

Abstract
Extracellular matrix remodeling has been proposed as one mechanism by which proximal pulmonary arteries stiffen during pulmonary arterial hypertension (PAH). Although some attention has been paid to the role of collagen and metallomatrix proteins in affecting vascular stiffness, much less work has been performed on changes in elastin structure-function relationships in PAH. Such work is warranted, given the importance of elastin as the structural protein primarily responsible for the passive elastic behavior of these conduit arteries. Here, we study structure-function relationships of fresh arterial tissue and purified arterial elastin from the main, left, and right pulmonary artery branches of normotensive and hypoxia-induced pulmonary hypertensive neonatal calves. PAH resulted in an average 81 and 72% increase in stiffness of fresh and digested tissue, respectively. Increase in stiffness appears most attributable to elevated elastic modulus, which increased 46 and 65%, respectively, for fresh and digested tissue. Comparison between fresh and digested tissues shows that, at 35% strain, a minimum of 48% of the arterial load is carried by elastin, and a minimum of 43% of the change in stiffness of arterial tissue is due to the change in elastin stiffness. Analysis of the stress-strain behavior revealed that PAH causes an increase in the strains associated with the physiological pressure range but had no effect on the strain of transition from elastin-dominant to collagen-dominant behavior. These results indicate that mechanobiological adaptations of the continuum and geometric properties of elastin, in response to PAH, significantly elevate the circumferential stiffness of proximal pulmonary arterial tissue.
Keywords: elastic modulus, stiffness, vascular remodeling
pulmonary arterial hypertension (PAH) in neonates, infants, and children is a progressive condition marked by substantial vascular remodeling of the distal pulmonary vasculature, which increases pulmonary vascular resistance (PVR) and pulmonary arterial (PA) pressure. PVR measurement has been the standard diagnostic for evaluating both the significance of PAH and the extent of vascular response to therapies. However, PVR is an inherently limited diagnostic in that it ignores pulmonary vascular stiffness (PVS), an especially important omission, given the inherently pulsatile nature of cardiac function and the importance of robust ventriculo-vascular coupling in maintaining hemodynamic efficiency through the pulmonary vasculature. In fact, many studies of vascular function in systemic hypertension are documenting the substantial role played by the elastic proximal arteries in maintaining systemic vascular hemodynamic efficiency and reducing cardiac workload. Several investigators have shown the significant mechanical advantages conveyed by the elasticity of systemic conduit arteries in reducing overall hydraulic impedance and cardiac workload (29, 32, 37, 38). Other authors have correlated proximal artery stiffness and cardiovascular mortality for patients with systemic hypertension (5, 13, 28). Although less work has been done regarding PA elasticity in PAH, several recent animal studies are beginning to address this topic. In particular, recent work by Kobs and Chesler (26) used fresh artery material testing to correlate increased collagen and elastin content with elevated elastic moduli for the collagen- and elastin-dominant regions of the stress-strain curve, respectively.
In addition, several clinical studies have shown that increased PVS is correlated with patient mortality (12, 18, 31, 43) and can account for as much as 30–40% of the increased load on the heart (40) due to elevated vascular hydraulic impedance and right ventricular afterload. Remodeling of the extracellular matrix (ECM), in particular, appears to be an important aspect of PVS, and a number of studies have examined various biological mechanisms, including inflammatory factors, upregulation of proliferative catalysts, and phenotypic changes in resident cells (2–4, 6, 17, 20–24, 40, 41, 44). Other studies have shown increased collagen content and vascular tone in stiffer arteries (7, 11, 14, 19, 21, 27, 34, 35). In elastin-specific studies, elastolytic activity has been shown to increase during PAH (33), and inhibition of this pathway by the serine elastase inhibitor elafin mitigates hypoxia-induced PAH (46). In recent work from our group coupling a novel orthotropic, hyperelastic, microstructural model of the PA wall to biomechanical studies of PAs from normotensive and hypertensive rats, we suggested that increased ECM structural protein cross-linking may be one mechanism by which these vessels stiffen (47, 48). The next step in addressing this hypothesis is to evaluate experimentally whether the increased stiffness seen in hypoxia-induced PAH can be attributable to increased stiffness of the structural protein matrix. Given that elastin is the ECM protein largely responsible for vascular elasticity, we evaluate here in a well-established PAH neonatal calf model the role of elastin in modulating the stiffness of upstream PA vessels.
In this study, we hypothesize that changes in the functional mechanical properties of elastin are one of the primary causes of the increased stiffness seen in the proximal PAs due to hypoxia-induced PAH. To test this hypothesis, we examined the biomechanics and structure of normotensive and hypertensive neonatal calf proximal PAs and their associated elastin tissue.
MATERIALS AND METHODS
Animal model.
All studies were performed after approval by institutional animal care and use committees. Arterial tissues from three normotensive and six hypertensive male Holstein calves (70–110 lbs.) were used. Hypertension was induced by hypobaric hypoxia, 2 wk, 430 mmHg (4,600-m equivalent air pressure). Hypoxic animals were killed at 430-mmHg pressure after 14 days at hypoxic conditions. All animals were studied and killed at 15 ± 1–2 days of age. PA pressures were obtained with a 5-French Swan-Ganz or Millar catheter, positioned in the main PA (MPA). After death, the right PA (RPA), left PA (LPA), and MPA were excised and stored in nutrient-balanced medium (4°C). Testing was conducted within 24 h of death; material properties of fresh tissues were unchanged for tests conducted up to 4 days postdeath.
Geometric and material property results were calculated using three control and six hypertensive tissues for the LPA and MPA. Two hypertensive RPA tissues were too small and irregularly shaped for stress-strain testing; therefore, three control and four hypertensive RPA tissues were used. We were also unable to accurately measure the PA pressure for one hypertensive animal. Due to this, three control and five hypertensive LPA and MPA and three control and four hypertensive RPA tissues were used for all material property comparisons made at physiological pressure/strain values.
