

Abstract
DNA methylation constitutes an important epigenetic regulation mechanism in many eukaryotes, although the extent of DNA methylation in the regulation of gene expression in the mammalian genome is poorly understood. We developed D-REAM, a genome-wide DNA methylation analysis method for tissue-dependent and differentially methylated region (T-DMR) profiling with restriction tag-mediated amplification in mouse tissues and cells. Using a mouse promoter tiling array covering a region from −6 to 2.5 kb (∼30,000 transcription start sites), we found that over 3000 T-DMRs are hypomethylated in liver compared to cerebrum. The DNA methylation profile of liver was distinct from that of kidney and spleen. This hypomethylation profile marked genes that are specifically expressed in liver, including key transcription factors such as Hnf1a and Hnf4a. Genes with T-DMRs, especially those lacking CpG islands and those with HNF-1A binding motifis in their promoters, showed good correlation between their tissue-specific expression and liver hypomethylation status. T-DMRs located downstream from their transcription start sites also showed tissue-specific gene expression. These data indicate that multilayered regulation of tissue-specific gene function could be elucidated by DNA methylation tissue profiling.
In multicellular organisms, cells, and tissues form as a result of differentiation of a single fertilized egg, and phenotypes are inherited over several cell generations without alteration in the DNA sequences. Epigenetic systems are recognized as memory systems for these inheritable gene functions and, in mammals, they comprise DNA methylation and histone modifications of chromatin. DNA methylation in tissue-dependent and differentially methylated regions (T-DMRs) is involved in expression of tissue-specific genes as well as expression of key transcription factors that constitute transcription networks governing tissue or cell specificity (Shen and Maniatis 1980; Cho et al. 2001; Imamura et al. 2001; Hattori et al. 2004b, 2007; Nishino et al. 2004). Abnormal methylation of T-DMRs has been implicated in the pathogenesis of certain diseases (Jones 2002; Ushijima 2005).
DNA methylation occurs at the cytosine residue of CpG dinucleotides, which are unevenly distributed within the mammalian genome (Bird 1980). CpG islands (CGIs) have been identified as CpG-rich regions that are associated with ∼50% of the promoter regions in the mouse genome (Bird et al. 1985; Gardiner-Garden and Frommer 1987). Previous genome-wide DNA methylation analyses, focusing on CGIs, have indicated that every cell and tissue type has a unique DNA methylation profile, comprising at least hundreds of T-DMRs (Ohgane et al. 1998; Shiota et al. 2002; Strichman-Almashanu et al. 2002), and these data suggested that a methylation profile could be used to identify cell types (Shiota 2004).
To identify genes with differentially methylated regions, several microarray technologies have been developed (Lieb et al. 2006), and microarray technology has been applied to identify aberrantly methylated regions in cancer cells and characterize cell lines such as human embryonic stem cells (Hatada et al. 2006; Keshet et al. 2006; Ordway et al. 2006; Rauch et al. 2006; Shen et al. 2006). However, the DNA methylation profiles obtained have not been directly related to gene function (Ching et al. 2005; Eckhardt et al. 2006; Khulan et al. 2006). The limited number of loci or regions available for genome-wide analysis of normal cells or tissues and the existence of method biases can affect the implementation of methylated profiles (for review, see Khulan et al. 2006).
We developed a novel, low-bias method for genome-wide DNA methylation analysis, and examined the DNA methylation profile of the promoter regions in normal mouse liver by comparing them with those of cerebrum, kidney, and spleen. The results indicate that the resultant methylation profile was implicated in tissue-specific function.
Results
Features of T-DMR profiling by restriction tag-mediated amplification
To illustrate the genome-wide mouse DNA methylation profile, we developed a method involving T-DMR profiling with restriction tag-mediated amplification (D-REAM), which combined microarray technology and modified ligation-mediated polymerase chain reaction (LM-PCR) (Fig. 1A; Supplemental Fig. S1A). D-REAM recognizes various combinations of microarrays and restriction enzymes for LM-PCR. In this study, we used the GeneChip DNA microarray, tiled with ∼4.4 million 25-nt oligomers corresponding to regions located from approximately −6 to 2.5 kb of transcription start sites (TSS) of 30,120 Ensembl mouse transcripts (Supplemental Table S1; Supplemental Fig. S2A). To screen unmethylated regions, we used HpyCH4IV, a methylation-sensitive restriction enzyme that recognizes ACGT residues. Such residues are distributed throughout the mouse genome in a less biased manner than HpaII sites, which are localized mainly at CGIs around the TSS on the promoter array (Supplemental Fig. S2B). The microarray probes covered regions comprising 10.1% of all the HpyCH4IV sites (1,751,098) in the mouse genome (Fig. 1C).
Figure 1.

DNA methylation profiles were analyzed by D-REAM. (A) Illustration of the D-REAM method. Genomic DNA was digested with methylation-sensitive restriction enzyme HpyCH4IV and amplified by modified LM-PCR (Supplemental Fig. S1). Amplified fragments (gray bars) were hybridized with mouse promoter tiling array (upper panel). Array signal intensities (vertical bars) were analyzed to identify regions corresponding to fragments in unmethylated HpyCH4IV loci. Comparison of signals from different samples enabled identification of differentially methylated regions (lower panel). HpyCH4IV loci overlapping with regions yielding differential signals were defined as T-DMRtags. (B) Agarose gel electrophoresis of undigested (lane 2), HpyCH4IV-digested (lane 3), and HpyCH4IV–TaqI-digested (lane 4) mouse liver DNA. Positions corresponding to 0.1, 0.5, 1.0, and 2.0 kbp (lanes 1,5) are indicated on one side of the gel image. (C) Venn diagram of DNA methylation status at HpyCH4IV sites in mouse liver and cerebrum. Numbers without parentheses represent numbers of HpyCH4IV sites, while Ensembl transcripts IDs are in parentheses. Outer and inner rectangles represent whole mouse genome and regions covered by the promoter tiling array, respectively. Ovals indicate unmethylated HpyCH4IV sites of liver and cerebrum identified by D-REAM. (D) Correlation of microarray probe intensities in duplicate mouse liver experiments, plotted on logarithmic axes (base 2). (E) MATscore distribution of array regions corresponding to the TaqI–TaqI fragments (gray) and HpyCH4IV-digested fragments (black). The dotted line represents the MATscore cutoff value. (F) Reliability of comparative MAT analysis. Bar-plots of MATscores of the hypomethylated regions identified by MAT (P < 10−3) using full .bpmap (Full) and subsets of .bpmap corresponding to HpyCH4IV fragments (Hpy) and TaqI–TaqI fragments (Taq). Shuffle column MATscores obtained by using both treatment and control samples containing both liver and cerebrum data from different mice. The boxes, and lines within the boxes, represent the interquartile ranges and medians of the ratios, respectively.
