

Abstract
Whole-genome hybridization studies have suggested that the nuclear genomes of accessions (natural strains) of Arabidopsis thaliana can differ by several percent of their sequence. To examine this variation, and as a first step in the 1001 Genomes Project for this species, we produced 15- to 25-fold coverage in Illumina sequencing-by-synthesis (SBS) reads for the reference accession, Col-0, and two divergent strains, Bur-0 and Tsu-1. We aligned reads to the reference genome sequence to assess data quality metrics and to detect polymorphisms. Alignments revealed 823,325 unique single nucleotide polymorphisms (SNPs) and 79,961 unique 1- to 3-bp indels in the divergent accessions at a specificity of >99%, and over 2000 potential errors in the reference genome sequence. We also identified >3.4 Mb of the Bur-0 and Tsu-1 genomes as being either extremely dissimilar, deleted, or duplicated relative to the reference genome. To obtain sequences for these regions, we incorporated the Velvet assembler into a targeted de novo assembly method. This approach yielded 10,921 high-confidence contigs that were anchored to flanking sequences and harbored indels as large as 641 bp. Our methods are broadly applicable for polymorphism discovery in moderate to large genomes even at highly diverged loci, and we established by subsampling the Illumina SBS coverage depth required to inform a broad range of functional and evolutionary studies. Our pipeline for aligning reads and predicting SNPs and indels, SHORE, is available for download at http://1001genomes.org.
The release of the first genome sequence of a plant, from a single inbred accession of Arabidopsis thaliana, was a major milestone for biology (The Arabidopsis Genome Initiative 2000). Apart from accelerating functional analyses and providing insights into gene content and genome evolution, the 119-Mb euchromatic reference sequence of the Columbia (Col-0) accession propelled A. thaliana to the forefront of efforts to understand microevolutionary processes, including the causes for the extensive phenotypic variation between accessions (Koornneef et al. 2004; Mitchell-Olds and Schmitt 2006). To characterize the corresponding patterns of whole-genome sequence variation, 20 accessions have recently been investigated using Perlegen high-density oligonucleotide arrays (Clark et al. 2007). Intriguingly, in the global sample used for the hybridization study, linkage disequilibrium (LD) decays to near background levels within 10 kb over much of the genome, although LD extends over moderately longer distances in regional population samples (Kim et al. 2007). These findings raise the possibility that LD (association) mapping can be used to localize the genetic causes for phenotypic variation to small chromosomal regions that include just a few genes.
While such efforts will often lead to the identification of haplotypes or alleles associated with a particular trait, they are generally insufficient to pinpoint causal sequence variants. Even in coding regions, only about half of all SNPs were detectable by array hybridization, and small deletions or insertions of any size were not identifiable at all (Clark et al. 2007). This is especially relevant, as indels commonly segregate in the global A. thaliana population (Nordborg et al. 2005), and have in some cases been linked to phenotypic differences among wild accessions (for review, see Mitchell-Olds and Schmitt 2006).
To develop tools for a more detailed understanding of genetic diversity in A. thaliana, we have examined the power of the Illumina sequencing-by-synthesis (SBS) technology (formerly known as Solexa sequencing) to describe sequence variation. While assembling short reads of ∼40 bp for a moderately sized genome presents a host of computational challenges, we show that even moderate coverage is sufficient to reveal a large fraction of sequence differences from read alignments. Using a targeted de novo assembly approach, we also demonstrate the potential of the method to resolve highly diverged regions (e.g., clusters of nearby polymorphisms or long indels) where hybridization-based techniques are typically uninformative.
Results
We employed the Illumina SBS technology to sequence two naturally inbred, and thus homozygous, A. thaliana accessions, Bur-0 and Tsu-1. To characterize the method, and to identify errors in the reference genome sequence (The Arabidopsis Genome Initiative 2000), we included the Col-0 accession in our study. The genetic distances of the two wild accessions to Col-0 are representative of much of the global population (Nordborg et al. 2005). Recombinant inbred lines (RILs) from crosses between Col-0 and Bur-0 (Simon et al. 2008) as well as Tsu-0, which is likely nearly identical to Tsu-1 as deduced from a genome-wide scan with 149 markers (Warthmann et al. 2007; http://naturalvariation.org/hapmap; http://dbsgap.versailles.inra.fr/vnat/Fichier_collection/Rech_rils_pop.php), have been established, making the Bur-0 and Tsu-1 sequences immediately useful in quantitative trait locus (QTL) cloning efforts with these RILs.
Bur-0, Tsu-1, and Col-0 have been sampled before by PCR and Sanger sequencing at 1213 fragments spaced throughout the genome (a total of 612–654 kb per accession), and the entire genomes have been analyzed with Perlegen resequencing arrays (Nordborg et al. 2005; Clark et al. 2007). We used these two data sets, the “dideoxy” and “resequencing array” data, in combination with the reference sequence as resources for our study. The Bur-0 and Tsu-1 seed stocks were the same as those used to generate the dideoxy and resequencing array data. The Col-0 lines (CS22625 and CS22681) examined in these studies were at most four generations apart.
Generation and alignment of SBS data to the reference genome
From genomic DNA libraries, we produced 120–173 million SBS reads per accession (Supplemental Methods). Most, 88%, were 36 bp in length, and the rest were 31 or 35 bp. Across all accessions, 75% of these raw reads were retained after applying a custom quality filter (Table 1). We aligned the filtered reads to the 119-Mb euchromatic Col-0 reference genome using the enhanced suffix array implementation Vmatch (http://www.vmatch.de; see Methods). We allowed up to four mismatches or indels of up to 3 bp, and reads were aligned through a series of increasing edit distances. A read that had been mapped at a given distance was removed from subsequent alignment attempts; where reads matched multiple genomic locations at a given distance, e.g., in repetitive regions, all alignments were recorded. For Col-0, 89% of filtered reads could be aligned to the reference sequence (Table 1).
Table 1.