Tissue dissection and processing.
Arteries were inspected for holes, tears, branch points, and localized thickening or other anomalies that may adversely affect test results. Loose connective tissue was removed, and gross anatomic measurements of diameter (1 distal, 1 proximal) and wall thickness (4 distal, 4 proximal) were obtained with digital calipers before axial bisection (see Fig. 1). Circumferential rectangular sections (∼8 × 20 mm) were taken for subsequent material testing. Biopsy samples were taken for histological analysis, three each in the longitudinal and circumferential directions.
Fig. 1.

Thickness measurements, tissue dissection, and biopsy locations (typical). ID, internal diameter; MC, biopsy taken in circumferential direction; ML, biopsy taken in longitudinal direction.
Materials testing.
Fresh tissue stress-strain data were acquired with an MTS, Insight 2 (MTS Systems, Eden Prairie, MN) material testing system, equipped with an ancillary environmental chamber and either 5-N or 2-kN load cell, depending on the maximum expected load for each test (Fig. 2). Stress-strain tests were conducted in calcium and magnesium-free NaCl-PBS buffer (0.01 mol/l, ionic strength 0.15, pH 7.4) at 36°C. Sample width, thickness, and gage length were measured using digital calipers. Stress-strain properties remained consistent for tissue tests performed up to 3 days postdeath.
Fig. 2.

Detail of material testing system (MTS) used to generate stress (σ)-strain (ɛ) data for arterial tissue samples.
Tissues were preconditioned by 10 full extension-relaxation cycles before data were taken. The strain rate for all mechanical tests was 10% strain per second. In this study, force and stress data refer to the loading region of the stress-strain curve. In an effort to avoid damage that may affect elastin-only material tests, tissues were stretched until the collagen-dependent, strain-stiffening response became fully developed, typically 60–80% strain (Fig. 3). Elastin tissue tests were conducted in the same manner as that outlined for fresh arteries, but with a 2- or 5-N load cell. Elastin samples were tested at strains ranging from 60 to 100% at 10% increments or until failure. Data processing was conducted using custom-written software (Matlab R2007a, The Math Works, Natick, MA).
Fig. 3.

Typical loading/unloading curves for fresh and elastin tissue tests. Fn, width-normalized force.
Tissue digestion using formic acid-cyanogen bromide solution.
After fresh artery material testing, tissues were processed into elastin-only scaffolds using the cyanogen bromide (CNBr)-formic acid digestion procedure outlined by Lu et al. (30). Briefly, fresh arteries were submerged in a 70% formic acid, 50 mg/ml CNBr solution and gently stirred for 19 h at room temperature followed by 1 h at 60°C. The samples were then boiled for 10 min to remove any residual CNBr. The digestion solution was then decanted, and the tissue was rinsed with distilled water.
CNBr-formic acid digestion was chosen for elastin purification due to its effectiveness for removing nonelastin components from arterial tissues. Other common elastin purification procedures using hot alkali or autoclaving were rejected due to possible network fragmentation and incomplete elastin purification, respectively (15).
Amino acid analysis was performed on LPA elastin samples (n = 7) to determine the effectiveness of the digestion procedure. After digestion, the samples retain formic acid, and subsequent material testing showed strong pH dependence. NaOH titration was, therefore, used to raise the pH from 2–3 to 7.4. After titration, samples were stored in NaCl-PBS buffer and were tested within 2 wk of animal death. Elastin mechanical properties were unchanged for tests conducted up to 1 mo postdeath.
Histology and elastin area fraction.
Biopsies were fixed in 10% buffered formalin, wax embedded, sectioned, and stained for elastin using Verhoeff's Van Gieson. Bright-field photomicrographs were taken with a Nikon TE-200 (Nikon Instruments, Melville, NY) microscope (×40 magnification, SPOT RT-900 camera; Diagnostic Instruments, Sterling Heights, MI). Elastin area fraction was determined using custom software (Matlab R2007a Image Processing Toolbox, The Math Works).
Calculations.
Fresh artery Cauchy stress (σfresh) was calculated with the assumption of incompressibility applied to the width (W) and thickness (T) directions to account for changes in cross-sectional area as a function of engineering strain (ɛ).
 |
(1) |
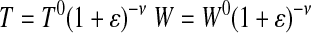 |
(2) |
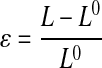 |
(3) |
Where F is the applied force; L is the loaded sample length in the direction of applied load; T0, W0, and L0 are the initial thickness, width, and length, respectively; Aelast is the area fraction of elastin; and ν is Poisson's ratio (0.5 for incompressible tissue). Due to the layered structure of the fenestrated elastic sheets, stresses for the digested elastin samples (σelast) were calculated with the condition of incompressibility applied to the width direction only. To facilitate modulus (E) calculations, stress-strain data for fresh and elastin samples were fit with ninth- and fourth-order polynomials, respectively. The ninth- and fourth-order polynomials were determined to be the lowest order polynomials necessary to achieve an average squared correlation coefficient R ≥ 0.995 for all fresh and elastin data sets, respectively.
≥ 0.995 for all fresh and elastin data sets, respectively.
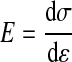 |
(4) |
Stiffness (Φ) is the derivative of the width-normalized force (Fn: force divided by current width) by strain, and, as such, stiffness is dependent on thickness geometry and represents the extensive equivalent of elastic modulus.
 |
(5) |
The condition of incompressibility was applied to the stiffness calculation to account for any variation in width as a function of imposed strain. Fn data were fit to polynomials in the same manner as was used for the stress. Unless otherwise noted, all results for the material parameters (E, Φ) were calculated at a strain of 35%, so that tissues could be compared. Strains higher than 35% resulted in one or more of the stress-strain curves extending into the collagen-dominant, strain-stiffening region. At strains >35%, any comparison between fresh and elastin stress-strain data would be complicated by the active loading of collagen. Since this collagen component cannot be removed from the fresh artery data set, any comparison of the mechanical properties made at strains >35% would include collagen and, therefore, not allow for the direct comparison of elastin mechanical property changes from the fresh to digested state.