When the mouse genome was digested with HpyCH4IV in silico, it generated fragments with median and average sizes of 907 and 1468 bp, respectively (Supplemental Fig. S2B). Because of the hypermethylation status of mouse genome, actual mouse liver HpyCH4IV fragments were deemed too large for efficient PCR amplification. To address this issue, the D-REAM method uses TaqI, a methylation-insensitive restriction enzyme, to reduce the size of the single-digested fragments (Fig. 1B). The modified LM-PCR protocol facilitates the selective amplification of unmethylated HpyCH4IV–TaqI and HpyCH4IV–HpyCH4IV, but not TaqI–TaqI fragments (Supplemental Fig. S1A,B). The selective amplification by this process was confirmed by using fragments digested by rarely occurring restriction enzymes, such as NotI (Supplemental Fig. S1C).
To analyze the D-REAM microarray data, we applied model-based analysis of tiling arrays (MAT) (Johnson et al. 2006). MATscores represent the enrichment of unmethylated fragments and are indicative of the relative methylation status of the HpyCH4IV site. They were visualized using the Integrated Genome Browser (IGB). Intra-genomic comparison can be affected by PCR sequences or fragment length biases; however, these effects were minimized by comparing the same regions in different tissues or cells. In this study, we designated these differentially methylated HpyCH4IV sites as T-DMRtags that represent T-DMRs (Fig. 1A).
T-DMRs hypomethylated in mouse liver compared to cerebrum
D-REAM assays of liver DNA obtained from two male mice were performed, and reproducible microarray results were obtained (Fig. 1D; Supplemental Fig. S1D). The number of fragments above the MATscore cutoff value of 2.44 was larger at regions corresponding to HpyCH4IV fragments than at those corresponding to TaqI–TaqI fragments (Fig. 1E). This indicated selective amplification of fragments with unmethylated HpyCH4IV sites. At least 20.9% of HpyCH4IV sites were estimated to be unmethylated within the regions covered by the microarray (Fig. 1C). In mouse cerebra (correlation coefficient of 0.955 of the biological duplicate), MAT analysis predicted 42,430 sites of unmethylated HpyCH4IV sites (P < 10−3), and 71.9% (30,521 loci) of these sites overlapped those in the liver tissue.
In a comparative analysis of liver and cerebrum, the differences in MATscores were significantly large at regions of HpyCH4IV fragments compared to those of HpyCH4IV fragments with a shuffled combination, and to those of TaqI–TaqI fragments (Fig. 1F). A total of 3774 of differentially hypomethylated T-DMRtags were identified in liver (P < 10−3). These tags were located in neighboring 10-kb regions of the TSS for 2194 (7.28% of all IDs) Ensembl transcripts (Fig. 1C).
Positions of T-DMRs relative to the TSS
We classified the genes for these 2194 transcripts into two types, CGI and non-CGI genes, according to TSS position. CGI gene TSS are located within 1 kb from the CGI, and non-CGI genes are located further than 1 kb from the CGI. The positions of T-DMRtags were distinct using this criterion (Fig. 2A). T-DMRtags neighboring non-CGI genes were observed up to −6 kb distal to and 2.5 kb downstream from TSS, and exhibited distribution patterns similar to the probes. No correlations were observed between T-DMR distribution and CpG density (observed/expected), GC percentage, or localization of HpyCH4IV sites (Fig. 2A; Supplemental Fig. S2A). Among the CGI genes, the distribution of T-DMRs seemed to negatively correlate with CpG density and the sequence conservation score among animals, suggesting that CGIs and first exons would be T-DMR-poor regions (Fig. 2A,B).
Figure 2.

T-DMR positions depend on the genomic context. (A) Distribution of positions relative to TSS of hypomethylated T-DMRtags in liver. Upper and lower panels display distributions in non-CGI and CGI genes, respectively. The width of histogram units is 250 bp. CpG densities are indicated by blue lines. (B) Center of phastCons track regions in all CGI genes on chromosomes 5, 12, and 15 (obtained from UCSC genome browser database) plotted with a histogram unit width of 125 bp. (C) Positions of T-DMRtags on liver-specific non-CGI genes with HNF1 motifs with expression levels in liver >2-fold those in cerebrum, plotted with a 500-bp histogram category width. (D) T-DMRs neighboring the Gnmt genes analyzed by COBRA. Upper panel displays the position of HpyCH4IV, the regions of restriction mapping with the analyzed HpyCH4IV site, indicated by small arrowheads on the top, and positions of T-DMRs plotted over the comparative MATscores on IGB browser from 6000 bp upstream to 2500 bp downstream from the TSS. Middle panel shows CpG density (blue) and GC percentage (gray line) in this region. Bottom panels show agarose-gel electrophoresis images of COBRA. Hypomethylated fragments converted by bisulfite treatment were resistant to HpyCH4IV digestion (+). L and C indicate liver and cerebrum samples, respectively.
T-DMRtags in eight liver-specific non-CGI genes were confirmed using combined bisulfite restriction analysis (COBRA) (Supplemental Fig. S3A,B; Xiong and Laird 1997). The degrees of methylation were not proportional to the differences in the MATscores that were observed in three sites of the Fga loci, but all 15 HpyCH4IV sites were confirmed to be differentially hypomethylated in liver. The T-DMRs were distributed from the 5′-upstream to the 3′-downstream region of TSS, similar to the distribution of all T-DMRtags in the non-CGI genes (Fig. 2A).
In liver-specific Gnmt gene, T-DMRtags were observed from −5.9 to −4.3 kb 5′ upstream and from 2.2 to 2.4 kb 3′ downstream from TSS. COBRA showed an unmethylated status of HpyCH4IV in CGIs of both liver and cerebrum, and a hypomethylated status of each T-DMRtags in liver (Fig. 2D). Similar methylation patterns of HpyCH4IV sites in five other CGI genes were confirmed by COBRA (Supplemental Fig. S3C,D).
