-fold vs. HDAC1 |
| 9 | HDAC6 | 14-fold vs. HDAC1 |
| 28 | HDAC4 | 3-fold vs. HDAC1,2,3 |
| Isoform-selective inhibitors within class II | ||
| 8d | HDAC6 | 46-fold vs. HDAC1 |
| 51-fold vs. HDAC4 | ||
| 24 | HDAC4 | 4-fold vs. HDAC1 |
| 15-fold vs. HDAC6 | ||
To guide future design efforts, several structural trends were observed in the known inhibitors. In particular, the sequence differences in the active site entrance (capping group modifications), residues adjacent to the catalytic metal (metal binding moiety modification), or hydrophobic active site channel (linker modifications) were cited to explain some observed selectivity. Certainly, the capping group has received considerable attention and has been modified successfully for the design of several class-selective inhibitors (section I.A). Bulky cyclic peptide capping groups demonstrate class I selectivity, possibly due to unfavorable interactions between the cyclic peptides and residues around the active site rim of class II HDAC proteins. However, large hydrophobic capping groups have also resulted in class II-specific HDACi. At this point, it is difficult to firmly predict the specific chemical features of the capping group required for class I or II selectivity, making systematic studies of the capping group structure a fruitful area of future study. Due to the high degree of conservation of the catalytic residues near the metal, few groups have targeted the metal binding moiety to identify selective HDAC inhibitors. However, the limited reports suggest that the metal-binding moiety can be modified to elicit selectivity (section I.B). Benzamides, for example, are exclusively class I selective. In recent efforts, HDAC inhibitors were targeted to the 14 Å internal cavity to impart selectivity, which represents an exciting avenue for future design. Finally, subtle structural differences in the hydrophobic active site channel have been exploited toward selective inhibitor design. Specifically, favorable interactions with a unique sub-pocket in the hydrophobic active site channel led to the creation of HDAC8-selective inhibitors (section I.C.1), although a similar strategy with additional isoforms has yet to be validated.
While modifying the capping group, linker, and metal binding moiety individually contributes to selectivity, the combination of modification at several of these regions is likely important for many of the selective inhibitors. For example, compounds exemplified by MS-275 display a benzamide metal binding moiety and an aromatic linker, which both likely contribute to the class I selectivity (sections I.B.1 and I.C.1). In addition, hydroxamic acids APHA (compounds 9 and 10) and SK-7041 maintain similar structures with subtle differences in their aromatic linkers and substituents on a phenyl capping group, yet display opposing class selectivities (APHA is class II selective while SK-7041 is class I selective, sections I.A.2 and I.C.1), suggesting that small structural differences in the capping group and aromatic linker can profoundly alter class selectivity. Towards creating robust isoform-selective inhibitors, understanding the interplay between each region of the inhibitor in dictating selectivity will be an important area of future work.
Future selective compounds should take advantage of the rational design based on analysis of sequence differences among the isoforms, as has been done previously. However, incomplete structural information for the isoforms makes rational design based on structure challenging. Clearly, an important area of future work will be the analysis of structural differences among the human HDAC isoforms. In the absence of additional structural information, however, creation of more selective inhibitors targeting all isoforms will likely require additional design approaches and methods. Possible design alternatives are exemplified by the compounds that contain atypical HDACi structures. Specifically, compounds 27, 28, and SB-429201 (section I.D) possess a high degree of selectivity, yet have unknown binding modes due to the lack of modular structure or crystallographic analysis. Future efforts to understand the binding mode and mechanism of inhibition of these atypical compounds may lead to novel approaches to selective inhibitor design. Alternatively, identification of substrate selectivities among the isoforms could also be exploited towards design of selective HDAC inhibitors, as was observed in the creation of compound 8d based on the HDAC6-selective substrate peptide 7 (section I.A.2). Finally, unlike the HDAC inhibitors discussed in this review, the small molecule parthenolide depletes the HDAC1 isoform in vivo through proteosomal degradation.56 The possibility of small molecule-mediated HDAC isoform depletion represents a novel method for modulating the activity of a single HDAC isoform.
