

Abstract
This study determined if altered vascular prostacyclin (PGI2) and/or thromboxane A2 (TxA2) production with reduced Po2 contributes to impaired hypoxic dilation of skeletal muscle resistance arterioles of obese Zucker rats (OZRs) versus lean Zucker rats (LZRs). Mechanical responses were assessed in isolated gracilis muscle arterioles following reductions in Po2 under control conditions and following pharmacological interventions inhibiting arachidonic acid metabolism and nitric oxide synthase and alleviating elevated vascular oxidant stress. The production of arachidonic acid metabolites was assessed using pooled arteries from OZRs and LZRs in response to reduced Po2. Hypoxic dilation, endothelium-dependent in both strains, was attenuated in OZRs versus LZRs. Nitric oxide synthase inhibition had no significant impact on hypoxic dilation in either strain. Cyclooxygenase inhibition dramatically reduced hypoxic dilation in LZRs and abolished responses in OZRs. Treatment of arterioles from OZRs with polyethylene glycol-superoxide dismutase improved hypoxic dilation, and this improvement was entirely cyclooxygenase dependent. Vascular PGI2 production with reduced Po2 was similar between strains, although TxA2 production was increased in OZRs, a difference that was attenuated by treatment of vessels from OZRs with polyethylene glycol-superoxide dismutase. Both blockade of PGH2/TxA2 receptors and inhibition of thromboxane synthase increased hypoxic dilation in OZR arterioles. These results suggest that a contributing mechanism underlying impaired hypoxic dilation of skeletal muscle arterioles of OZRs may be an increased vascular production of TxA2, which competes against the vasodilator influences of PGI2. These results also suggest that the elevated vascular oxidant stress inherent in metabolic syndrome may contribute to the increased vascular TxA2 production and may blunt vascular sensitivity to PGI2.
Keywords: skeletal muscle microcirculation, endothelium-dependent dilation, vascular reactivity, rodent models of obesity
metabolic syndrome represents a series of systemic pathologies that develop sequentially in afflicted individuals and can include obesity, insulin resistance/type II diabetes mellitus, dyslipidemia, and hypertension (25). While each of these pathologies in isolation can increase the future risk for the development of peripheral vascular disease, when present in combination, this risk increases dramatically (24) and can lead to numerous profound alterations to vascular structure/function relationships (26, 27). These vascular alterations can impair tissue perfusion-demand matching and can lead to a compromised function (27). An effective animal model for metabolic syndrome in humans is the obese Zucker rat (OZR), a rodent model characterized by its dysfunctional leptin receptor gene, resulting in abrogated leptin signaling and an impaired satiety reflex (10). As a result, the OZR experiences a chronic hyperphagia and sequentially develops each of the systemic pathologies listed above in addition to both prooxidant and proinflammatory states (1, 6, 22). Previous studies by multiple investigative groups have demonstrated negative vascular outcomes in OZRs with the development of metabolic syndrome, including alterations to vascular wall mechanics (4, 13, 28, 29), impairments to multiple indexes of dilator reactivity (9, 13, 15, 16, 33), signaling mechanisms underlying constrictor reactivity (17, 21, 30), and a rarefaction of microvascular networks within multiple tissues (7, 8, 31). The culmination of these alterations to microvascular structure and function within OZRs can result in profound impairments to the perfusion of tissue under an array of physiological and pathological conditions (8, 12, 33).
We have previously demonstrated that the dilator reactivity of skeletal muscle resistance arterioles in response to an acute reduction in Po2 is impaired in OZRs compared with lean Zucker rats (LZRs) and that a contributing mechanism to this impairment lies within the chronic elevation in vascular oxidant stress (9). However, while this previous study did not implicate specific mechanisms beyond the contribution of elevated vascular oxidant stress, recent studies from Hester and colleagues (32, 33) have provided data that may have considerable bearing on not only our previous observation but also on the integrated regulation of vascular reactivity in the skeletal muscle of OZRs. Specifically, the dilator response of in situ spinotrapezius muscle arterioles with increased metabolic demand, previously determined to be largely dependent on the vascular production of prostacyclin (PGI2) (11), was blunted in OZRs compared with LZRs. This observation may reflect an enhanced vascular production of thromboxane A2 (TxA2) or an elevated vasoconstrictor response to TxA2 within OZRs that is not present in lean animals (32, 33). Clearly, any increase in TxA2 production has the potential to compete against PGI2-mediated responses and impair dilator reactivity to multiple vasoactive stimuli.