Biased ontology annotations of genes with T-DMRs with regard to CGIs
To determine features represented by genes with T-DMRs hypomethylated in liver, we applied ontology analysis, using g:GOSt in the g:profiler web database (Table 1; Supplemental Table S2) (Reimand et al. 2007). Significantly enriched ontology terms for biological processes (BPs) (P < 10−5) were observed in 1817 g:profiler IDs that had been converted from 2194 Ensembl transcripts with T-DMRs for analysis. These BPs code for genes responsible for metabolism of organic acids and lipids and responses to defense and stress. Among these overrepresented terms, were found liver dominant tissue-specific functions present in the coagulation cascade (described in the Kyoto Encyclopedia of Genes and Genomes [KEGG] pathway database) (Kanehisa et al. 2006), and folate and methyl group metabolism (Fig. 3A,B) (Williams and Schalinske 2007).
Table 1.
Annotation analysis of genes with liver T-DMRtags

Detailed data are shown in Supplemental Table S3. χ2 tests were applied to examine the difference in the proportions of CGI and non-CGI genes for each criterion among all the genes (1817 genes) containing T-DMRtags. Percentage of non-CGI genes among all 1817 genes is 53.6%.
aOntology types of GO:BP, GO:CC, and GO:MF indicate biological process, cellular component, and molecular function in Gene Ontology criteria, respectively. KEGG represents KEGG pathway database.
bThe DAVID column indicates the overrepresentation of the terms in DAVID 2007 analysis among all genes classified into the same criterion according to the position of CGIs. NC, non-CGI genes; CGI, CGI genes.
Figure 3.

Genes with T-DMRtags (gray) in the complement and coagulation cascade in the modified KEGG pathway map (ID 04610) (A), and in the folate and methyl group metabolism pathway (Williams and Schalinske 2007) (B). (C) Transcription factor network for liver-specific gene expression. Arrows indicate that the gene expression is controlled by transcription factors. Dotted lines represent molecular interaction between factors.
The ratios of CGI genes to non-CGI genes were significantly biased among some ontology terms (P < 5 × 10−2, χ2 test), with non-CGI genes overrepresented among the genes encoding proteins exported to extracellular regions (Table 1). To avoid preexisting bias in the ontology terms in our criteria, we analyzed them using functional annotation tools in the DAVID Bioinformatics Resources 2007 website (Huang da et al. 2007) using lists of CGI or non-CGI genes as background. Most terms observed in the g:GOSt analysis were significantly overrepresented in the non-CGI genes with T-DMRs compared to CGI genes with T-DMRs. Among the CGI genes, only mitochondrial genes were significantly overrepresented, and one of the identified mitochondrial CGI genes with T-DMRs was Cpt2 (Supplemental Fig. S3C).
Genes encoding transcription factors responsible for expression of liver genes with T-DMRs
Transcription factor motifs were analyzed by referring to the MAPPER database in the 1-kb 5′-upstream region of TSS (Marinescu et al. 2005). When non-CGI genes with T-DMRs were analyzed, we found significantly enriched motifs, including HNF1A, HNF4, and RXRa (Table 2). In contrast, there were no overrepresented motifs among CGI genes with T-DMRs.
Table 2.
Transcription factor motifs overrepresented in genes with T-DMRs among non-CGI genes
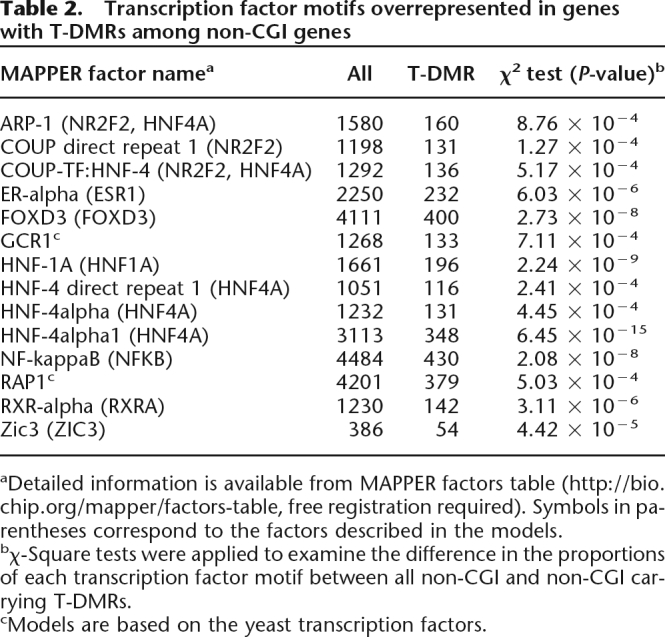
aDetailed information is available from MAPPER factors table (http://bio.chip.org/mapper/factors-table, free registration required). Symbols in parentheses correspond to the factors described in the models.
bχ-Square tests were applied to examine the difference in the proportions of each transcription factor motif between all non-CGI and non-CGI carrying T-DMRs.
cModels are based on the yeast transcription factors.
Hnf1a and Hnf4a are expressed in a tissue-specific manner and are involved in regulating liver-specific gene expression (Schrem et al. 2002). The MATscores and, hence, the methylation status of these regions were significantly different between liver and cerebrum (Fig. 4A,B). Bisulfite sequencing identified four CpGs hypomethylated in liver, corresponding to the T-DMRtags at −523 bp upstream of Hnf1a TSS (Fig. 4A). Among the six T-DMRtags of Hnf4a, two located at downstream regions of TSS and two at 5′ distal regions were present in the first exon of 0610008F07Rik, a gene in which transcription initiates from the 5-kb upstream region of Hnf4a TSS in the opposite direction. Bisulfite DNA sequencing revealed that 27 CpGs in these 5-kb regions were hypomethylated in liver but hypermethylated in cerebrum (Fig. 4B).
Figure 4.