Read, alignment, and coverage metrics
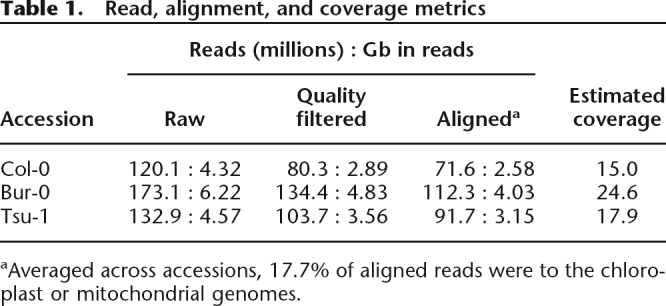
aAveraged across accessions, 17.7% of aligned reads were to the chloroplast or mitochondrial genomes.
Based on the reference sequence, we classified as repetitive any position for which all overlapping 36-bp sequences matched perfectly to multiple genome locations, and as moderately repetitive those for which at least one overlapping 36-bp sequence mapped to multiple locations. These sequences constitute 5% and 8% of the reference genome, respectively, leaving 87% (or 104 Mb) as nonrepetitive. For Col-0, 99.0% of nonrepetitive positions were covered by at least one read, and 95% were covered by three or more reads. Coverage depths by accession ranged from 15- to almost 25-fold (Table 1; Supplemental Methods).
We observed that read coverage was from about five- to 9500-fold higher than expected for 345–385 kb of the reference genome in a given accession (see Supplemental Methods). These regions, which corresponded to rDNA, centromeric, and other highly repetitive sequences whose copy number is not reflected by the reference genome assembly, were excluded from subsequent analyses. Disregarding these regions as mapping targets is expected to significantly reduce the runtime of our (or any related) read mapping method.
Read quality and coverage characteristics
For Col-0, nonperfect alignments resulted predominantly from sequencing errors. As assessed with uniquely aligned reads, incorrect base calls were infrequent at the 5′ end of reads, but increased to ∼5% at position 36 (Fig. 1A). In total, 67% of aligned reads were error-free, and 93% had at most two errors (Supplemental Fig. S1). By error type, insertion or deletion of bases accounted for <1% of disagreements with the reference (Table 2). Incorrectly called bases generally had low quality as measured by the probability (“prb”) and “chastity” values calculated by Illumina’s SolexaPipeline (Fig. 1B; Supplemental Fig. S2; see Methods).
Figure 1.

Errors by read position and prb values. (A) Distribution of observed errors by position in reads. (B) Relationship between prb values and observed errors with the frequency spectrum for prb values based on all called bases. All data are from uniquely aligned Col-0 reads.
Table 2.
Base call profiles as assessed with uniquely mapped Col-0 reads, before applying quality filtering

aDeleted bases, with 2-bp deletions counted twice (two “—“ instances) and 3-bp deletions counted three times (three “—“ instances) toward totals.
bInserted bases with counts analogous to those for deleted bases.
NA, Not applicable.
We also assessed coverage uniformity in nonrepetitive regions. Compared with the random (Poisson) expectation, a broader distribution in per-position coverage was observed in the Col-0 data (Supplemental Fig. S3). Coverage was positively correlated with local GC content, and the strongest correlation was observed with window sizes of ∼100 bp (Supplemental Fig. S4). This is longer than the read lengths we used, suggesting a potential bias during library construction using the protocol recommended by Illumina at the time (see Hillier et al. 2008 for a similar finding). Biases in base calling, although minor, may also have contributed to the observed patterns (Table 2). Similar relationships between local GC content and coverage were also apparent in the Bur-0 and Tsu-1 data (Supplemental Figs. S4, S5).
Optimization of sequence predictions
To optimize parameters for identification of polymorphic and conserved bases from the aligned reads, we used the genome-wide Bur-0 dideoxy data set, which covers 604 kb of the Bur-0 genome and includes 2806 SNPs and 326 indels of 1- to 3-bp in length at nonrepetitive positions (Nordborg et al. 2005; Clark et al. 2007). We examined the predictive power at coverage depths from about three- to 25-fold by subsampling the Bur-0 data (Table 1; Fig. 2).
Figure 2.

Performance evaluation for sequence predictions from aligned reads. Specificity (top) and sensitivity (bottom) for Bur-0 by genome coverage depth (see legend) for a range of minimum read supports. Approximate genome coverage estimates for each subsample of the data are based on the data in Table 1 (see also Supplemental Methods).
To lessen the ambiguities resulting from repeats, we predicted sequences only at nonrepetitive sites for which per-position read coverage was less than fivefold the expected sequencing depth (∼87% of the reference genome; see Supplemental Methods). Briefly, a sequence was predicted at a position subject to minimum read support and concordance of bases in the aligned reads (80% for base calls, 67% for indels). Minimum read support (Fig. 2) is the lowest number of reads required to accept a prediction for a given position, after reads with an aligned base of low quality at a position had been excluded, based on empirically determined prb and chastity score thresholds that removed ∼79% of sequencing errors while retaining ∼93% of all bases (see Methods). Moreover, as ambiguous alignments are inherently more likely at read ends (Supplemental Fig. S6), and because error rates are higher at the 3′ end of reads (Fig. 1A), we required all predictions to be supported by the core, which excluded four bases each at either end, of at least one read. When predictions were compared with the dideoxy data set, 16 false-positive SNP calls remained. The primary reason for these false predictions was ambiguous alignments at the edge of indel polymorphisms, and not the accumulation of sequence errors.