The purpose of the present study was to more fully elucidate mechanisms contributing to the impaired dilation of skeletal muscle arterioles of OZR in response to reduced Po2. Additionally, the present study was designed to more clearly determine the role of elevated vascular oxidant stress in contributing to the attenuated hypoxic dilation of arterioles from OZRs. Specifically, the hypothesis tested by the present experiments was that the compromised dilator reactivity of skeletal muscle arterioles of OZRs in response to reduced Po2 compared with responses in LZRs is due to a reduction in the vascular production of PGI2 and an increased vascular production of TxA2 as a result of the acute reductions in Po2. Furthermore, these effects on PGI2 and TxA2 production will be the result of an elevation in vascular oxidant stress, altering arachidonic acid metabolism within arterioles of OZRs.
MATERIALS AND METHODS
Animals.
Male LZRs and OZRs (15–17 wk old) were used for all experiments. Rats were fed standard chow and tap water ad libitum, and all protocols received prior Institutional Animal Care and Use Committee approval. After an overnight fast, rats were anesthetized with injections of pentobarbital sodium (50 mg/kg ip) and received tracheal intubation to facilitate maintenance of a patent airway. In all rats, a carotid artery and an external jugular vein were cannulated for the determination of arterial pressure and for the intravenous infusion of supplemental anesthetic, if necessary. While under anesthetic, an aliquot of blood was drawn from the jugular vein to be used for the biochemical determination of plasma glucose (Freestyle, Abbott Diabetes Care, Alameda, CA), insulin (Linco, St. Charles, MO), and nitrotyrosine concentrations (Linco, St. Charles, MO) as well as a plasma lipid profile (Stanbio, Boerne, TX) from each animal.
Preparation of isolated skeletal muscle resistance arterioles.
In anesthetized rats, the intramuscular continuation of the gracilis artery was identified and surgically removed. Arterioles were placed in a heated chamber (37°C) that allowed the vessel lumen and exterior to be perfused and superfused, respectively, with physiological salt solution (PSS; equilibrated with 21% O2-5% CO2-74% N2) from separate reservoirs. Vessels were cannulated at both ends and were secured to inflow and outflow glass micropipettes connected to a reservoir perfusion system allowing intralumenal pressure and gas concentrations to be controlled. Arterioles were extended to their in situ length and were equilibrated at 80% of the animal's mean arterial pressure (Table 1). Vessel diameters were measured using television microscopy and an on-screen video micrometer, and all mechanical responses of arterioles were assessed under pressurized conditions with no flow through the arteriolar lumen.
Table 1.
Baseline characteristics of LZRs and OZRs and for isolated arterioles from LZRs and OZRs in the present study
| LZRs | OZRs | |
|---|---|---|
| Body weight, g | 361±7 | 664±9* |
| Mean arterial pressure, mmHg | 108±4 | 124±4* |
| Blood glucose concentration, mg/dl | 102±5 | 184±11* |
| Plasma insulin concentration, ng/ml | 1.3±0.3 | 7.5±0.5* |
| Plasma total cholesterol concentration, mg/dl | 89±9 | 137±11* |
| Plasma triglyceride concentration, mg/dl | 154±10 | 367±22* |
| Plasma nitrotyrosine, ng/ml | 15±4 | 58±7* |
| Inner diameter − active diameter, μm | 104±4 | 102±5 |
| Inner diameter − passive diameter, μm | 172±5 | 156±4* |
| Active tone, % | 39±2 | 35±3 |
Values are means ± SE. LZRs, lean Zucker rats; OZRs, obese Zucker rats.
P < 0.05 vs. LZRs.
Subsequent to the initial equilibration period, the reactivity of isolated arterioles was assessed following a challenge with reduced Po2 [change in Po2 from ∼135 mmHg (21% O2) to ∼45 mmHg (0% O2)] under an array of physiological and pharmacological conditions (described below). In an additional series of experiments, isolated arterioles were also challenged with increasing concentrations of the selective TxA2 mimetic U-46619 (10−12–10−8 M, BioMol) and prostacyclin (PGI2·Na, 10−12–10−8 M, BioMol) to determine the intrinsic sensitivity of microvessels to these stimuli.