Bisulfite sequencing of T-DMRs of liver-specific transcription factors, as Hnf1a (A), Hnf4a (B), Nr1h3 (C), Nr1i2 (D), and Rxra (E). Genomic structures are presented at the top of each figure section. The graphs in boxes toward the center in A and B represent CpG density (blue) and GC percentages (gray). The bars visible along the top of the center lines in A and B represent CpG dinucleotide positions; bars below represent HpyCH4IV sites. Boxes and arrowheads represent T-DMRs and T-DMRtags, respectively. IGB plots of comparative microarray signals corresponding to the regions in the abovementioned figures are displayed toward the bottom of the middle sections. Bisulfite sequencing data obtained for 10 isolates from liver (L) and cerebrum (C) are summarized at the bottom or side of the figure section. Open and closed circles represent unmethylated and methylated CpG, respectively.
We further investigated T-DMRs in other liver-enriched transcription factors that support liver-specific gene expression (Giguere 1999; Handschin and Meyer 2005). In the cases of Nr1h3 (Lxr) and Nr1i2 (Pregnane X receptor, Pxr), the T-DMRs in liver were localized downstream from their TSS (Fig. 4C,D). T-DMRs in the Rxra gene were localized between exons 1 and 2 (Fig. 4E), where an alternative TSS for the testis-specific transcript, which is detected in liver, is located (Brocard et al. 1996). These data suggested that T-DMRs could be involved in gene regulation of transcription factors, aiding liver-specific gene expression (Fig. 3C).
Expression of genes with HNF1 motifs and T-DMRs
Previously, 222 HNF1-binding promoters were identified in human hepatocytes by chromatin immunoprecipitation combined with DNA microarray analysis (ChIP-chip) experiments (Odom et al. 2004). Assuming that mouse orthologs of these genes would be bound to HNF1 in mouse liver, we searched for these orthologs using g:Orth in the g:Profiler web database, and observed that 43 genes of 174 orthologs (unique Entrez gene IDs) were accompanied with T-DMRs (Supplemental Fig. S4A). Tissue specificity of expression was investigated in these 43 orthologs and in 180 Entrez gene IDs with HNF-1A motifs (TRANSFAC ID M00790) and T-DMRs (Supplemental Fig. S4B), which were selected by referring to the MAPPER database. The gene expression levels in liver were more than twofold of those in cerebrum in 32 of 43 orthologs and in 68 of 180 genes with HNF-1A motifs (Fig. 5A). In cerebrum, 75% of genes exhibited expression levels <229.6 (gcRMA preprocessed data), which indicated that gene hypermethylation might repress these genes in cerebrum (Fig. 5B). Among CGI genes with T-DMRs, liver-specific expressed genes were significantly overrepresented in genes with HNF-1A motifs, but less significantly in those with HNF4 motifs (Fig. 5C). These data supported the conclusion that HNF1 and T-DMRs are involved in the regulation of these genes in mice.
Figure 5.

The tissue-specific DNA methylation profiles and gene expression in mouse tissues. (A) Expression levels of human gene orthologs identified by the ChIP-Chip experiment using anti-HNF1 (closed rectangles) and of genes with HNF-1A motifs (model IDs T01211 and T00368) classified by the MAPPER database (open circles). (B) The distribution of expression levels of the genes listed in A, in cerebrum and liver represented by box plots. Expression levels were box-plotted with logarithmic scale (base = 10). (C) Box plots of log ratios (base = 2) of liver and cerebrum gene expression indicate factors affecting liver-specific expression of genes with T-DMRs. P-values obtained from Wilcoxon’s matched-pair signed rank test are indicated on the top of the plot. (D) Correlation of expression between two tissues. CGI genes containing T-DMRs are divided into two groups by the position of T-DMRs: T-DMRs within 0.5 to 2.5 kb downstream from the TSS (left panel) and those outside of this region (right panel). Gene expression levels and ratios of gene expression levels are expressed with logarithmic scale of base 10 and base 2, respectively. Numbers of liver-specific genes expressed, with expression levels <1000 in cerebral cortex and at a liver:cerebral cortex level ratio of >2, in the left and right panels are 60 out of 346 and 45 out of 685, respectively. (E) K-means clustering of regions corresponding to T-DMRs by Pearson’s correlations of their MATscores. The ranges of the MATscores represented in the plot are shown at the bottom of the panels. The MATscores were obtained by MAT analysis of D-REAM data from liver, kidney, and spleen using cerebrum data as the control. (F) χ2 test for distributions of genes, classified by the expression levels, first in kidney (>1000 or not) and those in the later set divided by their expression in liver (>2-fold of those in kidney or not), in each cluster. Statistically significant distributions are shadowed in pink.
Correlations between expression levels of genes and T-DMRs in somatic tissues
We examined correlations between T-DMRs and gene expression in liver (Fig. 5C). The expression levels of genes with T-DMRs in liver were significantly higher than those in cerebrum, especially with regard to non-CGI genes. In 77 genes (Ensembl gene IDs) with HNF1-binding motifs that were expressed in liver (Fig. 2C), the T-DMR distribution profile was similar to that of all non-CGI genes (Fig. 2A).
In liver-specific CGI genes, the positions of T-DMRs were biased toward the regions 0.5–2.5 kb, which are 3′ downstream from the TSS (Fig. 5C,D). These regions corresponded to the first introns, judged by the distribution of the gene conservation index and by CpG density (Fig. 2B). These data suggested that T-DMRs in noncore promoter regions could be involved in the regulation of gene plurality.
We analyzed the DNA methylation status at T-DMRs in mouse kidney and spleen in genes with expression levels higher in liver than in cerebrum. The regions corresponding to these T-DMRs were clustered into four groups according to their MATscores of comparative D-REAM analysis, representing relative methylation status (Fig. 5E). Distinct DNA methylation profiles were observed among tissues, with spleen hypomethylated T-DMRs classified into clusters 1 and 2. Cluster 3 contained genes that hypomethylated in both liver and kidney, while cluster 4 contained genes concerned with liver-specific hypomethylation. Although the expression of these genes was repressed in both spleen and cerebrum (Supplemental Fig. S5A), the expression levels in kidney were similar to those in liver in clusters 1 and 2, exhibited a greater distribution toward kidney in cluster 3, and toward liver in cluster 4 (Fig. 5F; Supplemental Fig. S5). In the genes with HNF-1A motifs, Hnf4a was classified into cluster 3, while Nr1h3, Nr1i2, and Serpina1e were placed in cluster 4 (Supplemental Figs. S4, S5B). These data indicated that tissue specificity of gene expression in these regions would reflect patterns of T-DMRs of the genes containing tissue-specific transcription factors and their targets.