Lame's equation for stress in thick-walled tubes (σL) (6) was used to determine the strains that correspond to the measured physiologic pressures.
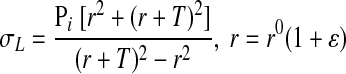 |
(6) |
Lame's equation was equated to the polynomial stress-strain function and solved iteratively for the strains that correspond to the in vivo measured systolic, diastolic, and mean pressures (Pi); r and r0 are the internal radii of the artery in the loaded and initial state, respectively. The estimated physiological strains were then used to calculate the systolic and diastolic moduli using Eq. 4.
Curvature (κ) of the stress-strain curve was used to determine the onset of collagen engagement (Fig. 4).
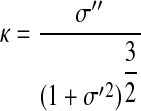 |
(7) |
Fig. 4.

Top: typical σ-ɛ behavior of fresh (σfresh) and elastin (σelast) tissue. ɛtrans is the ɛ of transition from the elastin-dominant region (A) to the transitional region (B), associated with increasing collagen engagement. Bottom: typical curvature (κ) plot of fresh tissue. H, height; κPSS, average prestrain-stiffening κ; κtrans, κ of the stress-strain curve at the transition strain; κmax, maximum κ calculated for a given tissue sample; ɛH, ɛ associated with maximum curvature minus 20% ɛ; ɛL, 20% ɛ; ɛPSS, prestrain-stiffening ɛ.
The second-order derivative in the numerator of the curvature equation led to large variances when calculated from the polynomial fit to the original stress-strain data. We, therefore, applied a zero-phase, low-pass, elliptic digital filter to smooth the data. A new ninth-order polynomial was then fit to the filtered data, resulting in reduced curvature variance.
Average prestrain-stiffening curvature (κPSS) was calculated for the strain range between ɛL and ɛH, where ɛL equals 20% strain and ɛH equals the strain associated with maximum curvature minus 20% strain (Fig. 4). The ɛL and ɛH were defined in this way so that any curvature associated with the low-strain loading region or high-strain strain-stiffening region would not influence the κPSS value. Transition strain (ɛtrans) was then determined by the relationship:
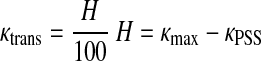 |
(8) |
such that ɛtrans is associated with the onset of curvature elevation; κtrans is the curvature of the stress-strain curve at the transition strain; and κmax is the maximum curvature calculated for a given tissue sample. The ɛtrans results compare favorably with those previously published using both experimental (36), as well as Lorentz distribution functions (45, 49). Stress-strain plots of elastin tissue do not exhibit curvature elevation behavior, and since strain-stiffening is associated with collagen engagement, we defined ɛtrans as the strain at which collagen begins to carry load. Strains below this value fall within the elastin-dominant region of the stress-strain curve (Fig. 4, top, A), while strains that are greater fall within the transitional or collagen-dominant region (Fig. 4, top, B). We further defined the collagen-dominant region as those strains that result in a fresh artery modulus greater than 10 times the artery modulus calculated within the elastin-dominant region at a strain of 35%. While this is an arbitrary definition for collagen dominance, it seemed logical to suggest that collagen is the dominant load-bearing structural protein when at least 90% of the material modulus can be attributed to collagen.
Equation 9 defines the percentage of the fresh tissue load that is carried by elastin (%Fn-C, %Fn-H) and the percentage of the increased load, due to PAH, that is attributable to the increased stiffness of elastin (%Fn-e,f), calculated at a strain of 35%.
 |
(9) |
where Fn-f,C and Fn-f,H are Fn for fresh tissue (control and hypertensive, respectively); and Fn-e,C and Fn-e,H are Fn for elastin tissue (control and hypertensive, respectively). Fn-C and Fn-H were similarly calculated at systolic and diastolic strains to determine the percent load-carrying capacity of elastin at physiological strains.
P values refer to the probability associated with the one-tail, two-sample equal variance, Student's t-test. Results were deemed statistically significant for P values ≤0.05. All error bars represent 1 SD above and below the mean. Standard deviations (SD) were calculated using the “n − 1” method. In all curve fitting, the average of the squared correlation coefficient was deemed statistically significant for R ≥ 0.995.
≥ 0.995.
RESULTS
Morphology.
PAH resulted in an increase in vessel wall thickness by 30% (SD = 12%, P = 0.02), 28% (SD = 11%, P = 0.04), and 26% (SD = 8%, P = 0.03) for the RPA, LPA, and MPA, respectively (Fig. 5). There was no statistically significant change in the internal diameter as a result of hypertension. PAH also caused a decrease in the area fraction of elastin by 17% (SD = 2%, P = 0.03), 16% (SD = 2%, P = 0.01), and 16% (SD = 3%, P = 0.03) for the RPA, LPA, and MPA, respectively (Fig. 5). The elastin content factor (ECF) is the product of the elastin area fraction and tissue thickness and is representative of the volume of elastin in a given tissue cross section. The increase in ECF, resulting from PAH, was calculated to be 7.6% (SD = 3.1%), 7.7% (SD = 3.1%), and 5.5% (SD = 2.0%) for the RPA, LPA, and MPA, respectively; average ECF = 6.9% (SD = 1.2%).
Fig. 5.

Comparison of average thickness values and elastin area fractions between control and hypoxic populations. Values are means ± SD. RPA, LPA, MPA: right, left, and main pulmonary artery, respectively. *P < 0.05.