Removal of the arteriolar endothelium was accomplished by passing an air bolus through the perfusate line into the isolated microvessel, the efficacy of which was determined from a loss of all dilator reactivity in response to an application of 10−6 M acetylcholine. To assess the contribution of nitric oxide (NO) production or the generation of metabolites via cyclooxygenase (COX) as mediators of arteriolar reactivity, isolated vessels were treated with the NO synthase (NOS) inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 10−4 M, Sigma) or the COX inhibitor indomethacin (Indo; 10−5 M, Sigma), respectively. To antagonize vascular PGH2/TxA2 receptors, vessels were treated with SQ-29548 (10−5 M, BioMol), whereas inhibition of thromboxane synthase was accomplished using carboxyheptyl imidazole (CHI; 10−5 M, BioMol). To reduce vascular oxidant stress, arterioles were treated with polyethylene glycol-superoxide dismutase (PEG-SOD; 200 U/ml, Sigma).
Determination of vascular metabolites of arachidonic acid.
Vascular production of 6-keto-PGF1α [the stable breakdown product of PGI2 (18, 23)] and 11-dehydro-thromboxane B2 [11-dehydro-TxB2; the stable plasma breakdown product of TxA2 (Ref. 5)] in response to a challenge with reduced Po2 was assessed using pooled conduit arteries (femoral, saphenous, and iliac arteries) from LZRs and OZRs. Pooled vessels from each animal were incubated in microcentrifuge tubes in 1 ml PSS for 30 min under control conditions (21% O2). After this time, the superfusate was removed, stored in a new microcentrifuge tube, and frozen in liquid N2 while a new aliquot of PSS was added to the vessels and the equilibration gas was switched to 0% O2 for the subsequent 30 min. After the second 30-min period, this new PSS was transferred to a fresh tube, frozen in liquid N2, and stored at −80°C. Metabolite release by the vessels was determined using commercially available EIA kits for 6-keto-PGF1α and 11-dehydro-TxB2 (Cayman Chemicals).
Data and statistical analyses.
The active tone of individual arterioles at the equilibration pressure was calculated as (ΔD/Dmax) × 100, where ΔD is the diameter increase from rest in response to Ca2+-free PSS and Dmax is the maximum diameter measured at the equilibration pressure in Ca2+-free PSS.
Mechanical responses of isolated arterioles following challenge with increasing concentrations of thromboxane or prostacyclin were fit with the following three-parameter logistic equation:
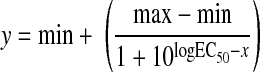 |
where y is the change in arteriolar diameter; “min” and “max” are the minimum and maximum bounds, respectively, of the change in arteriolar diameter with increasing agonist concentration; x is the logarithm of the agonist concentration; and log EC50 is the logarithm of the agonist concentration (x) at which the response (y) is halfway between the minimum and maximum bounds.
Data are presented as means ± SE. Statistically significant differences in the magnitude of hypoxic dilation, vascular production of 6-keto-PGF1α and 11-dehydro-TxB2, and the calculated parameters describing the thromboxane or prostacyclin concentration-response relationships were determined using ANOVA. In all cases, the Student-Newman-Keuls post hoc test was used when appropriate, and P < 0.05 was taken to reflect statistical significance.
RESULTS
Table 1 shows baseline characteristics of LZRs and OZRs in the present study. At 15–17 wk of age, OZRs demonstrated profound obesity, severe insulin resistance, and dyslipidemia characterized by moderate hypercholesterolemia and severe hypertriglyceridemia. In addition, OZRs also presented a moderate elevation in mean arterial pressure compared with LZRs. Plasma levels of nitrotyrosine, a protein marker of chronic elevations in oxidant stress, were significantly elevated in OZRs compared with LZRs. In regard to basal vascular tone, isolated arterioles from both rat strains demonstrated a comparable resting active diameter, although passive diameter was reduced in OZRs versus LZRs. However, this remodeling of the vessel wall did not translate into a difference in calculated active tone between the strains.
Data summarizing the hypoxic dilation of resistance arterioles from LZRs and OZRs are shown in Fig. 1. As shown in Fig. 1A, arterioles from OZRs exhibited a blunted dilator response to reduced Po2 compared with vessels from LZRs. However, arteriolar reactivity to reduced Po2 was abolished in both groups following removal of the vascular endothelium. Figure 1B shows data describing the contribution of NOS and COX products to arteriolar dilation in response to reduced Po2 in LZRs and OZRs. Whereas NOS inhibition had a consistently minor, albeit statistically insignificant, blunting of hypoxic dilation in arterioles of LZRs, treatment of vessels with l-NAME had no discernible impact on this response in OZRs. In contrast, incubation of vessels with Indo dramatically reduced hypoxic dilation in arterioles from both strains. Combined treatment with l-NAME and Indo abolished all vascular responses to reduced Po2 in both LZRs and OZRs. Figure 1C shows the impact of pretreatment of arterioles with the antioxidant PEG-SOD on both the magnitude of hypoxic dilation in these vessels and the contributions from NOS and COX. Treatment of vessels with PEG-SOD had no significant impact on either the magnitude of hypoxic dilation or the contribution of NOS and COX products to this response in LZRs. In contrast, following incubation with PEG-SOD, arterioles from OZRs exhibited an improved dilation in response to reduced Po2, and this enhanced reactivity was entirely dependent on COX metabolism.