One notable observation from the data listed in Table 1 is the limited number of HDAC isoforms that have been tested with the reported inhibitors. In some cases, only two of the eleven HDAC isoforms have been tested (see trapoxin and tubacin for examples). Even the most completely characterized compounds only test HDAC1–9, without analysis of HDAC10 and 11 (see apicidin and azumamide E for examples). Therefore, it is clear from available data that a significant hurdle toward development of selective HDAC inhibitors is the challenge of testing all HDAC isoforms simultaneously. Because of the likely influence on past and future screening of isoform-selective inhibitors, it is worth discussing the limitations of available HDAC activity assays here. The most widely used screen monitors deacetylase activity in vitro with colorimetric or fluorimetric substrates (Biomol or Calbiochem) and is easy to use, reproducible, and high throughput. Unfortunately, screening for isoform-selective HDAC inhibitors using the in vitro assay requires expressing and purifying the individual HDAC proteins. With the exception of HDAC8, all human HDAC proteins are catalytically inactive when expressed in bacteria and require human-derived or insectderived cell culture for expression.57 As a result, the expression of each human HDAC isoform is low yielding, costly, and consequently low throughput. Another strategy involves expressing each isoform in mammalian cells with subsequent immunoprecipitation.20,23,27 In addition to being laborious, low throughput, and costly, the results from immunoprecipitated proteins are unreliable, with error bars as large as 76%.20 With the significant expression and purification costs, the selectivity of known inhibitors is typically assessed with two to five purified HDAC proteins that often only provide representatives of each HDAC class (see Table 1).23,40 Therefore, it is arguable that a significant obstacle to creation of isoform-selective HDAC inhibitors is the limitations of current screening technologies. As a result, additional HDAC assays are needed to allow for the simultaneous screening of inhibitors with all eleven HDAC isoforms to establish the selectivity of known and future inhibitors.
While many challenges exist towards selective HDACi design, the inhibitors discussed in this review indicate that creation of both class-selective and isoform-selective HDAC inhibitors are possible. The medicinal chemistry problem encountered with HDAC proteins is analogous to that of other protein systems, such as the kinase enzyme family (see ref. 58 for a review of kinase structure and activity). In the case of kinases, roughly 510 family members exist with many crystal structures solved to aid in the drug design effort. As a result, many kinase inhibitors are in clinical use and development, 59 although selectivity remains a critical challenge. Compared to the kinase family, designing HDAC inhibitors is arguably manageable due to the fewer number of family members. However, the lack of structural data for the mammalian isoforms and the limitations of HDAC inhibitor screening tools constitute major challenges.
HDAC proteins have been linked to cancer and several HDAC inhibitors are in clinical trials as anti-cancer drugs.12,13 While it remains unclear whether selective inhibitors would provide advantages compared to the pan-inhibitor SAHA currently in clinical use, clinical trials of class-selective inhibitors, such as MS-275 and romidepsin, will test the relevance of selectivity in drug therapeutics. However, strictly isoform-selective inhibitors are needed to thoroughly test the efficacy and side effects observed with selective inhibitors. In addition to the clinical interest, HDACi drugs have been widely used to characterize HDAC proteins from cell culture to animal studies.60 Unfortunately, the pan-selectivity of most inhibitors limits their use in exploring the role of individual HDAC isoforms in basic biological processes and disease formation, as with cancer. With the temporal control and ease of use of small molecule reagents, isoform-selective HDAC inhibitors have the potential to quickly dissect the individual contributions of each HDAC isoform in carcinogenesis. Therefore, progress in cancer research related to HDAC proteins would be profoundly accelerated with the availability of selective inhibitors.
Acknowledgments
We apologize to the investigators whose important HDACi studies were not cited in this review due to space limitations and emphasis on selectivity. The work in our laboratory on HDAC proteins is supported by the National Institutes of Health (GM067657) and Wayne State University.
Biographies
 Anton V. Bieliauskas, Anton V. Bieliauskas began his research career with his undergraduate studies at Albion College in Albion, Michigan under Professor Clifford E. Harris. He currently is finishing his PhD research studies at Wayne State University in Detroit, Michigan under Professor Mary Kay H. Pflum. His graduate research interests include the design and synthesis of small molecule libraries of HDAC inhibitors.
Anton V. Bieliauskas, Anton V. Bieliauskas began his research career with his undergraduate studies at Albion College in Albion, Michigan under Professor Clifford E. Harris. He currently is finishing his PhD research studies at Wayne State University in Detroit, Michigan under Professor Mary Kay H. Pflum. His graduate research interests include the design and synthesis of small molecule libraries of HDAC inhibitors.