We used the dideoxy data set to examine the relationships between prediction specificity and sensitivity at different genome-wide coverage depths and minimum read support at a position (see Supplemental Table S1 for details). Even at about threefold coverage, specificity for SNPs was >99% when requiring a minimum read support of two, and more than half of all SNPs could be detected. Above ∼11-fold coverage, sensitivity increased only moderately, and reached 85% at ∼25-fold coverage. For 1-bp indels, which constitute half of the indels in the dideoxy data (Nordborg et al. 2005; Clark et al. 2007), the specificity and sensitivity of predictions was lower, with higher coverage needed to attain similar performance estimates (see also Supplemental Fig. S6). For 2- to 3-bp indels, 24 of 100 indels in the dideoxy data were correctly predicted with only two false predictions when employing a minimum read support of three and all data. For nonrepetitive bases identical to the Col-0 reference sequence, nearly 98.9% were predicted at a specificity of 99.985% when using all data and a minimum read support of three.
We also determined performance separately for coding and noncoding sequences (see Supplemental Methods). Coding regions are on average less polymorphic than noncoding regions (Nordborg et al. 2005; Clark et al. 2007), simplifying read alignment. Additionally, because our read coverage was higher in GC-rich regions (Supplemental Fig. S4), we would expect better performance in coding sequences, which tend to have higher GC content than noncoding sequences in A. thaliana (The Arabidopsis Genome Initiative 2000). Indeed, sensitivity for SNP prediction was higher for coding than for noncoding sequences (91% vs. 79%), with little difference in specificity (99.7% vs. 99.0%).
We obtained very similar performance estimates when applying our pipeline and parameter sets to the lower coverage Tsu-1 data (Supplemental Fig. S7).
Comparison with MAQ
For the Bur-0 data, we compared SHORE with the Mapping and Assembly with Quality (MAQ) software package (http://maq.sourceforge.net/), which also performs read alignment and SNP prediction, but identifies indels only from paired-end reads (Li et al. 2008). As input for MAQ, we supplied all Bur-0 reads together with their transformed prb values in FASTQ format (see Supplemental Information). Employing similar read mapping parameters as we used for SHORE (Supplemental Table S2), we predicted SNPs for regions of overlap to the Bur-0 dideoxy data, with and without MAQ’s SNPfilter option, which maximizes specificity (Table 3; see Supplemental Information; Supplemental Table S3).
Table 3.
Comparison of SHORE and MAQ for SNP prediction with all Bur-0 reads
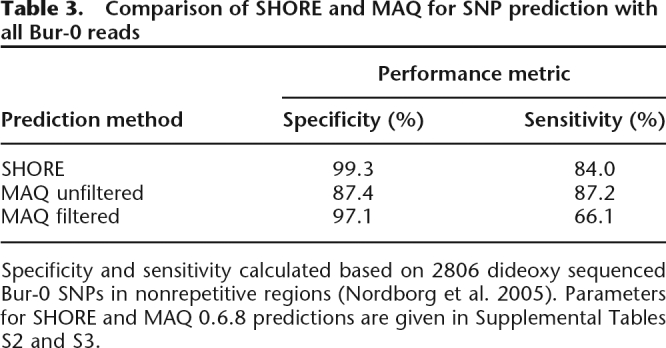
Specificity and sensitivity calculated based on 2806 dideoxy sequenced Bur-0 SNPs in nonrepetitive regions (Nordborg et al. 2005). Parameters for SHORE and MAQ 0.6.8 predictions are given in Supplemental Tables S2 and S3.
The error rate of MAQ-unfiltered predictions was >18-fold higher than those of SHORE, with only a small increase in sensitivity (Table 3; Supplemental Table S1). The MAQ SNPfilter option significantly decreased error rates, but they were still four times higher than those of SHORE, while sensitivity was then substantially lower (Table 3). SHORE therefore compares favorably with MAQ, at least for A. thaliana, which is considerably more polymorphic than human. One reason might be that SHORE, but not MAQ, allows for gapped alignments and prioritizes read core information. These optimizations reduce alignment ambiguities, e.g., mismatches where gaps should be introduced, which can lead to false SNP predictions. In addition, the standard implementation of SHORE does not rely on single read coverage, even if base qualities are optimal, thereby reducing the potential of false positives introduced by undetected sequencing errors.
Genome-wide predictions in the reference accession
Applying a minimum read support of three, we predicted SNPs, 1- to 3-bp indels, and reference bases genome-wide for each accession. For our Col-0 accession, which is closely related to the strain used for the reference sequence (The Arabidopsis Genome Initiative 2000), we predicted 1172 SNPs and 1287 indels. Although some of these predictions might be false, many identify potential errors in the reference sequence or reflect differences among the “Col-0” accessions used in various laboratories. Consistent with the latter possibilities, seven SNP and 11 indel predictions had also been identified in the 654 kb of Col-0 dideoxy data (Nordborg et al. 2005; Clark et al. 2007), for which we made only a single false prediction. Moreover, for 472 of the Col-0 SNP predictions, hybridization data were available from resequencing arrays (Clark et al. 2007). Here, the concordance with SBS calls was 97%. Finally, the majority of the Col-0 SNPs (82%) and indels (91%) were also predicted in either Bur-0 or Tsu-1. We conclude that many of these cases reflect errors in the reference sequence (Supplemental Table S4). To facilitate exclusion of potential reference errors in future sequencing projects, we provide these data in a dedicated Col-0 SNP database at http://1001genomes.org.
Genome-wide predictions in the divergent accessions
For Bur-0 and Tsu-1, we predicted 823,325 nonredundant SNPs and 79,961 nonredundant 1- to 3-bp indels after accounting for potential errors in the reference sequence (Table 4). In earlier work, we used resequencing array data for genome-wide SNP discovery at a specificity of ∼98% (Clark et al. 2007), with the resulting MBML2 data set serving as the basis for the A. thaliana 250 k SNP array design (Kim et al. 2007). The MBML2 data set contained SNP predictions for 153,218 and 120,524 positions in Bur-0 and Tsu-1, respectively, that we could also analyze with the Illumina SBS approach. At these positions, the concordance between the two methods was 99.0%.
Table 4.