Fig. 1.

Dilator reactivity of isolated skeletal muscle resistance arterioles from lean Zucker rats (LZRs) and obese Zucker rats (OZRs) in response to acute reductions in Po2. Data, presented as means ± SE, are shown for arterioles under control conditions and following removal of the vascular endothelium using air bolus perfusion (A); inhibition of nitric oxide synthase (NOS) with NG-nitro-l-arginine methyl ester (l-NAME) and/or inhibition of cyclooxygenase (COX) with indomethacin (Indo; B); and treatment with l-NAME and/or Indo following incubation of the arteriole with the antioxidant polyethylene glycol-superoxide dismutase (PEG-SOD; C). Please see the text for complete details. *P < 0.05 vs. control responses in that strain; †P < 0.05 vs. responses in LZR controls; ‡P < 0.05 vs. responses in OZRs + PEG-SOD.
Figure 2 shows data describing the production of 6-keto-PGF1α (the stable breakdown product of PGI2) and 11-dehydro-TxB2 (the stable breakdown product of TxA2) from pooled vessels of LZRs and OZRs in response to reduced Po2. As shown in Fig. 2A, vessels from both LZRs and OZRs demonstrated a significant increase in 6-keto-PGF1α production in response to reduced Po2, although this increase was greater in LZRs compared with OZRs. In both cases, treatment of vessels with Indo abolished 6-keto-PGF1α production in response to reduced Po2. In regard to the vascular production of TxA2, vessels from both LZRs and OZRs exhibited a significant increase in 11-dehydro-TxB2 production following an exposure to reduced Po2, although this level of production was dramatically elevated in vessels from OZRs (Fig. 2B). In both cases, the production of 11-dehydro-TxB2 was abolished following treatment of vessels with either Indo or CHI, suggesting that all production of TxA2 was mediated via the actions of thromboxane synthase distal to COX.
Fig. 2.

Vascular production of 6-keto-PGF1α (A; as an estimate of PGI2) and 11-dehydro-thromboxane B2 [11-dehydro-TxB2; B; as an estimate of thromboxane A2 (TxA2)] by pooled arteries of LZRs and OZRs in response to an acute reduction in Po2. Data, presented as means ± SE, are shown for arteries under control conditions and after pharmacological inhibition of COX with Indo, and, for 11-dehydro-TxB2, thromboxane synthase with carboxyheptyl imidazole (CHI). *P < 0.05 vs. control (21% O2) in that strain; †P < 0.05 vs. 0% O2 in that strain; ‡P < 0.05 vs. responses in LZRs (0% O2).
Data describing the production of 6-keto-PGF1α and 11-dehydro-TxB2 in response to reduced Po2 in vessels from OZRs subsequent to pretreatment with PEG-SOD are shown in Fig. 3. Treatment of vessels with PEG-SOD did not alter 6-keto-PGF1α production in vessels from OZRs compared with levels in untreated vessels (Fig. 3A). However, compared with the levels determined in untreated vessels, incubation of arteries from OZRs with PEG-SOD blunted the enhanced production of 11-dehydro-TxB2 (Fig. 3B). Treatment of vessels with Indo abolished the reduced Po2-induced production of 6-keto-PGF1α and 11-dehydro-TxB2.
Fig. 3.

Vascular production of 6-keto-PGF1α (A; as an estimate of PGI2) and 11-dehydro-TxB2 (B; as an estimate of TxA2) by pooled arteries from OZRs in response to an acute reduction in Po2. Data, presented as means ± SE, are shown for arteries under control conditions, following treatment of arteries with the antioxidant PEG-SOD, and following pharmacological inhibition of COX with Indo. *P < 0.05 vs. responses under control (21% O2) conditions; †P < 0.05 vs. responses under 0% O2; ‡P < 0.05 vs. responses determined under 0% O2 conditions + PEG-SOD.