Physiology.
Average systolic/diastolic pressures were 45/18 mmHg (SD = 12/7 mmHg) and 146/71 mmHg (SD = 29/18 mmHg) for control and hypoxic populations, respectively. Control and hypoxic mean pulmonary pressures were 31 mmHg (SD = 12 mmHg) and 106 mmHg (SD = 16 mmHg), respectively. We were unable to collect accurate diameter data for all tested tissues; therefore, the strain at systole and diastole was calculated using Lame's equation (6), with average unloaded internal radii calculated for each MPA, RPA, and LPA, measured from 4 control and 11 hypertensive tissues. The average systolic/diastolic strains for the PAs were 59% (SD = 10%)/34% (SD = 6%) and 72% (SD = 13%)/53% (SD = 11%) for the control and hypoxic populations, respectively. The average strain for the mean pulmonary pressures were 47% (SD = 8%) and 65% (SD = 12%) for the control and hypoxic populations, respectively. The average ɛtrans was unchanged by PAH: 49% (SD = 8%) for control and 51% (SD = 9%) for hypertensive tissues (P = 0.29). The ɛtrans values are comparable to the strain corresponding with the onset of collagen engagement published by Zulliger et al. (49). The strain associated with the onset of collagen dominance was calculated to be 69% (SD = 7%) and 74% (SD = 8%) for the control and hypoxic populations, respectively.
Mechanical properties of fresh tissue.
At 35% strain, the average increase in stiffness, due to PAH, was 72% (SD = 30%, P = 0.01), 107% (SD = 56%, P = 0.01), and 63% (SD = 14%, P = 0.01) for the RPA, LPA, and MPA, respectively (Fig. 6). The overall average increase in stiffness was 81% (SD = 23%, P ≪ 0.05). PAH also caused an increase in modulus of PA tissues by 28% (SD = 7%, P = 0.04), 45% (SD = 23%, P = 0.10), and 65% (SD = 18%, P = 0.02) for the RPA, LPA, and MPA, respectively. The average overall increase in fresh tissue modulus was 46% (SD = 18%, P = 0.02). Average stiffness values at physiological strains are shown in Fig. 7, and comparisons between physiological stiffness values are detailed in Table 1.
Fig. 6.

Comparison of mean values for artery stiffness (Φ) and modulus (E) of the RPA, LPA, and MPA for control and hypoxic populations. Values are means ± SD. *P < 0.05.
Fig. 7.

Average control and hypertensive tissue stiffness at systolic and diastolic pressures. Values are means ± SD. Comparative values are presented in Table 1.
Table 1.
Physiological stiffness comparison
| RPA | LPA | MPA | Average | |||||
|---|---|---|---|---|---|---|---|---|
| Fresh tissue | ||||||||
| ΦDias,Cont to PAH | 327* | 274* | 279* | 293* | ||||
| ΦSys,Cont to PAH | 584* | 512* | 435* | 510* | ||||
| ΦCont,Dias to Sys | 151* | 125* | 158 | 145* | ||||
| ΦPAH,Dias to Sys | 303* | 268* | 265* | 278* | ||||
| Elastin tissue | ||||||||
| ΦDias,Cont to PAH | 151* | 243* | 124* | 173* | ||||
| ΦSys,Cont to PAH | 102* | 186 | 81* | 123* | ||||
| ΦCont,Dias to Sys | 41 | 41 | 36 | 39 | ||||
| ΦPAH,Dias to Sys | 13 | 18 | 10 | 14 | ||||
| Percent load-carrying capacity of elastin | ||||||||
| Fn,Dias,Cont | 50 | 46 | 87 | 61 | ||||
| Fn,Dias,PAH | 45 | 43 | 56 | 48 | ||||
| Fn,Sys,Cont | 45 | 36 | 63 | 48 | ||||
| Fn,Sys,PAH | 30 | 31 | 35 | 32 | ||||
Average change (in %) in stiffness for control and pulmonary arterial hypotension (PAH) tissues at diastolic and systolic pressure and average load-carrying capacity of elastin. Comparative values are based on the data presented in Fig. 7. RPA, LPA, MPA: right, left, and main pulmonary artery, respectively; ΦDias,ConttoPAH and ΦSys,ConttoPAH: average increase in stiffness from control to PAH tissues measured at diastole or systole, respectively. ΦCont,DiastoSys and ΦPAH,DiastoSys: average increase in control or PAH tissue stiffness from diastole to systole, respectively. Fn,Dias,Cont, Fn,Sys,Cont: average percent load-carrying capacity of control tissue measured at diastole or systole, respectively; Fn,Dias,PAH, Fn,Sys,PAH: average percent load-carrying capacity of PAH tissue measured at diastole or systole, respectively.
P < 0.05.
Mechanical properties of purified arterial elastin.
At 35% strain, PAH resulted in an increase in the stiffness of the elastin by 57% (SD = 19%, P = 0.04), 109% (SD = 68%, P = 0.03), and 50% (SD = 17%, P = 0.05) for the RPA, LPA, and MPA, respectively (Fig. 8). The overall average increase in the stiffness of elastin was 72% (SD = 33%, P = 0.01). The modulus of elastin increased by 34% (SD = 7%, P = 0.04), 88% (SD = 43%, P = 0.01), and 73% (SD = 36%, P = 0.07) for the RPA, LPA, and MPA, respectively. The overall average increase in the modulus of elastin was 65% (SD = 28%, P ≪ 0.05). Elastin stiffness at physiological pressure is detailed in Fig. 7 and Table 1. Amino acid analysis confirmed that CNBr-formic acid digestion resulted in elastin tissue samples of high purity (25), as evidenced by the high number of residues per 1,000 for alanine (227, SD = 20), valine (127, SD = 6), and glycine (342, SD = 16), along with the lack of methionine and hydroxylysine residues (trace) and the low concentration of hydroxyproline (14, SD = 5).