SNPs, small indels, and reference base calls in nonrepetitive regions
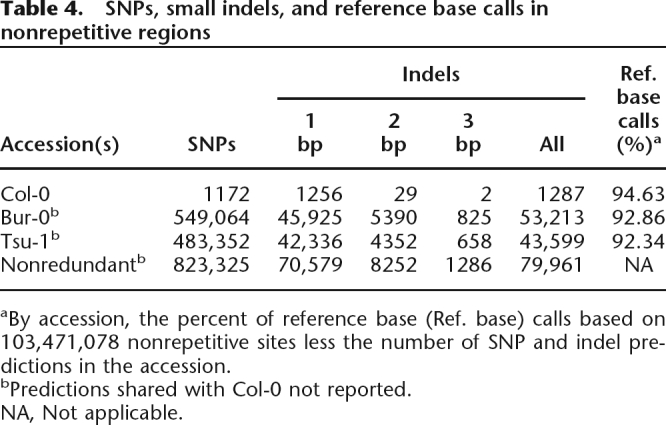
aBy accession, the percent of reference base (Ref. base) calls based on 103,471,078 nonrepetitive sites less the number of SNP and indel predictions in the accession.
bPredictions shared with Col-0 not reported.
NA, Not applicable.
With a modification to our base-calling algorithm (see Methods), we predicted 20,795 and 18,047 nonredundant SNPs in moderately repetitive positions for Bur-0 and Tsu-1, respectively, along with 1209 and 1026 1- to 3-bp indels (Supplemental Table S5). Moderately repetitive positions are largely absent from the dideoxy data obtained after PCR amplification (Nordborg et al. 2005), and the repeat composition in the dideoxy data differs from the genome at large, with a lower proportion of long repetitive tracks. Nevertheless, 56 of the 126 SNPs and indels in moderately repetitive regions that did overlap the dideoxy data were predicted correctly, with only three false predictions.
Identifying highly polymorphic regions and targets for de novo assembly
For Bur-0 and Tsu-1, 4.3 Mb of nonrepetitive or moderately repetitive regions in the reference genome were not covered by reads. In part, this is due to stochastic differences in coverage, although systematic biases, such as the reduced coverage of GC poor regions, likely contribute (Supplemental Fig. S3). However, low or no coverage can also be a consequence of sequences being highly dissimilar or deleted relative to the reference. To detect such polymorphic regions (PRs), we identified tracts of at least eight contiguous nonrepetitive or moderately repetitive positions covered by one or no read. Because of reduced read coverage for GC-poor regions, we made predictions only when GC content within PRs was at least 25% (see Methods).
For Col-0, 28 kb of the reference sequence was included in PRs (Table 5), and of this, 22 kb localized to eight genomic locations for which reduced or absent hybridization was apparent in the resequencing array data (Clark et al. 2007). Each of these regions overlapped PRs in Bur-0 and Tsu-1, and five of them overlapped known misassemblies in the reference genome that contain cloning vector sequences (Supplemental Table S6). In contrast to Col-0, 3.25 (3.13) Mb of sequences was included in PRs in Bur-0 (Tsu-1) (Table 5). As expected, these PRs strongly overlapped with a similar set of predictions made based on strongly reduced hybridization signal for Bur-0 or Tsu-1 on resequencing arrays (Supplemental Fig. S8; Zeller et al. 2008).
Table 5.
PRs by accession and length

We also identified short regions with fewer than eight contiguous positions with markedly reduced read coverage (see Methods). Many of these low-coverage tracks are expected to result from stochastic dips in coverage, and 80,912 were found in Col-0. Nonetheless, ∼1.6-fold more were identified in Bur-0 (134,544) and Tsu-1 (127,262). This suggests that many correspond to dissimilar sequences, such as clusters of SNPs, or insertions of any size. These regions, along with PRs, are therefore candidates for targeted de novo assembly (see below).
Targeted de novo assembly of dissimilar sequences
To characterize polymorphic sequences inaccessible to our mapping approach, we developed a targeted de novo assembly strategy that incorporates the Velvet short-read assembler (Zerbino and Birney 2008). As input, we included unmapped (leftover) reads of high quality and all reads that mapped to 100 bp of 5′ and 3′ flanking sequences of either PRs or low-coverage regions (Fig. 3; see Methods). To reduce the potential for incorrect assemblies, we excluded instances where >20% of flanking sequences were repetitive or had higher-than-expected coverage (see Methods). After assembly, we used several criteria to retain only high-confidence contigs spanning polymorphic regions. A contig was rejected if (1) it was <150 bp; (2) it harbored <80% of the 5′ and 3′ flanking reads for a given target; (3) it contained reads from unlinked flanking regions; or (4) <5% of the reads came from the pool of leftover reads (see Methods). The last criterion helped to identify contigs that resulted specifically from sequence dissimilarities, as it should exclude instances where reduced read coverage (one of the criteria used to select targets for de novo assembly) resulted solely from stochastic effects on coverage.
Figure 3.

Targeted de novo assembly. (A) Example of alignment of Bur-0 reads to the reference (“Ref.”) sequence. Columns of high-quality mismatches (red) identify SNPs. A stretch of nucleotides without overlapping reads defined a target for de novo assembly (gray shading). Masked mismatches are highlighted in yellow. (B) Targeted-assembly derived Bur-0 contig for the same region, with reads added from the pool of unmapped (leftover) reads (green). Flanking SNPs identified in the mapping were recovered in the assembly, as was a complex sequence, which included two adjacent insertions and four SNPs in Bur-0 compared with the reference. The Bur-0 sequence was validated by PCR amplification and dideoxy sequencing. Mismatches to the contig sequence are highlighted in light purple.