Figure 4 shows data summarizing the dilator responses of arterioles from LZRs and OZRs in response to reduced Po2 following antagonism of either the PGH2/TxA2 receptor with SQ-29548 or thromboxane synthase with CHI. While neither of these pharmacological interventions had a consistent and significant impact on response in vessels from LZRs, both SQ-29548 and CHI significantly increased hypoxic dilation in arterioles from OZRs.
Fig. 4.

Dilator reactivity of isolated skeletal muscle resistance arterioles of LZRs and OZRs in response to acute reductions in Po2. Data (means ± SE) are presented for each strain under control conditions and following pharmacological inhibition of the PGH2/TxA2 receptor with SQ-29548 or thromboxane synthase with CHI. *P < 0.05 vs. LZR controls; †P < 0.05 vs. OZR controls.
Arteriolar reactivity in response to challenges with increasing concentrations of thromboxane or prostacyclin is shown in Fig. 5. Arteriolar constriction in response to increasing concentrations of thromboxane was not different between LZRs and OZRs, and neither EC50 nor maximum bound were impacted in response to pretreatment of the vessels with PEG-SOD (Fig. 5A). Furthermore, arteriolar responses to thromboxane were abolished in vessels from both animal strains following incubation of the vessels with SQ-29548 (Fig. 5B). With increasing concentrations of prostacyclin, arterioles from OZRs exhibited a blunted dilator response compared with that determined in arterioles from LZRs, and treatment of vessels with PEG-SOD significantly improved dilator reactivity to prostacyclin, although not to the level determined in LZRs (Fig. 5C). EC50 was not significantly different between groups in regard to vascular reactivity in response to increasing concentrations of PGI2 (data not shown).
Fig. 5.

Vascular reactivity of isolated skeletal muscle resistance arterioles of LZRs and OZRs (means ± SE) in response to increasing concentrations of thromboxane under control conditions and following treatment of vessels with PEG-SOD (A) or SQ-29548 (B) as well as increasing concentrations of prostacyclin under control conditions and following treatment with PEG-SOD (C). The term “max” represents the maximum bound (the maximum change) in vessel diameter in response to increasing concentrations of either thromboxane or prostacyclin, which was estimated from the logistic regression equation in materials and methods. *P < 0.05 vs. control responses within that strain; †P < 0.05 vs. responses in LZRs under control conditions.
DISCUSSION
Inherent within the development of the multipathology state that defines metabolic syndrome is an array of alterations to vascular and microvascular structure and function that have the potential to profoundly impact the perfusion of tissues and organs as well as the contributing mechanisms that comprise this integrated process. Previous studies have clearly demonstrated that alterations to the patterns of vasodilator (8, 12) and vasoconstrictor (30) reactivity as well as vessel wall remodeling (28, 29) and a reduction in microvessel density (7) all represent avenues through which vascular function and tissue perfusion can be compromised. The purpose of the present study was to build on our previous observation of an impaired dilation of skeletal muscle resistance arterioles of OZRs in response to acute reductions in Po2 and the possible contributing role of elevated vascular oxidant stress in this process (9).
Comparable with our observations in the first study, hypoxic dilation of skeletal muscle resistance arterioles from OZRs was significantly reduced compared with that determined in LZRs (Fig. 1A). While this response was abolished following removal of the endothelium in both rat strains, comparable with the complete endothelium dependence of hypoxic dilation in other rodent models (14, 19, 20), our results also support previous observations of a strong dependence on COX products in mediating hypoxic dilation of these vessels (18–20). Interestingly, in our initial study, we determined a small but significant role for NOS activity in mediating hypoxic dilation in arterioles from LZRs. In the present study, this effect was not statistically significant, suggesting that it may represent a minor contributor to arteriolar dilation in response to reduced Po2 in LZRs. However, while the magnitude of this response is relatively minor in LZRs, both the results from the present study and our previous one (9) indicate that this is completely lost with the development of metabolic syndrome in OZRs. Additionally, pretreatment of arterioles from OZRs with the oxidative free radical scavenger PEG-SOD to lower the elevated oxidant stress in these animals resulted in a significant improvement to hypoxic dilation in OZRs (Fig. 1C). This improvement to hypoxic dilation was confined to activity involving COX only, as treatment with Indo abolished hypoxic dilation in OZRs and treatment with l-NAME was without effect.