Fig. 8.

Comparison of mean values for elastin stiffness and modulus for the RPA, LPA, and MPA for control and hypoxic populations. Values are means ± SD. *P < 0.05.
Average values for %Fn-C and %Fn-H are 51% (SD = 16%, P = 0.02) and 48% (SD = 13%, P = 0.003), respectively, meaning that, at a minimum, roughly one-half of the load applied to the fresh arteries is carried by elastin for both control and hypertensive conditions at a strain of 35%. Also, %Fn-e,f indicates that the change in the stiffness of the elastin accounts for an average of 43% (SD = 10%, P = 0.003) of the change in the stiffness of the fresh tissue as a result of hypertension. Changes in elastin load-carrying capacity at physiological strains are detailed in Table 1.
DISCUSSION
Several key issues arise from this study. First, both the physiological pressure and PA stiffness elevation for the hypoxia-induced neonatal calf model mimic the changes we have seen in the human (12) and confirm the value of this model for studying PA stiffness and impedance changes in PAH. Second, the increases in stiffness appear attributable to a significant extent on material rather than purely geometric factors. Third, the changes in material modulus due to PAH appear to be significantly attributable to changes in elastin modulus specifically. Fourth, the physiological tissue strain at diastolic pressure is largely dependent on the stiffness of elastin. Last, a significant component of the mechanical load for both normal and hypertensive conditions continues to be carried by elastin. We comment on each of these issues below.
Use of the neonatal bovine model.
The normal and pathological physiology of the bovine model more closely mimics that of humans than that of the available rodent models. Humans and young calves both have similar body mass, heart rate, blood pressure, and vascular wall thickness. In addition, the cellular composition of the proximal PA wall of the large mammals is similarly complex to humans, incorporating numerous phenotypically distinct smooth muscle cell populations that rapidly proliferate during PAH (40, 41). For these reasons and the fact that we have significant experience with this model, we chose the hypoxic neonatal calf as the in vivo model for understanding how the proximal PA stiffens during hypoxia-induced PAH.
Impact of PAH on physiological strain.
Hypoxic conditions successfully induced PAH, as evidenced by the significant increase in mean PA pressure, vascular wall thickness, and PVS. Due to the stress-strain nonlinearity of arterial tissue, the relative contribution of elastin and collagen is contingent on the strains associated with in vivo physiological pressures. Our results indicate that the physiological region of the stress-strain curve bounds the ɛtrans, with 62% of the curve residing in the elastin-dominant region and 38% in the transition region for the control population (Fig. 9). Hypertension shifts the physiological strain region to higher strain values, but has no effect on the ɛtrans. The resulting diastolic strain of hypertensive tissues is roughly equal to the ɛtrans, 53 and 50%, respectively, and the systolic strain of 72% nearly equals the strain associated with collagen-dominant, stress-strain behavior (74% in PAH tissues). Hypertensive stress-strain curves, therefore, begin at the ɛtrans and reside entirely within the transition region. This bounding of the ɛtrans in control tissues and the correlation of transition and diastolic strains for hypertensive tissues suggest that the diastolic strain, and, therefore, baseline slope, of the stress-strain curve is set by the stiffness of elastin, whereas the systolic strain incorporates collagen engagement.
Fig. 9.

Mean pressure-ɛ data for control and hypertensive tissues. Shaded regions indicate the predicted physiological conditions for control (A) and hypertensive (B) tissues. ɛcd, Collagen-dominant ɛ.
At diastolic strain, PAH causes a 293% (ΦDias,Cont to PAH) increase in artery stiffness (Fig. 7, Table 1). Since diastolic strain resides within the elastin-dominant region for control tissues and at the ɛtrans for PAH tissues, this increase in diastolic stiffness is likely the result of both increased elastin stiffness and the onset of collagen engagement. We calculate that PAH causes a 173% (ΦDias,Cont to PAH) increase in diastolic elastin stiffness for digested samples. The discrepancy in diastolic stiffness elevation between elastin and fresh tissue is a consequence of the removal of nonelastin material during elastin purification, which will necessarily change the mechanical properties of elastin between its natural and tested state. However, given that the minimum percent diastolic load-carrying capacity of elastin is 61% (%Fn Dias,Cont) and 48% (%Fn Dias,PAH) for the control and PAH tissues, respectively, and that elastin stiffness values are relatively strain independent, we feel confident in stating that the change in diastolic stiffness, due to PAH, is predominately a result of an increase in elastin stiffness.
The significant strain stiffening response of the transitional region acts to limit the maximum systolic strain at elevated pressure. Our data shows that PAH causes an increase in both fresh tissue and purified elastin systolic stiffness by 510% and 123% (ΦSys,Cont to PAH), respectively. By comparing the systolic and diastolic stiffness change from control to hypertensive tissues (ΦSys,Cont to PAH to ΦDias,Cont to PAH), we see that the systolic stiffness elevation of fresh tissue, resulting from PAH, is nearly double that of the diastolic stiffness elevation, while the elastin stiffness elevation remains relatively constant. This is not surprising, given the linear nature of the elastin stress-strain curve compared with the highly nonlinear fresh tissue curve. This highly nonlinear behavior coupled with the elevated PAH physiological strain results in an increase in stiffness for control and PAH tissue from diastolic to systolic strain of 145% (ΦCont,Dias to Sys) and 278% (ΦPAH,Dias to Sys), respectively, while the more linear elastin tissue only increased 39 and 14% for control and PAH tissues, respectively. Also, the minimum percent systolic load-carrying capacity of elastin is reduced from 48% (%Fn,Sys,Cont) for control tissues to 32% (%Fn,Sys,PAH) for PAH tissues, indicating that collagen carries more load in the diseased state. However, these data also show that elastin is an important load-carrying material, even at PAH systolic strains. Increased systolic tissue stiffness is, therefore, due to both elevated elastin stiffness, as well as a higher degree of collagen engagement, resulting from the increase in systolic strain due to PAH.