Discussion
D-REAM, performed using a high-density genome tiling array for the mouse promoter regions, revealed thousands of T-DMRs that are hypomethylated in liver and indicated that these T-DMRs represent the profiles of genes specifically expressed in liver, including those responsible for the liver phenotype and for transcription factors that regulate liver-specific gene expression.
A combination of transcription factors (e.g., HNF1 and HNF4a) is reported to confer liver-specific gene expression (Schrem et al. 2002). HNF1 affects the gene expression of HNF4a, and vice versa (Ktistaki and Talianidis 1997), and these factors, along with other liver-enriched transcription factors such as NR1H3 (LXR), NR1I2 (PXR), RXR, and PPARγ, affect each other’s expressions and functions (Geier et al. 2007). D-REAM indicated that T-DMR is involved in the expression of these transcription factors, and that many genes with HNF-1A motifs and T-DMRs are specifically expressed in liver and kidney (Figs. 3, 5F). D-REAM revealed coordinated DNA methylation in transcription factors and the target genes in somatic tissues including liver, kidney, spleen, and cerebrum, and that the combinations of them were distinct among tissues. These results indicated that the DNA methylation profile, comprising T-DMRs of transcription factors and their target genes, is responsible for tissue-specific gene expression in somatic tissues and, consequently, for their function.
Correlations between T-DMRs and transcriptional regulation have been revealed in several CGI and non-CGI genes by the previous investigations on T-DMRs focusing primarily on short core promoter regions (Imamura et al. 2001; Hattori et al. 2004a; Nishino et al. 2004). Genome-wide DNA methylation analysis of the human promoter regions (−700 to 200 bp from the TSS) suggested that the DNA methylation status of a limited number of genes correlated with transcriptional activity (Weber et al. 2005, 2007). A HELP assay, another genome-wide study, revealed plurality of T-DMRs within 1 kb in promoter regions, suggesting their involvement in tissue-specific expression (Khulan et al. 2006). In our study, D-REAM revealed that T-DMRs are localized at a few kilobases both upstream and downstream of the TSS and that the DNA methylation status of these T-DMRs correlates with the transcriptional activities of the neighboring genes. Preferential tissue-specific expression of non-CGI genes has been reported previously (Yamashita et al. 2005), but in the present study, hypomethylation and gene expression were also observed in CGI genes. Thus, the DNA methylation profile is expected to reflect the tissue-specific gene expression profile.
It has become feasible to identify the DNA methylation status of every CpG dinucleotide in plant (Cokus et al. 2008; Lister et al. 2008) and mammalian (Meissner et al. 2008) genomes; however, high redundancy in mammalian genome sequences narrows the window of analysis. All known analysis methods have bias windows, and different windows illustrate different results. In the mouse genome, the HpaII/MspI sites are concentrated in the region between −1 kb and +1 kb from the TSS. In the case of meDIP, the precipitation efficiency of methylated DNA depended on the density of CpG, whose distribution is biased (Keshet et al. 2006; Weber et al. 2007). We suggest that the different T-DMR profiles observed in this study were due to the relatively low bias of HpyCH4IV.
Microarray technology for the analysis of DNA methylation has advantages and disadvantages (Khulan et al. 2006). When compared with systems using isoschizomers, D-REAM is flexible because it enables selective amplification of DNA fragments digested with any restriction enzyme, e.g., HpyCH4IV and NotI, a methylation-sensitive enzyme with recognition sites without isoschizomers. One of the disadvantages in D-REAM is that intra-genomic comparison of MATscores between different genomic loci does not always represent differences in DNA methylation. However, a panel of profiles comprising several samples allowed us to conduct a broad intragenomic comparison due in part to the presence of numerous T-DMRs that could provide standard sample data.
A recent study revealed that the T-DMR distribution in randomly selected NotI sites was disproportionate in non-CGI loci, which were located both upstream of the TSS and in the intronic regions (Sakamoto et al. 2007). Present study demonstrates that the T-DMR distribution pattern appears to be independent of CpG density and GC content, except in regions around the TSS in CGI genes. In addition, numerous T-DMRs are localized at 3′ regions downstream from the TSS. T-DMRs were also identified in distal regions (up to 6 kb from the TSS). Furthermore, the distribution patterns of hypomethylated T-DMRs in cerebral and liver genes are similar (S. Yagi, K. Hirabayachi, T. Hirakawa, S. Sato, C. Maeda, J. Ohgane, S. Tanaka, and K. Shiota, unpubl.). These findings suggest that the function of T-DMRs might differ with respect to their positions in the genes.
In our analyses, especially those involving CGI genes, hypomethylation of T-DMRs in the 3′-regions downstream from the TSS was correlated with high gene expression levels in liver. The first exon and the first intron are hot spots of antisense RNA transcription, and antisense RNA TSS is preferentially observed in relatively long CGIs covering exon 1 and extending into intron 1 (Finocchiaro et al. 2007). Both sense and antisense RNA play a role in gene expression by affecting DNA methylation status (Sleutels et al. 2002; Pickford and Cogoni 2003; Imamura et al. 2004). ChIP-chip experiments on regions adjacent to the TSS have indicated that H3K4 methylation occurs in a 2-kb region downstream from the TSS and that the peaks in the region are located 1 kb downstream (Barski et al. 2007). In addition, histone modification has been reported to affect DNA methylation in a locus-specific manner (Ikegami et al. 2007). These data suggest that T-DMRs located in the 3′-region downstream from the TSS are involved in gene regulation.
In this study, we demonstrated T-DMR plurality to be involved in tissue-specific gene expression. DNA methylation regulates not only in gene expression, but also in other gene functions; therefore, T-DMRs identified by D-REAM could provide investigative insight into the roles of genome-wide DNA methylation. We conclude that T-DMR profiles are tissue specific and facilitate tissue identification by reflecting tissue-specific gene functions.