Recent studies from Hester and colleagues (32, 33) have suggested that functional dilation (i.e., arteriolar dilation in response to elevated metabolic demand) is impaired in OZRs as a result of alterations to arachidonic acid metabolism, which can result in the activation of thromboxane receptors, causing a competing constrictor influence that acts to partially constrain dilator responses. Given that both functional dilation for in situ spintotrapezius muscle of LZRs (11) and hypoxic dilation of skeletal muscle arterioles of LZRs (9) have previously been shown to be strongly dependent on the vascular production of PGI2, we sought to determine if an alteration in arachidonic acid metabolism, resulting in an increased vascular production of thromboxane or a decreased production of prostacyclin, may contribute to this impaired reactivity in arterioles of OZRs. As shown in Fig. 2, arteries from OZRs exhibited a significant increase in the production of PGI2 (estimated from measurements of 6-keto-PGF1α) in response to reductions in Po2. While this was not as robust a response as that determined for arteries of LZRs, it is unclear if this degree of attenuation in PGI2 production in OZRs is sufficient to manifest itself as a blunted mechanical response. In contrast, vascular production of TxA2 (estimated from measurements of 11-dehydro-TxB2), mildly elevated in arteries of LZR during reduced Po2, was dramatically increased in vessels from OZRs following exposure to reduced Po2. Finally, while treatment of arteries from both strains with Indo abolished the production of PGI2 and TxA2 in response to reduced Po2, treatment of vessels from OZRs with CHI, an inhibitor of thromboxane synthase, severely reduced thromboxane production in vessels from OZRs (Fig. 2B). These results suggest that reduced Po2 causes an increased production of TxA2 from vessels of OZRs, mediated via thromboxane synthase, and that this may compete with the dilator influences of vascular production of PGI2. However, it should be emphasized that these results must be interpreted cautiously, as the production of metabolites of arachidonic acid was assessed using conduit arteries and not resistance arterioles, whereas the reverse was true in regard to the study of the mechanical responses.
To better understand the impact of elevated vascular oxidant stress on the impaired dilator responses to reduced Po2 in vessels of OZRs, we treated vessels with PEG-SOD prior to exposure to reduced Po2 and determined the levels of 6-keto-PGF1α and 11-dehydro-TxB2 in the incubation superfusate (Fig. 3). These data suggest that while oxidant stress does not play a significant role in any reduction in the vascular production of prostacyclin (as the addition of PEG-SOD was without effect), elevated vascular oxidant stress may contribute to the increased production of thromboxane by these vessels, as incubation with the antioxidant significantly reduced the levels of 11-dehydro-TxB2 in the superfusate. However, this effect was not complete, as the levels of 11-dehydro-TxB2 production remained elevated in the superfusate despite the presence of PEG-SOD, indicating that additional factors may contribute to thromboxane production in vessels of OZRs that are independent of the effects of an acute reduction in vascular oxidant stress. This observation of a role for oxidant stress in shifting arachidonic acid metabolism toward an increased production of thromboxane has been identified previously (2, 3, 34), and the results of the present study suggest that a comparable effect may be occurring in the vasculature of OZRs, with the net result of a blunted vascular response to stimuli that are dependent on PGI2 production for their full manifestation.
To better evaluate this statement, isolated skeletal muscle resistance arterioles from LZRs and OZRs were exposed to acute reductions in Po2 under control conditions and in response to either PGH2/TxA2 receptor blockade with SQ-29548 or thromboxane synthase inhibition with CHI (Fig. 4). Although these pharmacological challenges had no consistent impact on hypoxic dilation in arterioles from LZRs, both interventions resulted in improved dilator reactivity in response to reduced Po2 in arterioles of OZRs. Taken with the previous results, these data clearly suggest that increased production of TxA2 contributes to the impaired hypoxic dilation in arterioles of OZRs. However, the data shown in Fig. 4 do not allow for discrimination between increased vascular production of TxA2, an increased vascular sensitivity to produced TxA2, or an altered vascular sensitivity to produced PGI2.