We aligned each contig to the homologous target in the reference genome. At this step, we further removed contig sequences for which alignments did not span the PR or low-coverage-region endpoints (see Methods). Genome-wide, 7396 (3525) contigs passed all filter criteria for Bur-0 (Tsu-1). The longest deletion was 641 bp, and the longest insertion was 408 bp. As assessed against the Bur-0 dideoxy data (Nordborg et al. 2005; Clark et al. 2007), 27% of the regions selected for targeted de novo assembly were closed, and for each of 80 contigs that overlapped the dideoxy data by at least 100 bp, the contig sequence was identical to the dideoxy sequence. Two additional sets of 96 short (≤250 bp) and 96 long (>250 bp) randomly chosen assemblies were validated by PCR and dideoxy sequencing (Supplemental Methods). All 188 successfully amplified and sequenced assemblies were found to be completely correct (Supplemental Table S7). Combined, the 268 validated assemblies revealed 287 indels. The longest validated deletion was 222 bp, and the longest insertion was 124 bp.
Genome-wide, for all but one of the 10,921 Bur-0 and Tsu-1 contigs, one or more sequence differences were observed relative to the reference genome. A large fraction of the targeted assemblies harbored polymorphisms inaccessible to our mapping strategy; in Bur-0 (Tsu-1) we identified 3597 (1690) deletions and 2989 (1257) insertions >3 bp (Supplemental Table S8). As expected, more deletions were identified, as de novo assembly of inserted sequences is a more complex task. In addition, the assemblies contained 13,599 (6997) SNPs and 4214 (2033) 1- to 3-bp indels that had not been predicted from the aligned reads alone (Supplemental Table S8).
The success of targeted assembly is expected to depend on coverage depth. This likely explains the higher number of contigs and polymorphisms detected for Bur-0, with ∼25-fold coverage, than for Tsu-1, with ∼18-fold coverage (Table 1).
When the targeted assembly method was applied to the Col-0 data, only 20 contigs passed the quality filters. The “assembly target regions” for Col-0 likely resulted from stochastic differences in coverage (i.e., no reads were present to contribute to assemblies). The coverage depths for Col-0 and Tsu-1 were similar, but 170 times as many assemblies were obtained for Tsu-1, indicating the robustness of our method.
Detection of duplications
Earlier work with microarrays has suggested that A. thaliana accessions vary substantially in copy number for many sequences (Borevitz et al. 2003, 2007). These finding are generally consistent with the high fraction of tandemly duplicated sequences observed in the Col-0 reference (The Arabidopsis Genome Initiative 2000), and with targeted sequencing of genomic clones from divergent accessions (e.g., Noël et al. 1999; Kroymann et al. 2003; Bomblies et al. 2007). In short-read data, higher-than-expected coverage can indicate such variants, and read coverage has been used as a metric for region-specific copy number in C. elegans (Hillier et al. 2008).
To identify sequences present in two or more copies relative to the reference, we identified regions of higher-than-expected coverage in which several reads supported more than one base at one or more positions (Fig. 4; Emrich et al. 2007). First, we estimated the expected coverage per position based on sequencing bias, local repetitiveness, and sequencing depth (see Supplemental Methods). The correlation between the expected coverage and the observed coverage is significant for Col-0 (Spearman’s test, ρ = 0.82, P < 10−10). Second, we used this estimated value to segment the genome into regions of contiguous, nonrepetitive positions of at least 250 bp for which the ratio of observed to expected coverage at all positions was between 1.2 and 3 (see Supplemental Methods). For Bur-0, 554 kb were included in these regions, and 499 kb for Tsu-1. A fraction of these is likely to result from stochastic effects or unrecognized biases in the method, as our criteria also identified a total of 199 kb in Col-0 (Supplemental Table S9). We therefore applied a second step to identify duplicated regions by searching for apparent “heterozygous” positions (Emrich et al. 2007). Here, reads from duplicated sequences map to the same location in the reference sequence, and the presence of two alternative high-quality bases at the same position defines sites that have diverged between duplicated sequences. Such copy variable positions (CVPs; see Supplemental Methods) were strongly overrepresented in the regions of higher-than-expected coverage in both Bur-0 and Tsu-1 (χ2-test, P < 10−10 for both Bur-0 and Tsu-1). The presence of two alternative bases within regions of elevated coverage was also readily apparent in resequencing array data (Fig. 4; Supplemental Fig. S9; Clark et al. 2007). While 332 and 364 kb of elevated coverage regions with one or more CVPs were identified in Bur-0 and Tsu-1, 30-fold less sequence was identified in Col-0, an indication of the high specificity of our method (Supplemental Table S9).
Figure 4.

Detection of duplicated sequences using read coverage and sequence criteria. (A) Expected vs. observed read coverage in Col-0 for a region on chromosome 4 (positions are given at bottom). (B) Analogous region in Bur-0 harboring a predicted duplication. (Vertical lines) Seven CVPs in the region with elevated observed-to-expected coverage. The relative support for each base at a CVP is indicated below (pie charts). (C) Pseudotraces (see Supplemental Fig. S1 in Clark et al. [2007]) from resequencing array data, for five of the seven CVPs for which data was available (see Supplemental Methods). Compared with Col-0, double peaks are apparent in Bur-0 that match, in sequence, bases identified in the short-read data.
Effects on genes
Our SBS-based predictions allow a finer scale characterization of the types of sequences and genes that vary among accessions than was previously possible. As assessed with polymorphisms predicted from aligned reads (Table 4), sequence variation is common within translated sequences. For example, we found 168,866 nonredundant SNPs in coding sequences, of which 80,660 were predicted to change amino acids, and 651 to introduce premature stop codons (Supplemental Table S10). Consistent with the expectation of selective constraint in coding regions, 1- or 2-bp indels, which introduce frameshifts, were about 12-fold underrepresented in coding relative to noncoding sequences. However, 1839 potential frameshift variants were identified in Bur-0 or Tsu-1 relative to the Col-0 reference (Supplemental Table S10). Within genes, the relative position of premature stops or frameshift variants was strongly biased to the 3′ end of open reading frames, which would be expected to have less dramatic effects on gene products (Fig. 5).
Figure 5.

Distribution of premature stop and frameshift mutations within coding regions. The frequency of premature stop codons and frameshift mutations was increased toward the 3′ ends of coding regions. In addition, frameshift mutations were overrepresented at the 5′ end, potentially compatible with alternative splicing or alternative use of initiation codons.