 Mary Kay H. Pflum, Prof. Mary Kay H. Pflum received a BA in chemistry from Carleton College in 1992 and a PhD in chemistry from Yale University in 1999 under the guidance of Prof. Alanna Schepartz. Her National Institutes of Health postdoctoral fellowship centered on histone deacetylase proteins in the laboratory of Prof. Stuart L. Schreiber. Since her appointment as an assistant professor at Wayne State University in 2001, her research interests have focused on using small molecule reagents to study histone deacetylase and kinase enzymes.
Mary Kay H. Pflum, Prof. Mary Kay H. Pflum received a BA in chemistry from Carleton College in 1992 and a PhD in chemistry from Yale University in 1999 under the guidance of Prof. Alanna Schepartz. Her National Institutes of Health postdoctoral fellowship centered on histone deacetylase proteins in the laboratory of Prof. Stuart L. Schreiber. Since her appointment as an assistant professor at Wayne State University in 2001, her research interests have focused on using small molecule reagents to study histone deacetylase and kinase enzymes.
Footnotes
Part of a thematic issue examining the interface of chemistry with biology.
References
- 1.Kornberg RD, Klug A. Sci Am. 1981;244:52. doi: 10.1038/scientificamerican0281-52. [DOI] [PubMed] [Google Scholar]
- 2.Wu J, Grunstein M. Trends Biochem Sci. 2000;25:619. doi: 10.1016/s0968-0004(00)01718-7. [DOI] [PubMed] [Google Scholar]
- 3.Gregoretti I, Lee YM, Goodson HV. J Mol Biol. 2004;338:17. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Pavletich NP. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 5.Grozinger CM, Schreiber SL. Chem Biol. 2002;9:3. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 6.Minucci S, Pelicci PG. Nat Rev Cancer. 2006;6:38. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 7.Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, Rice VM. Cancer Biol Ther. 2007;6:795. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
- 8.Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, Lee SH, Park WS, Yoo NJ, Lee JY, Nam SW. APMIS. 2005;113:264. doi: 10.1111/j.1600-0463.2005.apm_04.x. [DOI] [PubMed] [Google Scholar]
- 9.Bartling B, Hofmann HS, Boettger T, Hansen G, Burdach S, Silber RE, Simm A. Lung Cancer. 2005;49:145. doi: 10.1016/j.lungcan.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 10.KrennHrubec K, Marshall BL, Hedglin M, Verdin E, Ulrich SM. Bioorg Med Chem Lett. 2007;17:2874. doi: 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 11.Saji S, Kawakami M, Hayashi S, Yoshida N, Hirose M, Horiguchi SI, Itoh A, Funata N, Schreiber SL, Yoshida M, Toi M. Oncogene. 2005;24:4531. doi: 10.1038/sj.onc.1208646. [DOI] [PubMed] [Google Scholar]
- 12.Garber K. Nat Biotechnol. 2007;25:17. doi: 10.1038/nbt0107-17. [DOI] [PubMed] [Google Scholar]
- 13.Grant S, Easley C, Kirkpatrick P. Nat Rev Drug Discovery. 2007;6:21. doi: 10.1038/nrd2227. [DOI] [PubMed] [Google Scholar]
- 14.Mai A, Masse S, Rotili D, Cerbara I, Valente S, Pezzi R, Simeoni S, Ragno R. Med Res Rev. 2005;25:261. doi: 10.1002/med.20024. [DOI] [PubMed] [Google Scholar]
- 15.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Biochem J. 2008;409:581. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 16.Min J, Schuetz A, Loppnau P, Weigelt J, Sundstrom M, Arrowsmith CH, Edwards AM, Bochkarev A, Plotnikov AN Structural Genomics Consortium. 2008 pdb: 3C0Y, 3C0Z, 3C10. [Google Scholar]
- 17.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structure. 2004;12:1324. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, Francesco RD, Gallinari P, Steinkuhler C, Marco SD. Proc Natl Acad Sci U S A. 2004;101:15064. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen TK, Hildmann C, Dickmanns A, Schwienhorst A, Ficner R. J Mol Biol. 2005;354:107. doi: 10.1016/j.jmb.2005.09.065. [DOI] [PubMed] [Google Scholar]