To assess this final issue, isolated arterioles from both rat strains were challenged with increasing concentrations of thromboxane or prostacyclin (Fig. 5). Vasoconstrictor reactivity to thromboxane was very similar between arterioles of LZRs and OZRs, and, in both cases, constrictor responses to thromboxane were largely independent of oxidant stress (i.e., no identifiable impact of treatment with PEG-SOD) and were abolished by blockade of the PGH2/TxA2 receptor. These observations suggest that the vascular sensitivity to thromboxane is not significantly impacted by the presence of metabolic syndrome. In contrast, vascular reactivity to prostacyclin was significantly reduced in arterioles of OZRs, and this impairment was blunted following a reduction in oxidant stress with PEG-SOD. Whether this impaired response to PGI2 represents oxidant radical degradation of prostacyclin (leading to the production of isoprostanes), altered function at the level of the prostacyclin receptor, the impact of elevated oxidant stress on the intracellular signaling cascade distal to the receptor, or a combination of these effects remains to be determined.
In summary, with the evolution of metabolic syndrome in OZRs, the dilator responses of skeletal muscle resistance arterioles following acute reductions in Po2 are significantly attenuated. Both biochemical and pharmacological evidence suggests that this impaired dilator reactivity may be the result of an increase in vascular production of thromboxane with reduced Po2, which could represent a constrictor influence that competes against the dilator effects of prostacyclin (the production of which appears to be largely intact). Normalization of vascular oxidant stress blunts the increased reduced Po2-induced production of thromboxane in vessels from OZRs and also increases the responsiveness of arterioles from OZRs to exogenously supplied prostacyclin, thus leading to an improvement in the mechanical response of the vessel to reduced Po2. The present study provides no compelling evidence that skeletal muscle arteriolar sensitivity to thromboxane is altered with the progression of metabolic syndrome.
GRANTS
The authors gratefully acknowledge the support provided through the “Translational Research Initiative: Cardiorespiratory Health in Appalachia–From Mechanisms to Policy” at the West Virginia University Health Sciences Center in the performance of this study. This study was supported by American Heart Association Grant SDG 0330194N and National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-64668.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Asghar M, Monjok E, Kouamou G, Ohia SE, Bagchi D, Lokhandwala MF. Super CitriMax (HCA-SX) attenuates increases in oxidative stress, inflammation, insulin resistance, and body weight in developing obese Zucker rats. Mol Cell Biochem 304: 93–99, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Bachschmid M, Schildknecht S, Ullrich V. Redox regulation of vascular prostanoid synthesis by the nitric oxide-superoxide system. Biochem Biophys Res Commun 338: 536–542, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Bachschmid M, Thurau S, Zou MH, Ullrich V. Endothelial cell activation by endotoxin involves superoxide/NO-mediated nitration of prostacyclin synthase and thromboxane receptor stimulation. FASEB J 17: 914–916, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Bouvet C, de Chantemèle EB, Guihot AL, Vessières E, Bocquet A, Dumont O, Jardel A, Loufrani L, Moreau P, Henrion D. Flow-induced remodeling in resistance arteries from obese Zucker rats is associated with endothelial dysfunction. Hypertension 50: 248–254, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Catella F, Healy D, Lawson JA, FitzGerald GA. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc Natl Acad Sci USA 83: 5861–5865, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominguez J, Wu P, Packer CS, Temm C, Kelly KJ. Lipotoxic and inflammatory phenotypes in rats with uncontrolled metabolic syndrome and nephropathy. Am J Physiol Renal Physiol 293: F670–F679, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Frisbee JC, Samora JB, Peterson J, Bryner R. Exercise training blunts microvascular rarefaction in the metabolic syndrome. Am J Physiol Heart Circ Physiol 291: H2483–H2492, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Frisbee JC, Delp MD. Vascular function in the metabolic syndrome and the effects on skeletal muscle perfusion: lessons from the obese Zucker rat. Essays Biochem 42: 1451–1461, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Frisbee JC Impaired dilation of skeletal muscle microvessels to reduced oxygen tension in diabetic obese Zucker rats. Am J Physiol Heart Circ Physiol 281: H1568–H1574, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Guerre-Millo M Regulation of ob gene and overexpression in obesity. Biomed Pharmacother 51: 318–323, 1997. [DOI] [PubMed] [Google Scholar]