We also found that 1282 (1098) of Bur-0 (Tsu-1) PRs >99 bp overlapped coding regions. Many of these likely reflect genic deletions, and in total, 1871 (1694) of 26,819 protein-coding genes were predicted to be affected by major-effect changes (premature stops, frameshifts, or long PRs) (Supplemental Table S10). The distribution of these polymorphisms is highly nonrandom, with many genes harboring two or more such variants (χ2-tests, P < 10−10 for Bur-0 and Tsu-0). Although we expect predictions of major-effect changes to have moderately higher error rates (see discussion in Clark et al. 2007), our observations indicate that many genes annotated in Col-0 are either pseudogenes in other accessions, or alternatively vary substantially in structure. Consistent with earlier studies, the distribution of major-effect changes by gene family was also nonrandom, with NBS-LRR genes involved in pathogen response harboring exceptional polymorphism levels (Supplemental Tables S11, S1; Clark et al. 2007).
Discussion
A notable finding of our work is that even with short, single-end SBS data, a substantial fraction of the genetic variation in inbred genomes can be easily identified. For small bacterial genomes, as well as for bacterial artificial chromosomes containing genomic DNA from species with larger genomes, de novo assembly with deep SBS read data is becoming practical (Butler et al. 2008; Chaisson and Pevzner 2008; Hernandez et al. 2008; Zerbino and Birney 2008). In contrast, for larger genomes, complete de novo assembly with short single-end SBS reads is currently not feasible, although detection of polymorphisms from aligned reads has been reported (Hillier et al. 2008). Employing Illumina SBS data, we have developed efficient methods to map short SBS reads and to detect both SNPs and 1- to 3-bp indels from the resulting read alignments with high specificity and sensitivity. Further, we extended this approach by targeted de novo assembly to identify polymorphic sequences for which read alignment is not possible. Although we have studied only A. thaliana, our methods are general and should perform well with data from other species of moderate to large genome size for which homozygous (inbred) lines exist. Our methods for read alignment and for SNP and small indel prediction should be applicable, with modification, to heterozygous genomes as well, although the required coverage depth undoubtedly would be higher.
More generally, our SHORE pipeline can be readily adapted to handle longer reads, as well as paired-end read data. These improvements to SBS methods will be of particular value for sequencing A. thaliana accessions, for which average nucleotide diversity is higher than for human data (Wright and Gaut 2005). In addition, structural variation appears to be common (e.g., Borevitz et al. 2003, 2007; Zeller et al. 2008). While local deviations in coverage can be used to detect highly dissimilar sequences including deletions or duplications, paired-end SBS data will greatly simplify this task (e.g., Korbel et al. 2007). Paired-end data, as well as longer SBS read lengths, will also reduce the potential for incorrect read alignments.
Coverage requirements for evolutionary and functional studies
Strategies that employ de novo assembly of short SBS reads, even when using the targeted approach we describe, are expected to require high read coverage. Nonetheless, in aligned reads from polymorphic material, we identified SNPs and small indels with high specificity across a broad range of coverage depths. In fact, for SNPs, only modest gains in sensitivity and specificity were apparent with the homozygous material at coverage depths beyond ∼11-fold. As expected, sensitivity was reduced at lower coverage, as many positions were not covered by sufficient reads for polymorphism prediction. Even at threefold coverage, however, we could identify hundreds of thousands of sequence variants with near 99% specificity. Compared with genotyping arrays, which normally also capture only a fraction of SNPs, a disadvantage of low-fold SBS sequencing is that missing data will be randomly distributed between samples owing to the stochastic nature of read coverage. On the plus side, novel genetic variants can be easily detected with a modest investment in generating SBS data. As such, massively parallel SBS sequencing is highly applicable for cataloging sequence variants in hundreds of samples at relatively modest costs, and holds great promise for describing sequence variation for previously uncharacterized genetic material.
At the other extreme, our sequencing of the Col-0 reference accession suggests that as little as one run (∼11-fold coverage) on an Illumina Genome Analyzer instrument is sufficient to detect a small number of genetic differences when a sample is nearly identical to the reference, and hence the potential for incorrect read alignments is minimal. Indeed, SHORE has already been used to identify the causal mutations in EMS mutant lines of A. thaliana (S.O., K.S., and D.W., unpubl.).
Extensive variation in genic sequences in A. thaliana
Even with the experimental resources in A. thaliana, fine mapping and identification of causal genes and sequence variants underlying QTL is laborious. Here, we identified polymorphisms in hundreds of genes that are expected to have large effects on gene integrity. These findings are generally consistent with results from microarray studies (Borevitz et al. 2003; Clark et al. 2007). Notably, we identified premature stops and deletions, types of genetic variants already demonstrated to underlie diversity in flowering and other traits within the A. thaliana population (for review, see Mitchell-Olds and Schmitt 2006). While individual variants require validation, these major-effect changes, along with the more than 900,000 coding and noncoding polymorphisms we identified, are an immediate and rich resource for ongoing studies of phenotypic variation in Bur-0 and Tsu-1. Obtaining this information for additional accessions will greatly facilitate genetic studies in the hundreds to thousands of accessions to be sampled as part of the A. thaliana HapMap project (Kim et al. 2007), and sequencing of Bur-0 and Tsu-1 is the first step in the 1001 Genomes Project (http://1001genomes.org).