- 20.Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Proc Natl Acad Sci U S A. 2001;98:87. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hildmann C, Wegener D, Riester D, Hempel R, Schober A, Merana J, Giurato L, Guccione S, Nielsen TK, Ficner R, Schwienhorst A. J Biotechnol. 2006;124:258. doi: 10.1016/j.jbiotec.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 22.Maulucci N, Chini MG, DiMicco S, Izzo I, Cafaro E, Russo A, Gallinari P, Paolini C, Nardi MC, Casapullo A, Riccio R, Bifulco G, DeRiccardis F. J Am Chem Soc. 2007;129:3007. doi: 10.1021/ja0686256. [DOI] [PubMed] [Google Scholar]
- 23.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. Cancer Res. 2002;62:4916. [PubMed] [Google Scholar]
- 24.Yurek-George A, Cecil ARL, Mo AHK, Wen S, Rogers H, Habens F, Maeda S, Yoshida M, Packham G, Ganesan A. J Med Chem. 2007;50:5720. doi: 10.1021/jm0703800. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, Kapustin G, Etzkorn FA. J Med Chem. 2007;50:2003. doi: 10.1021/jm061082q. [DOI] [PubMed] [Google Scholar]
- 26.Jones P, Altamura S, Chakravarty PK, Cecchetti O, Francesco RD, Gallinari P, Ingenito R, Meinke PT, Petrocchi A, Rowley M, Scarpelli R, Serafini S, Steinkuhler C. Bioorg Med Chem Lett. 2006;16:5948. doi: 10.1016/j.bmcl.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Shinji C, Maeda S, Imai K, Yoshida M, Hashimoto Y, Miyachi H. Bioorg Med Chem. 2006;14:7625. doi: 10.1016/j.bmc.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Siliphaivanh P, Harrington P, Wittera DJ, Ottea K, Tempest P, Kattar S, Kral AM, Fleming JC, Deshmukh SV, Harsch A, Secrist PJ, Miller TA. Bioorg Med Chem Lett. 2007;17:4619. doi: 10.1016/j.bmcl.2007.05.080. [DOI] [PubMed] [Google Scholar]
- 29.Hamblett CL, Methot JL, Mampreian DM, Sloman DL, Stanton MG, Kral AM, Fleming JC, Cruz JC, Chenard M, Ozerova N, Hitz AM, Wang HM, Deshmukh SV, Nazef N, Harsch A, Hughes B, Dahlberg WK, Szewczak AA, Middleton RE, Mosley RT, Secrist JP, Miller TA. Bioorg Med Chem Lett. 2007;17:5300. doi: 10.1016/j.bmcl.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 30.Haggarty SJ, Koeller KM, Wong JC, Butcher RA, Schreiber SL. Chem Biol. 2003;10:383. doi: 10.1016/s1074-5521(03)00095-4. [DOI] [PubMed] [Google Scholar]
- 31.Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, Scognamiglio A, Altucci L, Loidl P, Brosch G. J Med Chem. 2005;48:3344. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]
- 32.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Proc Natl Acad Sci U S A. 2003;100:4389. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heltweg B, Dequiedt F, Marshall BL, Branch C, Yoshida M, Nishino N, Verdin E, Jung M. J Med Chem. 2004;47:5235. doi: 10.1021/jm0497592. [DOI] [PubMed] [Google Scholar]
- 34.Arts J, Angibaud P, Marien A, Floren W, Janssens B, King P, van Dun J, Janssen L, Geerts T, Tuman RW, Johnson DL, Andries L, Jung M, Janicot M, van Emelen K. Br J Cancer. 2007;97:1344. doi: 10.1038/sj.bjc.6604025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schafer S, Saunders L, Eliseeva E, Velena A, Jung M, Schwienhorst A, Strasser A, Dickmanns A, Ficner R, Schlimme S, Sippl W, Verdin E, Jung M. Bioorg Med Chem. 2008;16:2011. doi: 10.1016/j.bmc.2007.10.092. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki T, Kouketsu A, Itoh Y, Hisakawa S, Maeda S, Yoshida M, Nakagawa H, Miyata N. J Med Chem. 2006;49:4809. doi: 10.1021/jm060554y. [DOI] [PubMed] [Google Scholar]
- 37.Itoh Y, Suzuki T, Kouketsu A, Suzuki N, Maeda S, Yoshida M, Nakagawa H, Miyata N. J Med Chem. 2007;50:5425. doi: 10.1021/jm7009217. [DOI] [PubMed] [Google Scholar]
- 38.Mai A, Massa S, Pezzi R, Rotili D, Loidl P, Brosch G. J Med Chem. 2003;46:4826. doi: 10.1021/jm034167p. [DOI] [PubMed] [Google Scholar]