While our results indicate that PAH causes PA tissues to operate at higher strains, the elevated stiffness of the elastin-dominant region mitigates this effect and maintains the physiological strain range within the transitional region. Due to the large strains associated with control fresh tissues operating at PAH physiological pressures, we were unable to consistently stretch the control fresh tissues to strains large enough to attain hypoxic pressure-load values without risk of tissue damage. However, by projecting the fitted polynomial curves of control fresh tissues, we estimate that the PAH diastolic pressure intersects the control fresh tissue curve at a strain of ∼73%. At these high strains, the compliance of arterial tissue is significantly reduced due to elevated tissue modulus, resulting from increased collagen loading. Therefore, the elevated stiffness of the PAH elastin-dominant region of the stress-strain curve maintains the physiological strains of PAH arterial tissues within the transition region of the stress-strain curve by lowering the diastolic strain from ∼73 to 53%. Without this increased stiffness, PAH PA tissues would operate entirely within the high-modulus, collagen-dominant region of the stress-strain curve, which would result in a significant reduction of the windkessel effect. This further underlines the importance of understanding how elastin contributes to the mechanical properties and morphology of PA tissue in response to PAH.
PVS and elastin.
Typically, modulus is calculated with a cross-sectional area determined from microscopic analysis of arterial sections (8–10, 16, 27, 39, 42). This method assumes that the constituent artery materials maintain homogeneity, regardless of pathology or anatomical location. Since stiffness calculations do not require this assumption, the 72% increase in elastin stiffness is an independent result, indicating that elastin carries substantially more load in the pathological state. However, the removal of nonelastin tissue during the digestion process results in experimentally measured material properties that differ between the isolated elastin tissue and elastin in its native state.
Individual elastic fibers are arranged in a network and bound together to form fenestrated elastic sheets, which surround the artery in concentric layers and work to uniformly distribute arterial loads (38). While the individual elastic fibers do not have a linear stress-strain relationship, network effects allow the resulting elastin tissue to obey Hooke's law up to large strains (45). The resulting elastic sheets, which are bound to nonelastin ECM components and smooth muscle cells through glycoprotein and integrin bonds, imbue the artery with a Neo-Hookean load-carrying elastin component that behaves in a near perfectly elastic manner (1). Hypertensive adaptation of these intratissue bonding proteins is thought to play a role in the development of increased arterial stiffness (2–4, 6, 20–22); however, during the CNBr digestion process, the elastin intratissue bonds are broken, and all nonelastin material is extracted. In addition, the removal of bulk, nonelastin, artery material eliminates many of the constraints that resist the movement of elastin sheets relative to one another. The removal of both the intratissue bonds and nonelastin material likely results in experimentally measured stiffness and modulus values that are lower then those of the tissue in its native state. However, any discrepancy between the experimentally determined and native elastin stiffness should have little impact on material property comparisons between control and hypertensive elastin tissues, since both underwent the same digestion process. Due to this discrepancy between the experimentally measured and native stiffness properties, %Fn-C and %Fn-H should be considered as the lower bound for the fresh tissue load fraction carried by elastin, and %Fn-e,f defines the minimum fraction of increased load directly attributable to hypertensive elastin tissue. Hypertension, therefore, causes a significant increase in the stiffness of the elastin material responsible for carrying roughly one-half (%Fn-C, %Fn-H) of the arterial tissue load in the elastin dominant region, at a minimum.
PVS is a function of both the intrinsic material property of modulus and the extrinsic geometric consideration of thickness. Our data show a 46 and 28% average increase in fresh tissue modulus and thickness, respectively, and an 81% average increase in fresh tissue stiffness. For elastin tissues, our data show a 65 and 6.9% average increase in modulus and ECF, respectively, and a 72% average increase in stiffness. Since the width-normalized force, and hence stiffness, scale linearly with changes in both modulus and thickness, it is evident that modulus, rather than geometric tissue thickening, is the dominant factor in determining the stiffness of hypertensive arterial and elastin tissues. Also, it is interesting to note that the stiffness elevation predicted by our fresh tissue modulus and thickness data is 87%, which is close to the measured stiffness increase of 81%, and the stiffness elevation predicted by our increase in elastin modulus and ECF is 76%, which is close to the 72% average increase in measured elastin stiffness.
Elastin modulus.
Our biomechanical tests show that hypertension causes an increase in the modulus of both the fresh and elastin arterial tissues by an average of 46 and 65%, respectively. This increase in elastin modulus supports our thinking that one of the primary structural targets of proximal vascular remodeling during hypoxia-induced PAH is arterial elastin. Changes in fresh artery modulus are comparable to the 30% average increase in the modulus of the PAs in mice published by Kobs et al. (26, 27) and 40% decrease in compliance, identified as the threshold for pediatric hypertension in humans, by Dyer et al. (12).
The increased modulus of elastin, resulting from hypertension, suggests either a change in elastin fibril modulus or a structural adaptation in fibril orientation/interelastin binding. The turnover rate for collagen and elastin is low in healthy arteries, but vascular pathology upsets the regulatory pathways that maintain this balance. In response to hypertension, the overexpression of both proinflammatory and proteinase-inhibitory molecules dramatically increases arterial ECM synthesis (2, 23, 24). Although little is known about the ECM synthesis pathway of hypertensive arterial tissue (2, 40), the upregulation of the tissue inhibitor of matrix metalloproteinase-1 is one method by which ECM synthesis may be enhanced during hypertension (23, 24). However, the ECM proteins synthesized in response to hypertension have a three-dimensional architecture that is functionally less optimal then those deposited during fetal development and may play an important role in determining the modulus of pathological elastin tissue (2, 23, 24). More experimentation is required to determine the underlying cause for the increase in elastin modulus.