Methods
Mice and genomic DNA extraction
Male mice (C57BL/6NCrj, 12- to 13-wk-old mice for liver, cerebrum, and kidney; 6-wk-old for spleen) were euthanized after fasting for 16 h. Tissue samples were collected and frozen at −80°C until use. The samples (<20 mg) were thawed, homogenized, and incubated with 300 μL of lysis solution (10 mM Tris-HCl at pH 8.0, 5 mM EDTA, 200 mM NaCl, 0.2% SDS, and 200 μg/mL proteinase K) at 55°C for 30 min. Samples were extracted with a phenol/chloroform/isoamyl alcohol (PCI) mixture, incubated with RNase for 30 min, and re-extracted with PCI. DNA was precipitated with ethanol and dissolved in 20 μL of Tris-EDTA (TE) buffer (pH 8.0).
Combined bisulfite restriction analysis (COBRA) and bisulfite sequencing
Genomic DNA was digested with PstI. Digested DNA (3 μg) was denatured with 0.3 N NaOH. Sodium metabisulfite (pH 5.0) and hydroquinone were added to a final concentration of 2.0 M and 0.5 mM, respectively. The reaction mixture was incubated in the dark at 55°C for 16 h. DNA was purified using the Wizard DNA Clean-up System (Promega KK), treated with 0.3 M NaOH at 37°C for 15 min, and precipitated with ethanol. It was then dissolved in 20 μL of TE buffer (pH 8.0) and used in a concentration range of 1/100 to 1/20 for PCR analysis with Immolase Taq DNA polymerase (Bioline). During the bisulfite reaction, unmethylated CpGs are converted to TpGs, while methylated CpGs remain intact. For restriction mapping, 10% of the PCR product was digested with HpyCH4IV at 37°C overnight and electrophoresed with the undigested product (control) on a 1% agarose gel. The CpG methylation status within the HpyCH4IV restriction sites was assessed according to the proportion of cleaved fragments. For bisulfite sequencing, 50% of the PCR product was gel-extracted and subcloned into the pGEM-T easy vector (Promega KK). A minimum of 10 clones was sequenced, and the methylation status of individual CpGs was determined.
D-REAM
Genomic DNA (5 μg) was digested with HpyCH4IV (New England BioLabs) overnight. The digestion was monitored by gel electrophoresis. Digested DNA was recovered by ethanol precipitation following extraction with PCI and chloroform, and was dissolved in TE buffer (pH 8.0). Fifty nanograms of the DNA sample were ligated to the R-adaptor pair (Supplemental Table S2) using T4 DNA ligase (New England BioLabs). Following treatment with the Klenow fragment, the DNA was digested with TaqI at 65°C for at least 1 h and purified using a Microspin S-300 HR column (GE Healthcare UK Ltd.). DNA samples were then ligated to the N-adaptor pair (Supplemental Table S2) and purified using the Wizard SV Gel and PCR Clean-up System (Promega KK). PCR was performed using Immolase Taq DNA polymerase and the R18 and N18 primers in the presence of dUTP under the following conditions: denaturation at 95°C for 7 min and 20 cycles, each cycle comprising 95°C for 30 sec, 62°C for 30 sec, and 72°C for 2 min. A total of 10 μg of amplified DNA was used for microarray analysis. When NotI was used as the first methylation sensitive restriction enzyme, we used the R-adaptor pair for NotI instead of that for HpyCH4IV.
Microarray analysis was conducted using the GeneChip System (Affymetrix), and all procedures were performed according to the Affymetrix chromatin immunoprecipitation assay protocol provided by the manufacturer. DNA samples were labeled using the GeneChip WT Double-Stranded DNA Terminal Labeling Kit (Affymetrix) and hybridized with Affymetrix GeneChip mouse promoter 1.0R arrays. The arrays were stained and washed with GeneChip Fluidics Station 450 and scanned with the GeneChip 3000 7G Scanner to obtain a “.CEL” file describing the probe intensities. The instruments were operated using the GeneChip operating software version 1.4.
Bioinformatics
Data flow is summarized in Supplemental Fig. S6. To satisfy gene ID requirements of the bioinformatics analysis, we converted gene IDs under certain circumstances. MAT (Johnson et al. 2006) (bandwidth, 300 bp) was used to analyze the tiling array .CEL files and identify the hypomethylated regions based on tiling probe signals, probe sequences, and copy numbers. xMAN (Li et al. 2008) was used to remap the original tiling probes according to the mouse genome assembly of version mm8 (March 2006 build) from the UCSC genome database (Kuhn et al. 2007). A separate “.bpmap” file, containing a subset of probes for the HpyCH4IV–HpyCH4IV and HpyCH4IV–TaqI fragments, was used to verify the MAT analysis. The data were visualized using the Integrated Genome Browser (http://www.affymetrix.com/support/-developer/tools/download_igb-.affx).
Statistical analysis was performed using the R software package and BioConductor package (Gentleman et al. 2004). The tiling array package in BioConductor was used to examine the reproducibility of the microarray data. MultiExperiment Viewer (MeV in TM4 Microarray Software Suite) (http://www.tm4.org/mev.html) was used for K-means clustering of MATscores (Saeed et al. 2003). Genomic annotations, including Ensembl gene assignments (Birney et al. 2004), were obtained from the Galaxy website (http://g2.bx.psu.edu; Giardine et al. 2005). Transcriptome data were obtained from the GNF SymAtlas website (http://symatlas.gnf.org/SymAtlas/; Su et al. 2002), and annotation and ontology analyses were conducted using g:profiler (http://biit.cs.ut.ee/gprofiler/; Reimand et al. 2007), DAVID 2007 (http://niaid.abcc.ncifcrf.gov/; Huang da et al. 2007), and KEGG pathway database (http://www.genome.jp/kegg/kegg2.html; Kanehisa et al. 2006). EMBOSS (Rice et al. 2000) was applied for DNA sequence analysis, and the BIQ analyzer (Bock et al. 2005) was used to analyze the bisulfite sequencing data. Mouse gene symbols were confirmed by referring to the MGI database (http://www.informatics.jax.org/). Transcription factor motifs 1 kb upstream of TSS were analyzed on the MAPPER database website (http://bio.chip.org/mapper/; Marinescu et al. 2005).
Acknowledgments
We thank Dr. Bruce Murphy (University of Montreal) for critically reviewing the manuscript. Our work was supported by the Program for Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN), Japan, and a Grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan 20062003 (S.T.) and 15080202 (K.S.)
Footnotes
[Supplemental material is available online at www.genome.org and at http://www.vm.a.u-tokyo.ac.jp/seika/D-REAM/. The array data from this study have been submitted to ArrayExpress (http://www.ebi.ac.uk/microarray-as/ae/) under accession no. E-TABM-551.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.074070.107.