- 39.Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, Maier T, Sanders K. Int J Cancer. 2007;121:1138. doi: 10.1002/ijc.22751. [DOI] [PubMed] [Google Scholar]
- 40.Hu E, Dul E, Sung C-M, Chen Z, Kirkpatrick R, Zhang G-F, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, De Los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. J Pharmacol Exp Ther. 2003;307:720. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- 41.Wong JC, Hong R, Schreiber SL. J Am Chem Soc. 2003;125:5586. doi: 10.1021/ja0341440. [DOI] [PubMed] [Google Scholar]
- 42.Bouchain G, Delorme D. Curr Med Chem. 2003;10:2359. doi: 10.2174/0929867033456585. [DOI] [PubMed] [Google Scholar]
- 43.Wang DF, Wiest O, Helquist P, Lan-Hargest HY, Wiech NL. J Med Chem. 2004;47:3409. doi: 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- 44.Wang DF, Helquist P, Wiech NL, Wiest O. J Med Chem. 2005;48:6936. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- 45.Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, Fournel M, Bonfils C, Trachy-Bourget MC, Liu JH, Yan TP, Lu AH, Rahil J, Wang J, Lefebvre S, Li ZM, Vaisburg AF, Besterinan JM. J Med Chem. 2007;50:5543. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 46.Witter DJ, Harrington P, Wilson KJ, Chenard M, Fleming JC, Haines B, Kral AM, Secrist JP, Miller TA. Bioorg Med Chem Lett. 2008;18:726. doi: 10.1016/j.bmcl.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 47.Methot JL, Chakravarty PK, Chenard M, Close J, Cruz JC, Dahlberg WK, Fleming J, Hamblett CL, Hamill JE, Harrington P, Harsch A, Heidebrecht R, Hughes B, Jung J, Kenific CM, Kral AM, Meinke PT, Middleton RE, Ozerova N, Sloman DL, Stanton MG, Szewczak AA, Tyagarajan S, Witter DJ, Paul Secrist J, Miller TA. Bioorg Med Chem Lett. 2008;18:973. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 48.Bhuiyan MPI, Kato T, Okauchi T, Nishino N, Maeda S, Nishino TG, Yoshida M. Bioorg Med Chem. 2006;14:3438. doi: 10.1016/j.bmc.2005.12.063. [DOI] [PubMed] [Google Scholar]
- 49.Nishino N, Jose B, Okamura S, Ebisusaki S, Kato T, Sumida Y, Yoshida M. Org Lett. 2003;5:5079. doi: 10.1021/ol036098e. [DOI] [PubMed] [Google Scholar]
- 50.Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, Kim DK, Lee JS, Kim NK, Kim TY, Bang YJ. Clin Cancer Res. 2004;10:5271. doi: 10.1158/1078-0432.CCR-03-0709. [DOI] [PubMed] [Google Scholar]
- 51.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. Leukemia. 2008 doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 52.Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimp A, Giavara S, Sleeman JP, Coco FL, Nervi C, Pelicci PG, Heinzel T. EMBO J. 2001;20:6969. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Cancer Res. 2004;64:1079. doi: 10.1158/0008-5472.can-03-0799. [DOI] [PubMed] [Google Scholar]
- 54.Guardiola AR, Yao TP. J Biol Chem. 2002;277:3350. doi: 10.1074/jbc.M109861200. [DOI] [PubMed] [Google Scholar]
- 55.Perez-Balado C, Nebbioso A, Rodriguez-Grana P, Minichiello A, Miceli M, Altucci L, de Lera AR. J Med Chem. 2007;50:2497. doi: 10.1021/jm070028m. [DOI] [PubMed] [Google Scholar]
- 56.Gopal YNV, Arora TS, Van Dyke MW. Chem Biol. 2007;14:813. doi: 10.1016/j.chembiol.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Staver MJ, Curtin ML, Holms JH, Frey RR, Edalji R, Smith R, Michaelides MR, Davidsen SK, Glaser KB. Life Sci. 2004;74:2693. doi: 10.1016/j.lfs.2003.09.070. [DOI] [PubMed] [Google Scholar]
- 58.Adams JA. Chem Rev. 2001;101:2271. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- 59.Cohen P. Nat Rev Drug Discovery. 2002;1:309. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 60.Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Nat Neurosci. 2004;7:847. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]