Limitations.
Several limitations in these studies must be acknowledged. First, the tissue digestion procedure may alter the mechanical properties of elastin through chemical attack or thermal degradation, although no evidence of this was found in our studies or in the literature. Second, the use of continuum mechanics for discontinuous materials will lead to some degree of error in calculated mechanical properties due to simplifying assumptions; however, this should have little impact on comparison of those properties. Third, uniaxial rather than fully biaxial testing was performed. However, given that the objective of this study was not to investigate the anisotropic behaviors of artery tissues, but rather to determine how such properties of fresh and digested tissue samples change as a result of hypertension, the uniaxial test was deemed sufficient.
Conclusion.
Via biomechanical testing of both fresh and digested elastin tissues of control and hypoxia-induced pulmonary hypertensive animals, we have confirmed that vascular remodeling results in a significant increase in the modulus and stiffness of both the fresh and elastin tissues of the proximal PAs in the circumferential direction. We have further shown that, at 35% strain, a minimum 48% of the load imposed on fresh arterial tissue is carried by elastin, and that, at a minimum, 43% of the change in stiffness of fresh arterial tissue is directly attributable to the change in stiffness of the elastin component of those arteries. We have also shown that PAH causes an increase in the strains associated with the physiological pressure range but had no effect on the strain of transition from elastin-dominant to collagen-dominant behavior. Stress-strain behavior of fresh arterial tissues further revealed that the diastolic strain and baseline slope of the stress-strain curve are determined by elastin mechanics, and the strain at systole becomes increasingly dependent on collagen mechanics as a result of hypertension. The final conclusion drawn from the above statements is that elastin is an important passive material for carrying the load imposed on the proximal PAs at physiological pressure ranges, and that the increase in stiffness of elastin acts to maintain the physiological strain range within the more elastin-dependent region of the stress-strain curve.
GRANTS
This study was supported by National Institutes of Health Grants P50HL84923, T32HL072738, and K24HL081506.
Acknowledgments
The authors thank Dr. Conrad Stoldt for the use of laboratory equipment and Colorado State University for providing access to their hypobaric chamber.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Aaron BB, Gosline JM. Elastin as a random-network elastomer–a mechanical and optical analysis of single elastin fibers. Biopolymers 20: 1247–1260, 1981. [Google Scholar]
- 2.Arribas SM, Hinek A, Gonzalez MC. Elastic fibres and vascular structure in hypertension. Pharmacol Ther 111: 771–791, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bezie Y, Lacolley P, Laurent S, Gabella G. Connection of smooth muscle cells to elastic lamellae in aorta of spontaneously hypertensive rats. Hypertension 32: 166–169, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Bezie Y, Lamaziere JMD, Laurent S, Challande P, Cunha RS, Bonnet J, Lacolley P. Fibronectin expression and aortic wall elastic modulus in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 18: 1027–1034, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation 99: 2434–2439, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Boumaza S, Arribas SM, Osborne-Pellegrin M, McGrath JC, Laurent S, Lacolley P, Challande P. Fenestrations of the carotid internal elastic lamina and structural adaptation in stroke-prone spontaneously hypertensive rats. Hypertension 37: 1101–1107, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Chamiot Clerc P, Renaud JF, Blacher J, Legrand M, Samuel JL, Levy BI, Sassard J, Safar ME. Collagen I and III and mechanical properties of conduit arteries in rats with genetic hypertension. J Vasc Res 36: 139–146, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Chesler NC, Thompson-Figueroa J, Millburne K. Measurements of mouse pulmonary artery biomechanics. J Biomech Eng 126: 309–314, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Dobrin PB Vascular mechanics. In: Handbook of Physiology. The Cardiovascular System. Peripheral Circulation and Organ Blood Flow. Bethesda, MD: Am. Physiol. Soc., 1983, sect. 2, vol. III, pt. 1, chapt. 3, p. 65–102.
- 10.Drexler ES, Quinn TP, Slifka AJ, McCowan CN, Bischoff JE, Wright JE, Ivy DD, Shandas R. Comparison of mechanical behavior among the extrapulmonary arteries from rats. J Biomech 40: 812–819, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Durmowicz AG, Orton EC, Stenmark KR. Progressive loss of vasodilator responsive component of pulmonary-hypertension in neonatal calves exposed to 4,570 m. Am J Physiol Heart Circ Physiol 265: H2175–H2183, 1993. [DOI] [PubMed] [Google Scholar]
- 12.Dyer K, Lanning C, Das B, Lee PF, Ivy DD, Valdes-Cruz L, Shandas R. Noninvasive Doppler tissue measurement of pulmonary artery compliance in children with pulmonary hypertension. J Am Soc Echocardiogr 19: 403–412, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Et-Taouil K, Safar M, Plante GE. Mechanisms and consequences of large artery rigidity. Can J Physiol Pharmacol 81: 205–211, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L656–L664, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Gundiah N, Ratcliffe MB, Pruitt LA. Determination of strain energy function for arterial elastin: experiments using histology and mechanical tests. J Biomech 40: 586–594, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Huang W, Delgado-West D, Wu JT, Fung YC. Tissue remodeling of rat pulmonary artery in hypoxic breathing. II. Course of change of mechanical properties. Ann Biomed Eng 29: 552–562, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Hunter KS, Lee PF, Lanning CJ, Ivy DD, Kirby KS, Claussen LR, Chan KC, Shandas R. Pulmonary vascular input impedance is a combined measure of pulmonary vascular resistance and stiffness and predicts clinical outcomes better than pulmonary vascular resistance alone in pediatric patients with pulmonary hypertension. Am Heart J 155: 166–174, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 97: 185–191, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Intengan HD, Schiffrin EL. Structure and mechanical properties of resistance arteries in hypertension–role of adhesion molecules and extracellular matrix determinants. Hypertension 36: 312–318, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 38: 581–587, 2001. [DOI] [PubMed] [Google Scholar]
- 22.Intengan HD, Thibault G, Li JS, Schiffrin EL. Resistance artery mechanics, structure, and extracellular components in spontaneously hypertensive rats–effects of angiotensin receptor antagonism and converting enzyme inhibition. Circulation 100: 2267–2275, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Jacob MP Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 57: 195–202, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Jacob MP, Badier-Commander C, Fontaine V, Benazzoug Y, Feldman L, Michel JB. Extracellular matrix remodeling in the vascular wall. Pathol Biol (Paris) 49: 326–332, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Jacob MP, Ladislas R. Isolation, characterization and biochemical properties of elastin. In: Elastin and Elastases, edited by Ladislas R and Hornebeck W. Boca Raton, FL: CRC, 1989, p. 45–69.