- 11.Hammer LW, Ligon AL, Hester RL. Differential inhibition of functional dilation of small arterioles by indomethacin and glibenclamide. Hypertension 37: 599–603, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Hodnett BL, Hester RL. Regulation of muscle blood flow in obesity. Microcirculation 14: 273–288, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Jonk AM, Houben AJ, de Jongh RT, Serné EH, Schaper NC, Stehouwer CD. Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiology (Bethesda) 22: 252–260, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Kerkhof CJ, Bakker EN, Sipkema P. Role of cytochrome P-450 4A in oxygen sensing and NO production in rat cremaster resistance arteries. Am J Physiol Heart Circ Physiol 277: H1546–H1552, 1999. [DOI] [PubMed] [Google Scholar]
- 15.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113: 1888–1904, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Knudson JD, Dincer UD, Bratz IN, Sturek M, Dick GM, Tune JD. Mechanisms of coronary dysfunction in obesity and insulin resistance. Microcirculation 14: 317–338, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Lesniewski LA, Donato AJ, Behnke BJ, Woodman CR, Laughlin MH, Ray CA, Delp MD. Decreased NO signaling leads to enhanced vasoconstrictor responsiveness in skeletal muscle arterioles of the ZDF rat prior to overt diabetes and hypertension. Am J Physiol Heart Circ Physiol 294: H1840–H1850, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Fredricks KT, Roman RJ, Lombard JH. Response of resistance arteries to reduced Po2 and vasodilators during hypertension and elevated salt intake. Am J Physiol Heart Circ Physiol 273: H869–H877, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Lombard JH, Liu Y, Fredricks KT, Bizub DM, Roman RJ, Rusch NJ. Electrical and mechanical responses of rat middle cerebral arteries to reduced Po2 and prostacyclin. Am J Physiol Heart Circ Physiol 276: H509–H516, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Messina EJ, Sun D, Koller A, Wolin MS, Kaley G. Role of endothelium-derived prostaglandins in hypoxia-elicited arteriolar dilation in rat skeletal muscle. Circ Res 71: 790–796, 1992. [DOI] [PubMed] [Google Scholar]
- 21.Naik JS, Xiang L, Hester RL. Enhanced role for RhoA-associated kinase in adrenergic-mediated vasoconstriction in gracilis arteries from obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 290: R154–R161, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Naka Y, Bucciarelli LG, Wendt T, Lee LK, Rong LL, Ramasamy R, Yan SF, Schmidt AM. RAGE axis: animal models and novel insights into the vascular complications of diabetes. Arterioscler Thromb Vasc Biol 24: 1342–1349, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Nies AS Prostaglandins and the control of the circulation. Clin Pharmacol Ther 39: 481–488, 1986. [DOI] [PubMed] [Google Scholar]
- 24.Rosin BL The progression of cardiovascular risk to cardiovascular disease. Rev Cardiovasc Med 8: S3–S8, 2007. [PubMed] [Google Scholar]
- 25.Saely CH, Rein P, Drexel H. The metabolic syndrome and risk of cardiovascular disease and diabetes: experiences with the new diagnostic criteria from the International Diabetes Federation. Horm Metab Res 39: 642–650, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Shammas NW Epidemiology, classification, and modifiable risk factors of peripheral arterial disease. Vasc Health Risk Manag 3: 229–234, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shammas NW, Dippel EJ. Evidence-based management of peripheral vascular disease. Curr Atheroscler Rep 7: 358–363, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Sista AK, O'Connell MK, Hinohara T, Oommen SS, Fenster BE, Glassford AJ, Schwartz EA, Taylor CA, Reaven GM, Tsao PS. Increased aortic stiffness in the insulin-resistant Zucker fa/fa rat. Am J Physiol Heart Circ Physiol 289: H845–H851, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Stepp DW, Pollock DM, Frisbee JC. Low-flow vascular remodeling in the metabolic syndrome X. Am J Physiol Heart Circ Physiol 286: H964–H970, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Stepp DW, Frisbee JC. Augmented adrenergic vasoconstriction in hypertensive diabetic obese Zucker rats. Am J Physiol Heart Circ Physiol 282: H816–H820, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Toblli JE, Cao G, DeRosa G, Di Gennaro F, Forcada P. Angiotensin-converting enzyme inhibition and angiogenesis in myocardium of obese Zucker rats. Am J Hypertens 17: 172–180, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Xiang L, Dearman J, Abram SR, Carter C, Hester RL. Insulin resistance and impaired functional vasodilation in obese Zucker rats. Am J Physiol Heart Circ Physiol 294: H1658–H1666, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Xiang L, Naik JS, Hodnett BL, Hester RL. Altered arachidonic acid metabolism impairs functional vasodilation in metabolic syndrome. Am J Physiol Regul Integr Comp Physiol 290: R134–R138, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Zou MH Peroxynitrite and protein tyrosine nitration of prostacyclin synthase. Prostaglandins Other Lipid Mediat 82: 119–127, 2007. [DOI] [PubMed] [Google Scholar]