References
- Barski A., Cuddapah S., Cui K., Roh T., Schones D., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8:1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A., Taggart M., Frommer M., Miller O., Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. doi: 10.1016/0092-8674(85)90312-5. [DOI] [PubMed] [Google Scholar]
- Birney E., Andrews T., Bevan P., Caccamo M., Chen Y., Clarke L., Coates G., Cuff J., Curwen V., Cutts T., et al. An overview of Ensembl. Genome Res. 2004;14:925–928. doi: 10.1101/gr.1860604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C., Reither S., Mikeska T., Paulsen M., Walter J., Lengauer T. BiQ Analyzer: Visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21:4067–4068. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- Brocard J., Kastner P., Chambon P. Two novel RXR alpha isoforms from mouse testis. Biochem. Biophys. Res. Commun. 1996;229:211–218. doi: 10.1006/bbrc.1996.1782. [DOI] [PubMed] [Google Scholar]
- Ching T.T., Maunakea A.K., Jun P., Hong C., Zardo G., Pinkel D., Albertson D.G., Fridlyand J., Mao J.H., Shchors K., et al. Epigenome analyses using BAC microarrays identify evolutionary conservation of tissue-specific methylation of SHANK3. Nat. Genet. 2005;37:645–651. doi: 10.1038/ng1563. [DOI] [PubMed] [Google Scholar]
- Cho J., Kimura H., Minami T., Ohgane J., Hattori N., Tanaka S., Shiota K. DNA methylation regulates placental lactogen I gene expression. Endocrinology. 2001;142:3389–3396. doi: 10.1210/endo.142.8.8347. [DOI] [PubMed] [Google Scholar]
- Cokus S.J., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C.D., Pradhan S., Nelson S.F., Pellegrini M., Jacobsen S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt F., Lewin J., Cortese R., Rakyan V., Attwood J., Burger M., Burton J., Cox T., Davies R., Down T., et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finocchiaro G., Carro M., Francois S., Parise P., DiNinni V., Muller H. Localizing hotspots of antisense transcription. Nucleic Acids Res. 2007;35:1488–1500. doi: 10.1093/nar/gkm027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M., Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Geier A., Wagner M., Dietrich C.G., Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim. Biophys. Acta. 2007;1773:283–308. doi: 10.1016/j.bbamcr.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Gentleman R., Carey V., Bates D., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B., Riemer C., Hardison R., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J., et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere V. Orphan nuclear receptors: From gene to function. Endocr. Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- Handschin C., Meyer U.A. Regulatory network of lipid-sensing nuclear receptors: Roles for CAR, PXR, LXR, and FXR. Arch. Biochem. Biophys. 2005;433:387–396. doi: 10.1016/j.abb.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Hatada I., Fukasawa M., Kimura M., Morita S., Yamada K., Yoshikawa T., Yamanaka S., Endo C., Sakurada A., Sato M., et al. Genome-wide profiling of promoter methylation in human. Oncogene. 2006;25:3059–3064. doi: 10.1038/sj.onc.1209331. [DOI] [PubMed] [Google Scholar]
- Hattori N., Abe T., Hattori N., Suzuki M., Matsuyama T., Yoshida S., Li E., Shiota K. Preference of DNA methyltransferases for CpG islands in mouse embryonic stem cells. Genome Res. 2004a;14:1733–1740. doi: 10.1101/gr.2431504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori N., Nishino K., Ko Y., Hattori N., Ohgane J., Tanaka S., Shiota K. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J. Biol. Chem. 2004b;279:17063–17069. doi: 10.1074/jbc.M309002200. [DOI] [PubMed] [Google Scholar]
- Hattori N., Imao Y., Nishino K., Hattori N., Ohgane J., Yagi S., Tanaka S., Shiota K. Epigenetic regulation of Nanog gene in embryonic stem and trophoblast stem cells. Genes Cells. 2007;12:387–396. doi: 10.1111/j.1365-2443.2007.01058.x. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M.W., Lane H.C., et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K., Iwatani M., Suzuki M., Tachibana M., Shinkai Y., Tanaka S., Greally J., Yagi S., Hattori N., Shiota K. Genome-wide and locus-specific DNA hypomethylation in G9a deficient mouse embryonic stem cells. Genes Cells. 2007;12:1–11. doi: 10.1111/j.1365-2443.2006.01029.x. [DOI] [PubMed] [Google Scholar]
- Imamura T., Ohgane J., Ito S., Ogawa T., Hattori N., Tanaka S., Shiota K. CpG island of rat sphingosine kinase-1 gene: Tissue-dependent DNA methylation status and multiple alternative first exons. Genomics. 2001;76:117–125. doi: 10.1006/geno.2001.6607. [DOI] [PubMed] [Google Scholar]
- Imamura T., Yamamoto S., Ohgane J., Hattori N., Tanaka S., Shiota K. Non-coding RNA directed DNA demethylation of Sphk1 CpG island. Biochem. Biophys. Res. Commun. 2004;322:593–600. doi: 10.1016/j.bbrc.2004.07.159. [DOI] [PubMed] [Google Scholar]
- Johnson W., Li W., Meyer C., Gottardo R., Carroll J., Brown M., Liu X. Model-based analysis of tiling-arrays for ChIP-chip. Proc. Natl. Acad. Sci. 2006;103:12457–12462. doi: 10.1073/pnas.0601180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. DNA methylation and cancer. Oncogene. 2002;21:5358–5360. doi: 10.1038/sj.onc.1205597. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Hattori M., Aoki-Kinoshita K.F., Itoh M., Kawashima S., Katayama T., Araki M., Hirakawa M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet I., Schlesinger Y., Farkash S., Rand E., Hecht M., Segal E., Pikarski E., Young R., Niveleau A., Cedar H., et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 2006;38:149–153. doi: 10.1038/ng1719. [DOI] [PubMed] [Google Scholar]
- Khulan B., Thompson R., Ye K., Fazzari M., Suzuki M., Stasiek E., Figueroa M., Glass J., Chen Q., Montagna C., et al. Comparative isoschizomer profiling of cytosine methylation: The HELP assay. Genome Res. 2006;16:1046–1055. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistaki E., Talianidis I. Modulation of hepatic gene expression by hepatocyte nuclear factor 1. Science. 1997;277:109–112. doi: 10.1126/science.277.5322.109. [DOI] [PubMed] [Google Scholar]