- 26.Kobs RW, Chesler NC. The mechanobiology of pulmonary vascular remodeling in the congenital absence of eNOS. Biomech Model Mechanobiol 5: 217–225, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Kobs RW, Muvarak NE, Eickhoff JC, Chesler NC. Linked mechanical and biological aspects of remodeling in mouse pulmonary arteries with hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol 288: H1209–H1217, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 37: 1236–1241, 2001. [DOI] [PubMed] [Google Scholar]
- 29.London GM, Blacher J, Pannier B, Guerin AP, Marchais SJ, Safar ME. Arterial wave reflections and survival in end-stage renal failure. Hypertension 38: 434–438, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Lu Q, Ganesan K, Simionescu DT, Vyavahare NR. Novel porous aortic elastin and collagen scaffolds for tissue engineering. Biomaterials 25: 5227–5237, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Mahapatra S, Nishimura RA, Jja PS, Cha S, McGoon MD. Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J Am Coll Cardiol 47: 799–803, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Mahmud A, Feely J. Arterial stiffness and the renin-angiotensin-aldosterone system. J Renin Angiotensin Aldosterone Syst 5: 102–108, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Maruyama K, Ye CL, Woo M, Venkatacharya H, Lines LD, Silver MM, Rabinovitch M. Chronic hypoxic pulmonary-hypertension in rats and increased elastolytic activity. Am J Physiol Heart Circ Physiol 261: H1716–H1726, 1991. [DOI] [PubMed] [Google Scholar]
- 34.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med 171: 494–499, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Olsen MH, Christensen MK, Wachtell K, Tuxen C, Fossum E, Bang LE, Wiinberg N, Devereux RB, Kjeldsen SE, Hildebrandt P, Dige-Petersen H, Rokkedal J, Ibsen H. Markers of collagen synthesis is related to blood pressure and vascular hypertrophy: a LIFE substudy. J Hum Hypertens 19: 301–307, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Roach MR, Burton AC. The reason for the shape of the distensibility curves of arteries. Can J Biochem Physiol 35: 681–690, 1957. [PubMed] [Google Scholar]
- 37.Safar ME, Levy BI, Struijker-Boudier H. Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation 107: 2864–2869, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Shadwick RE Mechanical design in arteries. J Exp Biol 202: 3305–3313, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Sokolis DP, Boudoulas H, Karayannacos PE. Assessment of the aortic stress-strain relation in uniaxial tension. J Biomech 35: 1213–1223, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling–cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. [DOI] [PubMed] [Google Scholar]
- 41.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: a time for reappraisal? Circ Res 97: 95–98, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Van Gorp AW, Schenau DSV, Hoeks APG, Boudier H, De Mey JGR, Reneman RS. In spontaneously hypertensive rats alterations in aortic wall properties precede development of hypertension. Am J Physiol Heart Circ Physiol 278: H1241–H1247, 2000. [DOI] [PubMed] [Google Scholar]
- 43.Weinberg C, Hertzberg J, Valdes-Cruz LM, Shandas R. Extraction of pulmonary vascular compliance, PVR and RV work from single-pressure and Doppler flow measurements in children with pulmonary hypertension–a new method for evaluating reactivity: in vitro and clinical studies. Circulation 110: 2609–2617, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Wohrley JD, Frid MG, Moiseeva EP, Orton EC, Belknap JK, Stenmark KR. Hypoxia selectively induces proliferation in a specific subpopulation of smooth-muscle cells in the bovine neonatal pulmonary arterial media. J Clin Invest 96: 273–281, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wuyts FL, Vanhuyse VJ, Langewouters GJ, Decraemer WF, Raman ER, Buyle S. Elastic properties of human aortas in relation to age and atherosclerosis: a structural model. Phys Med Biol 40: 1577–1597, 1995. [DOI] [PubMed] [Google Scholar]
- 46.Zaidi SHE, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation 105: 516–521, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Dunn ML, Hunter KS, Lanning C, Ivy DD, Claussen L, Chen SJ, Shandas R. Application of a microstructural constitutive model of the pulmonary artery to patient-specific studies: validation and effect of orthotropy. J Biomech Eng 129: 193–201, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang YH, Dunn ML, Drexler ES, McCowan CN, Slifka AJ, Ivy DD, Shandas R. A microstructural hyperelastic model of pulmonary arteries under normo- and hypertensive conditions. Ann Biomed Eng 33: 1042–1052, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Zulliger MA, Fridez P, Hayashi K, Stergiopulos N. A strain energy function for arteries accounting for wall composition and structure. J Biomech 37: 989–1000, 2004. [DOI] [PubMed] [Google Scholar]