Methods
Image analysis and quality filtering
We used the Illumina SolexaPipeline version 0.2.2.5 software for image analysis, calculation of normalized per cycle intensities and prb values, and for base calling (the Firecrest and Bustard programs). We extracted the first 36 bases for all runs, except for two runs of 35 and 31 bases. Reads were excluded from further analysis by a custom quality filter if they harbored more than two violations of the chastity rule as defined in the SolexaPipeline in the first 12 bases and, at the same time, more than five violations in the first 25 bases of a read. Briefly, chastity at a given position p is defined as
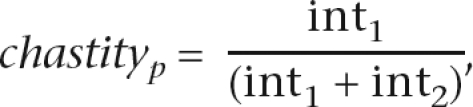 |
with int1 and int2 being the highest and second highest intensities recorded from the SolexaPipeline image analysis at p. Relative to the default Illumina SolexaPipeline read filter, our custom filter allowed us to retain reads for which a small number of bases early in the read were of low quality (e.g., owing to transient technical problems during a sequencing run). All reads with more than four ambiguous base calls were removed from subsequent analyses.
Mapping of reads to the reference sequence
Quality filtered reads for each sample were mapped against the A. thaliana reference genome (TIGR version 5) using enhanced suffix arrays (Vmatch at http://www.vmatch.de) with a seed length of 9 (parameters used were “–d –p –l 36 –seedlength 9 –s abbrev –showdesc 30 –noscore –noidentity”). Reads were aligned according to a best-match strategy by iteratively increasing the allowed number of mismatches and gaps at each round. Briefly, perfect alignments were first identified, followed by alignments through the series H (Hamming distance) = 1, L (Levenshtein distance) = 1, H = 2, L = 2, H = 3, L = 3, and H = 4. Thus, a read could be mapped with up to four mismatches without gaps, or with up to 3 bp in gaps without mismatches. Evaluation of the Hamming distance (mismatches) before the Levenshtein distance (mismatches and gaps) at each step introduced a gap penalty in making alignments (note that indels are less common than SNPs; Nordborg et al. 2005). After each step, reads that aligned to at least one location were removed from subsequent alignment attempts. At the given distances, 50 million reads could be mapped in ∼12 h on a server with 2 Quad-Core Intel Xeon 2.3 GHz processors and 32 GB memory. SHORE is readily adaptable to read mapping with greater Hamming and Levenshtein distances; however, run times increase markedly.
SNP and 1- to 3-bp indel prediction
Given our parameters for read mapping, SNPs and indels up to 3 bp can be inferred from nonperfect alignments. To detect such polymorphisms, we implemented a consensus caller that employed a position-wise majority vote to assign base calls subject to a minimum read support. If a base in a read at a position was supported by a prb value of <5 or a chastity value of <0.57, or if an “N” was called by the SolexaPipeline software, the read was not used in calculating read support or for consensus base calling. Based on our Col-0 comparison with the nonrepetitive positions of the reference genome, these thresholds removed ∼79.0% of sequencing errors while retaining ∼92.5% of all bases (see also Fig. 1; Supplemental Fig. S2).
In making SNP (indel) predictions at nonrepetitive positions, ≥80% (≥67%) of the reads used to calculate minimum read support at a position had to agree to generate a base call. Indels were conceptually treated as single sites. As read ends are prone to misalignment (Supplemental Fig. S6), and because the 3′ ends of reads have high base calling error rates (Fig. 1A), we required polymorphism predictions to be supported by the core of at least one read (the central n − 8 bp portion of a read of length n). If fewer than three read cores overlapped a position, all full-length reads overlapping the respective position were used in making the prediction subject to the requirement of one core overlap. These core criteria drastically decreased the false-positive rates.
For moderately repetitive regions, we reduced the potential for false predictions resulting from repeats by requiring ≥90% of base calls in aligned reads to agree, and read support from the cores of at least three reads.
Detecting regions of no or low read coverage
We applied two model-based methods to detect regions of low coverage that are likely to result from sequences so dissimilar to the reference that read alignment is either impossible or error-prone. First, we identified polymorphic regions (PRs) corresponding to continuous tracts of at least eight positions for which no reads overlapped (for nonrepetitive positions), or for which at most one read overlapped (for moderately repetitive positions). To take the effects of reduced coverage of GC poor regions into account, such a region had to have a minimum GC content of 25% (see Supplemental Fig. S4 for selection of this threshold). Given the edit distances employed for read mapping, PRs are expected to correspond primarily to sequences that on average harbor at least four sequence differences every 36 bp (the read length), or that are altogether absent from the reference.
We also implemented a second algorithm to identify polymorphic, low-coverage regions that failed to meet the requirements above for inclusion as PRs. Where such regions result from clusters of SNPs, small deletions, or insertions of any size, a high frequency of mismatches in overlying alignments is expected to accompany a local coverage drop (Supplemental Fig. S6). We therefore identified positions p for which the observed read coverage for eight or more of the 18 bp upstream or downstream of p was at least twice the observed coverage at p, and for which seven of the 15 positions centered on p were covered by reads that averaged at least two mismatches in alignments. We excluded from consideration positions p for which the GC content was <25% in the surrounding 100 bp, or that corresponded to 1- to 3-bp indels or PRs as identified from the mapped data (Tables 4, 5). To construct low-coverage regions for targeted de novo assembly, adjacent positions satisfying these criteria were joined.
Targeted de novo assembly
All PRs and low-coverage regions <20% repetitive and with no oversampled positions (see Supplemental Methods) in their flanking regions were targeted for de novo assembly. Leftover reads were first subjected to an additional filter; reads were removed if they had (1) more than two bases with a chastity value <0.60 within the first 12 bases; or (2) more than eight bases flagged by our low quality-base masking (see above).
We applied the Velvet assembler (version 0.5.05; Zerbino and Birney 2008) with default parameters and without coverage or length cutoffs, using all filtered leftover reads and the mapped reads in the 100 bp on either side of all assembly targets. After assemblies had been trimmed by 6 bp at each end, they were subjected to several additional filters. For a given target, at least 80% of (and at minimum 10) reads from both the 5′ and 3′ flanking regions had to be present in the corresponding assembled contig. In addition, at least 5% of the assembled reads had to originate from the collection of leftover reads. All contigs conforming to these criteria were aligned to their target region using the global alignment program needle implemented in the EMBOSS package (gap opening penalty = 10, gap extension penalty = 0.5) (Rice et al. 2000). Alignments had to cover the entire flanking regions, except for the 11 bp at either extreme end.