- Kuhn R., Karolchik D., Zweig A., Trumbower H., Thomas D., Thakkapallayil A., Sugnet C., Stanke M., Smith K., Siepel A., et al. The UCSC genome browser database: Update 2007. Nucleic Acids Res. 2007;35:D668–D673. doi: 10.1093/nar/gkl928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Carroll J., Brown M., Liu X. xMAN: Extreme mapping of oligonucleotides. BMC Genomics. 2008;9:S20. doi: 10.1186/1471-2164-9-S1-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb J., Beck S., Bulyk M., Farnham P., Hattori N., Henikoff S., Liu X., Okumura K., Shiota K., Ushijima T., et al. Applying whole-genome studies of epigenetic regulation to study human disease. Cytogenet. Genome Res. 2006;114:1–15. doi: 10.1159/000091922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., O'Malley R.C., Tonti-Filippini J., Gregory B.D., Berry C.C., Millar A.H., Ecker J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinescu V.D., Kohane I.S., Riva A. The MAPPER database: A multi-genome catalog of putative transcription factor binding sites. Nucleic Acids Res. 2005;33:D91–D97. doi: 10.1093/nar/gki103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A., Mikkelsen T.S., Gu H., Wernig M., Hanna J., Sivachenko A., Zhang X., Bernstein B.E., Nusbaum C., Jaffe D.B., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K., Hattori N., Tanaka S., Shiota K. DNA methylation-mediated control of Sry gene expression in mouse gonadal development. J. Biol. Chem. 2004;279:22306–22313. doi: 10.1074/jbc.M309513200. [DOI] [PubMed] [Google Scholar]
- Odom D.T., Zizlsperger N., Gordon D.B., Bell G.W., Rinaldi N.J., Murray H.L., Volkert T.L., Schreiber J., Rolfe P.A., Gifford D.K., et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgane J., Aikawa J., Ogura A., Hattori N., Ogawa T., Shiota K. Analysis of CpG islands of trophoblast giant cells by restriction landmark genomic scanning. Dev. Genet. 1998;22:132–140. doi: 10.1002/(SICI)1520-6408(1998)22:2<132::AID-DVG3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Ordway J., Bedell J., Citek R., Nunberg A., Garrido A., Kendall R., Stevens J., Cao D., Doerge R., Korshunova Y., et al. Comprehensive DNA methylation profiling in a human cancer genome identifies novel epigenetic targets. Carcinogenesis. 2006;27:2409–2423. doi: 10.1093/carcin/bgl161. [DOI] [PubMed] [Google Scholar]
- Pickford A.S., Cogoni C. RNA-mediated gene silencing. Cell. Mol. Life Sci. 2003;60:871–882. doi: 10.1007/s00018-003-2245-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch T., Li H., Wu X., Pfeifer G. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–7947. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]
- Reimand J., Kull M., Peterson H., Hansen J., Vilo J. g:Profiler—A web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden I., Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Saeed A.I., Sharov V., White J., Li J., Liang W., Bhagabati N., Braisted J., Klapa M., Currier T., Thiagarajan M., et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Sakamoto H., Suzuki M., Abe T., Hosoyama T., Himeno E., Tanaka S., Greally J.M., Hattori N., Yagi S., Shiota K. Cell-type specific methylation profiles occuring disproportionately in CpG-less regions that delineate developmetal similarity. Genes Cells. 2007;12:1123–1132. doi: 10.1111/j.1365-2443.2007.01120.x. [DOI] [PubMed] [Google Scholar]
- Schrem H., Klempnauer J., Borlak J. Liver-enriched transcription factors in liver function and development. Part I: The hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol. Rev. 2002;54:129–158. doi: 10.1124/pr.54.1.129. [DOI] [PubMed] [Google Scholar]
- Shen C., Maniatis T. Tissue-specific DNA methylation in a cluster of rabbit beta-like globin genes. Proc. Natl. Acad. Sci. 1980;77:6634–6638. doi: 10.1073/pnas.77.11.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Chow J., Wang Z., Fan G. Abnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cells. Hum. Mol. Genet. 2006;15:2623–2635. doi: 10.1093/hmg/ddl188. [DOI] [PubMed] [Google Scholar]
- Shiota K. DNA methylation profiles of CpG islands for cellular differentiation and development in mammals. Cytogenet. Genome Res. 2004;105:325–334. doi: 10.1159/000078205. [DOI] [PubMed] [Google Scholar]
- Shiota K., Kogo Y., Ohgane J., Imamura T., Urano A., Nishino K., Tanaka S., Hattori N. Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes Cells. 2002;7:961–969. doi: 10.1046/j.1365-2443.2002.00574.x. [DOI] [PubMed] [Google Scholar]
- Sleutels F., Zwart R., Barlow D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002;415:810–813. doi: 10.1038/415810a. [DOI] [PubMed] [Google Scholar]
- Strichman-Almashanu L.Z., Lee R.S., Onyango P.O., Perlman E., Flam F., Frieman M.B., Feinberg A.P. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 2002;12:543–554. doi: 10.1101/gr.224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su A.I., Cooke M.P., Ching K.A., Hakak Y., Walker J.R., Wiltshire T., Orth A.P., Vega R.G., Sapinoso L.M., Moqrich A., et al. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer. 2005;5:223–231. doi: 10.1038/nrc1571. [DOI] [PubMed] [Google Scholar]
- Weber M., Davies J., Wittig D., Oakeley E., Haase M., Lam W., Schübeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- Weber M., Hellmann I., Stadler M., Ramos L., Pääbo S., Rebhan M., Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- Williams K.T., Schalinske K.L. New insights into the regulation of methyl group and homocysteine metabolism. J. Nutr. 2007;137:311–314. doi: 10.1093/jn/137.2.311. [DOI] [PubMed] [Google Scholar]
- Xiong Z., Laird P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R., Suzuki Y., Sugano S., Nakai K. Genome-wide analysis reveals strong correlation between CpG islands with nearby transcription start sites of genes and their tissue specificity. Gene. 2005;350:129–136. doi: 10.1016/j.gene.2005.01.012. [DOI] [PubMed] [Google Scholar]