Acknowledgments
We thank R. Keller for help with initial data analysis, D.R. Zerbino for updating the Velvet short read assembler to facilitate our targeted assembly approach, J. Fitz for developing the 1001 genomes data repository, and J.R. Ecker, R. O’Malley, and R. Lister for helpful discussions. This work was funded by the BMBF (ERA-PG ARABRAS and GABI-GNADE), a Gottfried Wilhelm Leibniz Award (DFG), and the Max Planck Society.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRA001168 and GenBank (http://www.ncbi.nlm.nih.gov/Genbank/index.html) under accession nos. FI160450–FI160637. Polymorphism and reference base predictions, contigs from targeted de novo assembly, and a version of the reference sequence masked for oversampled regions are available at http://1001genomes.org; polymorphism predictions are also available at TAIR (http://www.arabidopsis.org).]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.080200.108.
References
- The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Bomblies K., Lempe J., Epple P., Warthmann N., Lanz C., Dangl J.L., Weigel D. Autoimmune response as a mechanism for a Dobzhansky–Muller-type incompatibility syndrome in plants. PLoS Biol. 2007;5:e236. doi: 10.1371/journal.pbio.0050236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz J., Liang D., Plouffe D., Chang H., Zhu T., Weigel D., Berry C., Winzeler E., Chory J. Large-scale identification of single-feature polymorphisms in complex genomes. Genome Res. 2003;13:513–523. doi: 10.1101/gr.541303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz J.O., Hazen S.P., Michael T.P., Morris G.P., Baxter I.R., Hu T.T., Chen H., Werner J.D., Nordborg M., Salt D.E., et al. Genome-wide patterns of single-feature polymorphism in Arabidopsis thaliana. Proc. Natl. Acad. Sci. 2007;104:12057–12062. doi: 10.1073/pnas.0705323104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J., MacCallum I., Kleber M., Shlyakhter I., Belmonte M., Lander E., Nusbaum C., Jaffe D. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson M., Pevzner P. Short read fragment assembly of bacterial genomes. Genome Res. 2008;18:324–330. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R.M., Schweikert G., Toomajian C., Ossowski S., Zeller G., Shinn P., Warthmann N., Hu T.T., Fu G., Hinds D.A., et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007;317:338–342. doi: 10.1126/science.1138632. [DOI] [PubMed] [Google Scholar]
- Emrich S.J., Li L., Wen T.J., Yandeau-Nelson M.D., Fu Y., Guo L., Chou H.H., Aluru S., Ashlock D.A., Schnable P.S. Nearly identical paralogs: Implications for maize (Zea mays L.) genome evolution. Genetics. 2007;175:429–439. doi: 10.1534/genetics.106.064006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez D., Francois P., Farinelli L., Osteras M., Schrenzel J. De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. Genome Res. 2008;18:802–809. doi: 10.1101/gr.072033.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier L.W., Marth G.T., Quinlan A.R., Dooling D., Fewell G., Barnett D., Fox P., Glasscock J.I., Hickenbotham M., Huang W., et al. Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- Kim S., Plagnol V., Hu T.T., Toomajian C., Clark R.M., Ossowski S., Ecker J.R., Weigel D., Nordborg M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007;39:1151–1155. doi: 10.1038/ng2115. [DOI] [PubMed] [Google Scholar]
- Koornneef M., Alonso-Blanco C., Vreugdenhil D. Naturally occurring genetic variation in Arabidopsis thaliana. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2004;55:141–172. doi: 10.1146/annurev.arplant.55.031903.141605. [DOI] [PubMed] [Google Scholar]
- Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L., et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroymann J., Donnerhacke S., Schnabelrauch D., Mitchell-Olds T. Evolutionary dynamics of an Arabidopsis insect resistance quantitative trait locus. Proc. Natl. Acad. Sci. 2003;100 (Suppl. 2):14587–14592. doi: 10.1073/pnas.1734046100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell-Olds T., Schmitt J. Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature. 2006;441:947–952. doi: 10.1038/nature04878. [DOI] [PubMed] [Google Scholar]
- Noël L., Moores T.L., van der Biezen E.A., Parniske M., Daniels M.J., Parker J.E., Jones J.D. Pronounced intraspecific haplotype divergence at the RPP5 complex disease resistance locus of Arabidopsis. Plant Cell. 1999;11:2099–2112. [PMC free article] [PubMed] [Google Scholar]
- Nordborg M., Hu T.T., Ishino Y., Jhaveri J., Toomajian C., Zheng H., Bakker E., Calabrese P., Gladstone J., Goyal R., et al. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005;3:e196. doi: 10.1371/journal.pbio.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden I., Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Simon M., Loudet O., Durand S., Bérard A., Brunel D., Sennesal F.X., Durand-Tardif M., Pelletier G., Camilleri C. Quantitative trait loci mapping in five new large recombinant inbred line populations of Arabidopsis thaliana genotyped with consensus single-nucleotide polymorphism markers. Genetics. 2008;178:2253–2264. doi: 10.1534/genetics.107.083899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warthmann N., Fitz J., Weigel D. MSQT for choosing SNP assays from multiple DNA alignments. Bioinformatics. 2007;23:2784–2787. doi: 10.1093/bioinformatics/btm428. [DOI] [PubMed] [Google Scholar]
- Wright S.I., Gaut B.S. Molecular population genetics and the search for adaptive evolution in plants. Mol. Biol. Evol. 2005;22:506–519. doi: 10.1093/molbev/msi035. [DOI] [PubMed] [Google Scholar]
- Zeller G., Clark R.M., Schneeberger K., Bohlen A., Weigel D., Rätsch G. Detecting polymorphic regions in the Arabidopsis thaliana genome with resequencing microarrays. Genome Res. 2008;18:918–929. doi: 10.1101/gr.070169.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D., Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]